User login
Periprocedural Management of Chronically Anticoagulated Patients: A Practical Approach to Use of Novel Anticoagulants in Orthopedic Surgery
Chronic anticoagulation is a common preexisting condition in patients undergoing total joint arthroplasty (TJA). Atrial fibrillation (AF), the most common underlying disorder requiring chronic anticoagulation, affects more than 3 million patients in the United States—a number that is projected to increase to 16 million by 2050.1,2 Other common indications for anticoagulation are deep vein thrombosis (DVT) treatment, presence of a prosthetic heart valve, and venous thromboembolism (VTE) prevention after hip or knee arthroplasty. These patients face the additional risks of hemorrhage, persistent wound drainage, hematoma formation, transfusion requirements, periprosthetic joint infection, and longer hospital stay.1 Chronic anticoagulation traditionally has been managed with warfarin, which inhibits production of the vitamin K–dependent clotting factors II, VII, IX, and X. However, the new novel oral anticoagulants (NOACs), which target individual factors in the clotting cascade, are gaining favor as chronic anticoagulant agents because of their ease of use and improved efficacy and safety. These agents include the factor IIA inhibitor dabigatran (Pradaxa) and the direct factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis).
Management of patients at risk for thromboembolism and bleeding issues, particularly within the context of elective, urgent, or emergent orthopedic surgeries, is an evolving area. Understanding the pharmacokinetics, conventional laboratory tests, dosing, and reversal methods for NOACs is important, especially because clinical data are limited and the treatment itself can cause clinically significant harm.
In this article, we review the medical literature on these medications, their mechanism of action, and their reversal agents, and outline a practical approach for managing patients during the perioperative period.
Dabigatran
In October 2010, dabigatran became the first NOAC approved by the US Food and Drug Administration (FDA) for the prevention of arterial thromboembolic events in patients with nonvalvular AF, on the basis of the results of the RELY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial. Dabigatran is an oral factor IIA (thrombin) inhibitor. From time of ingestion, dabigatran takes 1.25 to 3 hours to reach peak plasma concentration. It has a half-life of 12 to 14 hours, is excreted predominantly by the kidneys (80%), and is renally dosed. The usual dose is 150 mg 2 times daily if creatinine clearance (CrCl) is >30 mL/minute, or 75 mg 2 times daily if CrCl is 15 to 30 mL/minute.3 Dabigatran is not recommended for patients with CrCl <15 mL/minute.
Dabigatran affects prothrombin time (PT), activated partial thromboplastin time (aPTT), ecarin clotting time, and thrombin time, with the latter 2 providing the most accurate means of monitoring appropriate drug levels.3,4 Of the tests commonly used to assess coagulation hemostasis in hospitals, normalization of thrombin time and aPTT provide the most accurate results (Table 1). The pharmacokinetics of dabigatran mandate consideration of dose, time of ingestion relative to time of blood sampling, and renal function in the assessment of coagulation hemostasis.
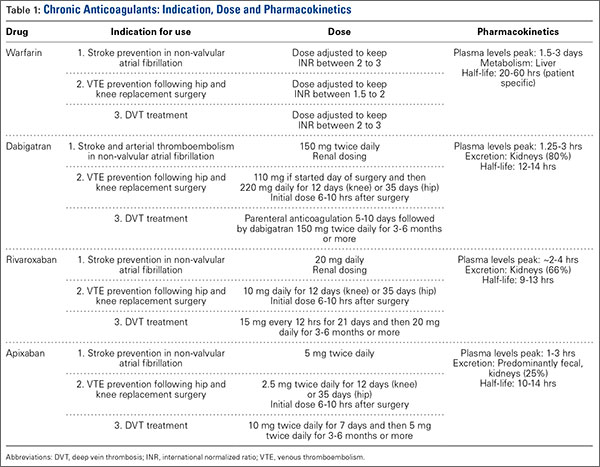
For elective surgeries, the periprocedure recommendation for patients being treated with dabigatran is to discontinue the medication 3 to 4 days before an operation if CrCl is ≥50 mL/minute, or 4 to 5 days beforehand if CrCl is <50 mL/minute.3 There is no antidote for dabigatran. In an in vitro model, activated charcoal reduced 99.9% of dabigatran absorption after recent ingestion.3 According to case reports, acute hemodialysis successfully removed 60% of the medication after 6 hours.5 In patients with end-stage renal disease, hemodialysis removed up to 68% of active dabigatran after 4 hours.3
Pernod and colleagues6 proposed that urgent surgeries can proceed if the concentration of dabigatran is ≤30 ng/mL—equivalent to normal aPTT. Their dictum was extrapolated from the data of patients who underwent elective surgeries while being treated with dabigatran, as recorded during the RELY trial. According to Pernod and colleagues,6 if aPTT is increased (probable drug level, ≥30 ng/mL), surgery should be postponed for up to 12 hours, with aPTT checked again and the process repeated if the concentration of dabigatran is still elevated and surgery can continue to be delayed. In patients who require urgent surgical interventions, we previously utilized nanofiltered activated prothrombin complex concentrate (aPCC; Feiba NF) 30 to 50 IU/kg over prothrombin complex concentrate (PCC; Kcentra or Bebulin) 25 to 50 IU/kg, as supported by in vitro and animal model studies and anecdotal case reports. However, neither aPCC nor PCC fully corrects the abnormalities evident on hemostasis tests.3,6 In October 2015, the FDA approved Idarucizumab (Praxbind), an injectable monoclonal antibody fragment that binds to dabigatran, as a reversing agent for use in urgent/emergent settings. Recommendation is to administer two 50-ml bolus infusions, each containing 2.5 g of idarucizumab, no more than 15 minutes apart.7 Additionally, hemodialysis could be discussed before surgery, with the understanding that it will take a long time to reach the threshold of 30 ng/mL in these patients (Table 2).
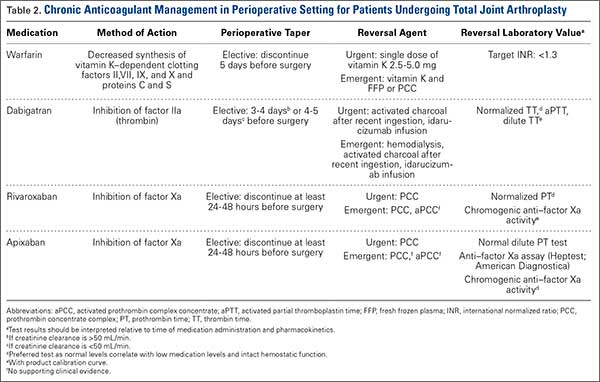
Rivaroxaban
Rivaroxaban is an oral direct factor Xa inhibitor that was initially approved in November 2011 for the prevention of stroke and systemic embolism in patients with nonvalvular AF. Since then, clinical use of rivaroxaban has been expanded to include prevention of VTE after elective hip or knee arthroplasty as well as treatment of DVT and prevention of recurrent VTE after acute DVT. In the phase 3 ROCKET AF (Rivaroxaban Once-Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) study, rivaroxaban 20 mg daily (CrCl, ≥50 mL/min) and rivaroxaban 15 mg daily (CrCl, 15-49 mL/min) were equally effective as warfarin. Compared with warfarin, rivaroxaban had a similar safety rate for bleeding and adverse events but fewer intracranial hemorrhage and fatal bleeding events.8 On the basis of the outcomes of the RECORD (Regulation of Coagulation in Orthopedic Surgery to Prevent Deep Venous Thrombosis and Pulmonary Embolism) studies comparing rivaroxaban and enoxaparin sodium, rivaroxaban 10 mg daily was approved for the prevention of VTE and pulmonary embolism after elective hip or knee arthroplasty.8
The half-life of rivaroxaban is 5 to 9 hours in the young and 11 to 13 hours in the elderly.8 As rivaroxaban takes 2 to 4 hours after ingestion to reach peak plasma concentration, it is important to know the timing and the dose taken. Because of the short half-life and rapid onset of action of this medication, bridging with another anticoagulant is not required when rivaroxaban is discontinued before surgery or initiated after surgery.8 The recommendation is to withhold rivaroxaban for 24 to 48 hours before surgery and then to administer the first postoperative dose 6 to 10 hours after surgery, or when hemostasis is achieved (Table 1).
PT is recommended for rivaroxaban detection. Conventional assays are not sensitive at low concentrations, and degree of prolongation does not reliably predict amount of medication present.3,9 However, normal PT corresponds to a drug concentration of about 30 ng/mL and is considered safe for patients undergoing surgical intervention without increased risk for bleeding.6 This recommendation was extrapolated from data in the ROCKET AF study of patients who underwent elective surgeries while on rivaroxaban.6 Commercially available chromogenic anti–factor Xa assays, used with a rivaroxaban calibration curve, are sensitive and specific for rivaroxaban plasma concentrations.3,8 However, these assays are not widely available.
If a bleeding complication occurs in a patient who is being treated with rivaroxaban, the next rivaroxaban dose should be delayed, or treatment should be discontinued, as appropriate.8 Urgency of surgery should be weighed against risk for bleeding complications on a case-by-case basis. This decision is deferred to the clinical judgment of the surgeon. In the case of a patient with severe, life-threatening bleeding or a patient who requires emergent surgery, PCC 25-50 IU/kg is the recommended reversal agent.9 Recombinant factor VIIa and aPCC have been used in experimental settings, but there is concern about the greater prothrombotic potential of these agents compared with PCC8 (Table 2).
Apixaban
Apixaban is the second factor Xa inhibitor introduced in the United States and the first to show—in the ARISTOTLE (Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation) study—efficacy superior to that of warfarin for the prevention of stroke and systemic embolism, all-cause mortality, and major bleeding. Furthermore, in the AVERROES (Apixaban Versus Acetylsalicylic Acid to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment) study, apixaban used in AF patients who were deemed not suitable for warfarin proved to be more effective than aspirin for stroke prevention, and had a similar rate of major bleeding.10 Apixaban is administered in a 5-mg dose 2 times daily. It has a half-life of 10 to 14 hours, is highly protein-bound, and has predominantly fecal excretion (27% is renal). Apixaban can prolong PT, but the correlation is nonlinear. Barrett and colleagues11 found that chromogenic anti–factor Xa assays provided the most accurate readings of apixaban plasma concentrations. Normal anti–factor Xa activity in patients being treated with apixaban suggests low drug levels and an intact hemostatic function, which are indicators of low bleeding risk with surgical intervention3 (Table 1).
Similar to other NOACs, apixaban has no antidote. In vitro testing showed that PCC improved thrombin generation when added to the blood of healthy donors who had received apixaban. Despite the lack of clinical experience, use of PCC 50 IU/kg may be reasonable for apixaban patients with severe or life-threatening bleeding3 (Table 2). Unlike dabigatran, apixaban cannot be eliminated with dialysis because of its high degree of protein binding. In nonemergent circumstances, delaying surgery 24 to 48 hours is considered effective in reducing the concentration of apixaban to a range that does not cause additional risk for bleeding.
Conclusion
Compared with warfarin, the NOACs dabigatran, rivaroxaban, and apixaban are efficacious and safe. Because of their steady pharmacokinetics, they do not require regular coagulation testing, as is the case with warfarin. These NOACs have been approved for the prevention of stroke and thromboembolic events in patients with nonvalvular AF; rivaroxaban has also been approved for VTE prevention after total hip or knee arthroplasty, for DVT treatment, and for prevention of recurrent VTE after acute DVT. Other options for VTE prophylaxis after hip and knee surgery are addressed in the guidelines issued by the American Academy of Orthopaedic Surgeons in 2011.12 As the incidence of chronic anticoagulation continues to increase among patients undergoing TJA, orthopedic surgeons need to be aware of the mechanism of action of these NOACs, as well as their pharmacokinetics and available reversal agents. Aggarwal and colleagues1 found that AF patients undergoing TJA had longer hospital stays, increased transfusion requirements, and increased risk for periprosthetic joint infection and unplanned hospital readmission.
The anticoagulation tests recommended for evaluation of hemostasis and drug reversal are normalization of aPTT for dabigatran; PT for rivaroxaban; and chromogenic anti–factor Xa activity for apixaban3 (Table 2). Although several research projects are being planned to develop an antidote for these medications, no antidote has been approved for human trials. The coagulation agents currently being used for reversal of NOACs are nonactivated PCC (Kcentra, Bebulin) and aPCC. Kcentra is a 4-factor PCC (II, VII, IX, X), and Bebulin is a 3-factor PCC (II, IX, X). Most authors recommend using 4-factor PCC 25 to 50 IU/kg. In vivo studies and animal studies have shown that nanofiltered aPCC (Feiba NF) at doses of 30 to 50 IU/kg can to some extent reverse anticoagulation in patients receiving NOACs. The current, limited data support use of reversal agent PCC for rivaroxaban and apixaban (no human studies for apixaban) and use of aPCC for dabigatran.3,6,8 Activated charcoal can be used for patients who have taken dabigatran <6 hours before presentation.3 Hemodialysis is another option for dabigatran removal. Hemodialysis, however, takes 4 to 6 hours or longer to remove about 60% of the medication (Table 2).3,5
In major orthopedic surgeries, such as TJA, bleeding is a critical concern. Using reversal agents to overcome the anticoagulation effect adds to the potential concern for thromboembolism secondary to these agents. Therefore, in cases in which surgery cannot be delayed any longer, the decision to use reversal agents should be made on a case-by-case basis. For most patients on rivaroxaban or apixaban, it is sufficient to delay for 24 to 48 hours before proceeding safely with surgery; for dabigatran, a delay of 3 to 4 days is recommended. Delay before surgery may need to be extended for the elderly and for patients with renal failure. The pharmacokinetics of these medications is summarized in Table 1.
There are no guidelines for perioperative management of patients undergoing elective, urgent, or emergent surgeries while on NOACs. As discussed, Pernod and colleagues6 proposed better perioperative management of major bleeding risks in patients receiving rivaroxaban or dabigatran. Adapting their approach, and using the data available from the medical literature, we propose a perioperative algorithm that can guide practicing orthopedic surgeons performing urgent and emergent surgeries (Figure).
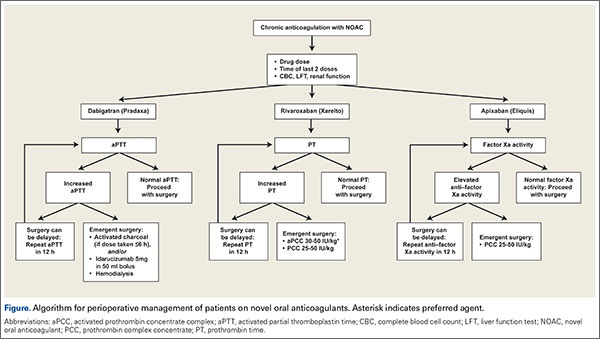
The population of patients receiving chronic anticoagulation therapy is growing, and anticoagulant and antiplatelet options are increasing in the United States and around the world. We propose a team approach for patient care, with orthopedic surgeon and cardiologist or vascular medicine specialist collaborating to ensure the safety and effectiveness of this treatment.
1. Aggarwal VK, Tischler EH, Post ZD, Kane I, Orozco FR, Ong A. Patients with atrial fibrillation undergoing total joint arthroplasty increase hospital burden. J Bone Joint Surg Am. 2013;95(17):1606-1611.
2. Curtis AB. Practice implications of the atrial fibrillation guidelines. Am J Cardiol. 2013;111(11):1660-1670.
3. Siegal DM, Crowther MA. Acute management of bleeding in patients on novel oral anticoagulants. Eur Heart J. 2013;34(7):489-498b.
4. van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost. 2010;103(6):1116-1127.
5. Lillo-Le Louët A, Wolf M, Soufir L, et al. Life-threatening bleeding in four patients with an unusual excessive response to dabigatran: implications for emergency surgery and resuscitation. Thromb Haemost. 2012;108(3):583-585.
6. Pernod G, Albaladejo P, Godier A, et al; Working Group on Perioperative Haemostasis. Management of major bleeding complications and emergency surgery in patients on long-term treatment with direct oral anticoagulants, thrombin or factor-Xa inhibitors: proposals of the Working Group on Perioperative Haemostasis (GIHP) - March 2013. Arch Cardiovasc Dis. 2013;106(6-7):382-393.
7. Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med. 2015;373(6):511-520.
8. Turpie AG, Kreutz R, Llau J, Norrving B, Haas S. Management consensus guidance for the use of rivaroxaban: an oral, direct factor Xa inhibitor. Thromb Haemost. 2012;108(5):876-886.
9. Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011;124(14):1573-1579.
10. Yates SW. Apixaban for stroke prevention in atrial fibrillation: a review of the clinical trial evidence. Hosp Pract. 2011;39(4):7-16.
11. Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost. 2010;104(6):1263-1271.
12. American Academy of Orthopaedic Surgeons. American Academy of Orthopaedic Surgeons clinical practice guideline on preventing venous thromboembolic disease in patients undergoing elective hip and knee arthroplasty. Agency for Healthcare Research and Quality website. http://www.guideline.gov/content.aspx?id=35173. Released 2007. Revised 2011. Accessed March 21, 2016.
Chronic anticoagulation is a common preexisting condition in patients undergoing total joint arthroplasty (TJA). Atrial fibrillation (AF), the most common underlying disorder requiring chronic anticoagulation, affects more than 3 million patients in the United States—a number that is projected to increase to 16 million by 2050.1,2 Other common indications for anticoagulation are deep vein thrombosis (DVT) treatment, presence of a prosthetic heart valve, and venous thromboembolism (VTE) prevention after hip or knee arthroplasty. These patients face the additional risks of hemorrhage, persistent wound drainage, hematoma formation, transfusion requirements, periprosthetic joint infection, and longer hospital stay.1 Chronic anticoagulation traditionally has been managed with warfarin, which inhibits production of the vitamin K–dependent clotting factors II, VII, IX, and X. However, the new novel oral anticoagulants (NOACs), which target individual factors in the clotting cascade, are gaining favor as chronic anticoagulant agents because of their ease of use and improved efficacy and safety. These agents include the factor IIA inhibitor dabigatran (Pradaxa) and the direct factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis).
Management of patients at risk for thromboembolism and bleeding issues, particularly within the context of elective, urgent, or emergent orthopedic surgeries, is an evolving area. Understanding the pharmacokinetics, conventional laboratory tests, dosing, and reversal methods for NOACs is important, especially because clinical data are limited and the treatment itself can cause clinically significant harm.
In this article, we review the medical literature on these medications, their mechanism of action, and their reversal agents, and outline a practical approach for managing patients during the perioperative period.
Dabigatran
In October 2010, dabigatran became the first NOAC approved by the US Food and Drug Administration (FDA) for the prevention of arterial thromboembolic events in patients with nonvalvular AF, on the basis of the results of the RELY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial. Dabigatran is an oral factor IIA (thrombin) inhibitor. From time of ingestion, dabigatran takes 1.25 to 3 hours to reach peak plasma concentration. It has a half-life of 12 to 14 hours, is excreted predominantly by the kidneys (80%), and is renally dosed. The usual dose is 150 mg 2 times daily if creatinine clearance (CrCl) is >30 mL/minute, or 75 mg 2 times daily if CrCl is 15 to 30 mL/minute.3 Dabigatran is not recommended for patients with CrCl <15 mL/minute.
Dabigatran affects prothrombin time (PT), activated partial thromboplastin time (aPTT), ecarin clotting time, and thrombin time, with the latter 2 providing the most accurate means of monitoring appropriate drug levels.3,4 Of the tests commonly used to assess coagulation hemostasis in hospitals, normalization of thrombin time and aPTT provide the most accurate results (Table 1). The pharmacokinetics of dabigatran mandate consideration of dose, time of ingestion relative to time of blood sampling, and renal function in the assessment of coagulation hemostasis.
For elective surgeries, the periprocedure recommendation for patients being treated with dabigatran is to discontinue the medication 3 to 4 days before an operation if CrCl is ≥50 mL/minute, or 4 to 5 days beforehand if CrCl is <50 mL/minute.3 There is no antidote for dabigatran. In an in vitro model, activated charcoal reduced 99.9% of dabigatran absorption after recent ingestion.3 According to case reports, acute hemodialysis successfully removed 60% of the medication after 6 hours.5 In patients with end-stage renal disease, hemodialysis removed up to 68% of active dabigatran after 4 hours.3
Pernod and colleagues6 proposed that urgent surgeries can proceed if the concentration of dabigatran is ≤30 ng/mL—equivalent to normal aPTT. Their dictum was extrapolated from the data of patients who underwent elective surgeries while being treated with dabigatran, as recorded during the RELY trial. According to Pernod and colleagues,6 if aPTT is increased (probable drug level, ≥30 ng/mL), surgery should be postponed for up to 12 hours, with aPTT checked again and the process repeated if the concentration of dabigatran is still elevated and surgery can continue to be delayed. In patients who require urgent surgical interventions, we previously utilized nanofiltered activated prothrombin complex concentrate (aPCC; Feiba NF) 30 to 50 IU/kg over prothrombin complex concentrate (PCC; Kcentra or Bebulin) 25 to 50 IU/kg, as supported by in vitro and animal model studies and anecdotal case reports. However, neither aPCC nor PCC fully corrects the abnormalities evident on hemostasis tests.3,6 In October 2015, the FDA approved Idarucizumab (Praxbind), an injectable monoclonal antibody fragment that binds to dabigatran, as a reversing agent for use in urgent/emergent settings. Recommendation is to administer two 50-ml bolus infusions, each containing 2.5 g of idarucizumab, no more than 15 minutes apart.7 Additionally, hemodialysis could be discussed before surgery, with the understanding that it will take a long time to reach the threshold of 30 ng/mL in these patients (Table 2).
Rivaroxaban
Rivaroxaban is an oral direct factor Xa inhibitor that was initially approved in November 2011 for the prevention of stroke and systemic embolism in patients with nonvalvular AF. Since then, clinical use of rivaroxaban has been expanded to include prevention of VTE after elective hip or knee arthroplasty as well as treatment of DVT and prevention of recurrent VTE after acute DVT. In the phase 3 ROCKET AF (Rivaroxaban Once-Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) study, rivaroxaban 20 mg daily (CrCl, ≥50 mL/min) and rivaroxaban 15 mg daily (CrCl, 15-49 mL/min) were equally effective as warfarin. Compared with warfarin, rivaroxaban had a similar safety rate for bleeding and adverse events but fewer intracranial hemorrhage and fatal bleeding events.8 On the basis of the outcomes of the RECORD (Regulation of Coagulation in Orthopedic Surgery to Prevent Deep Venous Thrombosis and Pulmonary Embolism) studies comparing rivaroxaban and enoxaparin sodium, rivaroxaban 10 mg daily was approved for the prevention of VTE and pulmonary embolism after elective hip or knee arthroplasty.8
The half-life of rivaroxaban is 5 to 9 hours in the young and 11 to 13 hours in the elderly.8 As rivaroxaban takes 2 to 4 hours after ingestion to reach peak plasma concentration, it is important to know the timing and the dose taken. Because of the short half-life and rapid onset of action of this medication, bridging with another anticoagulant is not required when rivaroxaban is discontinued before surgery or initiated after surgery.8 The recommendation is to withhold rivaroxaban for 24 to 48 hours before surgery and then to administer the first postoperative dose 6 to 10 hours after surgery, or when hemostasis is achieved (Table 1).
PT is recommended for rivaroxaban detection. Conventional assays are not sensitive at low concentrations, and degree of prolongation does not reliably predict amount of medication present.3,9 However, normal PT corresponds to a drug concentration of about 30 ng/mL and is considered safe for patients undergoing surgical intervention without increased risk for bleeding.6 This recommendation was extrapolated from data in the ROCKET AF study of patients who underwent elective surgeries while on rivaroxaban.6 Commercially available chromogenic anti–factor Xa assays, used with a rivaroxaban calibration curve, are sensitive and specific for rivaroxaban plasma concentrations.3,8 However, these assays are not widely available.
If a bleeding complication occurs in a patient who is being treated with rivaroxaban, the next rivaroxaban dose should be delayed, or treatment should be discontinued, as appropriate.8 Urgency of surgery should be weighed against risk for bleeding complications on a case-by-case basis. This decision is deferred to the clinical judgment of the surgeon. In the case of a patient with severe, life-threatening bleeding or a patient who requires emergent surgery, PCC 25-50 IU/kg is the recommended reversal agent.9 Recombinant factor VIIa and aPCC have been used in experimental settings, but there is concern about the greater prothrombotic potential of these agents compared with PCC8 (Table 2).
Apixaban
Apixaban is the second factor Xa inhibitor introduced in the United States and the first to show—in the ARISTOTLE (Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation) study—efficacy superior to that of warfarin for the prevention of stroke and systemic embolism, all-cause mortality, and major bleeding. Furthermore, in the AVERROES (Apixaban Versus Acetylsalicylic Acid to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment) study, apixaban used in AF patients who were deemed not suitable for warfarin proved to be more effective than aspirin for stroke prevention, and had a similar rate of major bleeding.10 Apixaban is administered in a 5-mg dose 2 times daily. It has a half-life of 10 to 14 hours, is highly protein-bound, and has predominantly fecal excretion (27% is renal). Apixaban can prolong PT, but the correlation is nonlinear. Barrett and colleagues11 found that chromogenic anti–factor Xa assays provided the most accurate readings of apixaban plasma concentrations. Normal anti–factor Xa activity in patients being treated with apixaban suggests low drug levels and an intact hemostatic function, which are indicators of low bleeding risk with surgical intervention3 (Table 1).
Similar to other NOACs, apixaban has no antidote. In vitro testing showed that PCC improved thrombin generation when added to the blood of healthy donors who had received apixaban. Despite the lack of clinical experience, use of PCC 50 IU/kg may be reasonable for apixaban patients with severe or life-threatening bleeding3 (Table 2). Unlike dabigatran, apixaban cannot be eliminated with dialysis because of its high degree of protein binding. In nonemergent circumstances, delaying surgery 24 to 48 hours is considered effective in reducing the concentration of apixaban to a range that does not cause additional risk for bleeding.
Conclusion
Compared with warfarin, the NOACs dabigatran, rivaroxaban, and apixaban are efficacious and safe. Because of their steady pharmacokinetics, they do not require regular coagulation testing, as is the case with warfarin. These NOACs have been approved for the prevention of stroke and thromboembolic events in patients with nonvalvular AF; rivaroxaban has also been approved for VTE prevention after total hip or knee arthroplasty, for DVT treatment, and for prevention of recurrent VTE after acute DVT. Other options for VTE prophylaxis after hip and knee surgery are addressed in the guidelines issued by the American Academy of Orthopaedic Surgeons in 2011.12 As the incidence of chronic anticoagulation continues to increase among patients undergoing TJA, orthopedic surgeons need to be aware of the mechanism of action of these NOACs, as well as their pharmacokinetics and available reversal agents. Aggarwal and colleagues1 found that AF patients undergoing TJA had longer hospital stays, increased transfusion requirements, and increased risk for periprosthetic joint infection and unplanned hospital readmission.
The anticoagulation tests recommended for evaluation of hemostasis and drug reversal are normalization of aPTT for dabigatran; PT for rivaroxaban; and chromogenic anti–factor Xa activity for apixaban3 (Table 2). Although several research projects are being planned to develop an antidote for these medications, no antidote has been approved for human trials. The coagulation agents currently being used for reversal of NOACs are nonactivated PCC (Kcentra, Bebulin) and aPCC. Kcentra is a 4-factor PCC (II, VII, IX, X), and Bebulin is a 3-factor PCC (II, IX, X). Most authors recommend using 4-factor PCC 25 to 50 IU/kg. In vivo studies and animal studies have shown that nanofiltered aPCC (Feiba NF) at doses of 30 to 50 IU/kg can to some extent reverse anticoagulation in patients receiving NOACs. The current, limited data support use of reversal agent PCC for rivaroxaban and apixaban (no human studies for apixaban) and use of aPCC for dabigatran.3,6,8 Activated charcoal can be used for patients who have taken dabigatran <6 hours before presentation.3 Hemodialysis is another option for dabigatran removal. Hemodialysis, however, takes 4 to 6 hours or longer to remove about 60% of the medication (Table 2).3,5
In major orthopedic surgeries, such as TJA, bleeding is a critical concern. Using reversal agents to overcome the anticoagulation effect adds to the potential concern for thromboembolism secondary to these agents. Therefore, in cases in which surgery cannot be delayed any longer, the decision to use reversal agents should be made on a case-by-case basis. For most patients on rivaroxaban or apixaban, it is sufficient to delay for 24 to 48 hours before proceeding safely with surgery; for dabigatran, a delay of 3 to 4 days is recommended. Delay before surgery may need to be extended for the elderly and for patients with renal failure. The pharmacokinetics of these medications is summarized in Table 1.
There are no guidelines for perioperative management of patients undergoing elective, urgent, or emergent surgeries while on NOACs. As discussed, Pernod and colleagues6 proposed better perioperative management of major bleeding risks in patients receiving rivaroxaban or dabigatran. Adapting their approach, and using the data available from the medical literature, we propose a perioperative algorithm that can guide practicing orthopedic surgeons performing urgent and emergent surgeries (Figure).
The population of patients receiving chronic anticoagulation therapy is growing, and anticoagulant and antiplatelet options are increasing in the United States and around the world. We propose a team approach for patient care, with orthopedic surgeon and cardiologist or vascular medicine specialist collaborating to ensure the safety and effectiveness of this treatment.
Chronic anticoagulation is a common preexisting condition in patients undergoing total joint arthroplasty (TJA). Atrial fibrillation (AF), the most common underlying disorder requiring chronic anticoagulation, affects more than 3 million patients in the United States—a number that is projected to increase to 16 million by 2050.1,2 Other common indications for anticoagulation are deep vein thrombosis (DVT) treatment, presence of a prosthetic heart valve, and venous thromboembolism (VTE) prevention after hip or knee arthroplasty. These patients face the additional risks of hemorrhage, persistent wound drainage, hematoma formation, transfusion requirements, periprosthetic joint infection, and longer hospital stay.1 Chronic anticoagulation traditionally has been managed with warfarin, which inhibits production of the vitamin K–dependent clotting factors II, VII, IX, and X. However, the new novel oral anticoagulants (NOACs), which target individual factors in the clotting cascade, are gaining favor as chronic anticoagulant agents because of their ease of use and improved efficacy and safety. These agents include the factor IIA inhibitor dabigatran (Pradaxa) and the direct factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis).
Management of patients at risk for thromboembolism and bleeding issues, particularly within the context of elective, urgent, or emergent orthopedic surgeries, is an evolving area. Understanding the pharmacokinetics, conventional laboratory tests, dosing, and reversal methods for NOACs is important, especially because clinical data are limited and the treatment itself can cause clinically significant harm.
In this article, we review the medical literature on these medications, their mechanism of action, and their reversal agents, and outline a practical approach for managing patients during the perioperative period.
Dabigatran
In October 2010, dabigatran became the first NOAC approved by the US Food and Drug Administration (FDA) for the prevention of arterial thromboembolic events in patients with nonvalvular AF, on the basis of the results of the RELY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial. Dabigatran is an oral factor IIA (thrombin) inhibitor. From time of ingestion, dabigatran takes 1.25 to 3 hours to reach peak plasma concentration. It has a half-life of 12 to 14 hours, is excreted predominantly by the kidneys (80%), and is renally dosed. The usual dose is 150 mg 2 times daily if creatinine clearance (CrCl) is >30 mL/minute, or 75 mg 2 times daily if CrCl is 15 to 30 mL/minute.3 Dabigatran is not recommended for patients with CrCl <15 mL/minute.
Dabigatran affects prothrombin time (PT), activated partial thromboplastin time (aPTT), ecarin clotting time, and thrombin time, with the latter 2 providing the most accurate means of monitoring appropriate drug levels.3,4 Of the tests commonly used to assess coagulation hemostasis in hospitals, normalization of thrombin time and aPTT provide the most accurate results (Table 1). The pharmacokinetics of dabigatran mandate consideration of dose, time of ingestion relative to time of blood sampling, and renal function in the assessment of coagulation hemostasis.
For elective surgeries, the periprocedure recommendation for patients being treated with dabigatran is to discontinue the medication 3 to 4 days before an operation if CrCl is ≥50 mL/minute, or 4 to 5 days beforehand if CrCl is <50 mL/minute.3 There is no antidote for dabigatran. In an in vitro model, activated charcoal reduced 99.9% of dabigatran absorption after recent ingestion.3 According to case reports, acute hemodialysis successfully removed 60% of the medication after 6 hours.5 In patients with end-stage renal disease, hemodialysis removed up to 68% of active dabigatran after 4 hours.3
Pernod and colleagues6 proposed that urgent surgeries can proceed if the concentration of dabigatran is ≤30 ng/mL—equivalent to normal aPTT. Their dictum was extrapolated from the data of patients who underwent elective surgeries while being treated with dabigatran, as recorded during the RELY trial. According to Pernod and colleagues,6 if aPTT is increased (probable drug level, ≥30 ng/mL), surgery should be postponed for up to 12 hours, with aPTT checked again and the process repeated if the concentration of dabigatran is still elevated and surgery can continue to be delayed. In patients who require urgent surgical interventions, we previously utilized nanofiltered activated prothrombin complex concentrate (aPCC; Feiba NF) 30 to 50 IU/kg over prothrombin complex concentrate (PCC; Kcentra or Bebulin) 25 to 50 IU/kg, as supported by in vitro and animal model studies and anecdotal case reports. However, neither aPCC nor PCC fully corrects the abnormalities evident on hemostasis tests.3,6 In October 2015, the FDA approved Idarucizumab (Praxbind), an injectable monoclonal antibody fragment that binds to dabigatran, as a reversing agent for use in urgent/emergent settings. Recommendation is to administer two 50-ml bolus infusions, each containing 2.5 g of idarucizumab, no more than 15 minutes apart.7 Additionally, hemodialysis could be discussed before surgery, with the understanding that it will take a long time to reach the threshold of 30 ng/mL in these patients (Table 2).
Rivaroxaban
Rivaroxaban is an oral direct factor Xa inhibitor that was initially approved in November 2011 for the prevention of stroke and systemic embolism in patients with nonvalvular AF. Since then, clinical use of rivaroxaban has been expanded to include prevention of VTE after elective hip or knee arthroplasty as well as treatment of DVT and prevention of recurrent VTE after acute DVT. In the phase 3 ROCKET AF (Rivaroxaban Once-Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) study, rivaroxaban 20 mg daily (CrCl, ≥50 mL/min) and rivaroxaban 15 mg daily (CrCl, 15-49 mL/min) were equally effective as warfarin. Compared with warfarin, rivaroxaban had a similar safety rate for bleeding and adverse events but fewer intracranial hemorrhage and fatal bleeding events.8 On the basis of the outcomes of the RECORD (Regulation of Coagulation in Orthopedic Surgery to Prevent Deep Venous Thrombosis and Pulmonary Embolism) studies comparing rivaroxaban and enoxaparin sodium, rivaroxaban 10 mg daily was approved for the prevention of VTE and pulmonary embolism after elective hip or knee arthroplasty.8
The half-life of rivaroxaban is 5 to 9 hours in the young and 11 to 13 hours in the elderly.8 As rivaroxaban takes 2 to 4 hours after ingestion to reach peak plasma concentration, it is important to know the timing and the dose taken. Because of the short half-life and rapid onset of action of this medication, bridging with another anticoagulant is not required when rivaroxaban is discontinued before surgery or initiated after surgery.8 The recommendation is to withhold rivaroxaban for 24 to 48 hours before surgery and then to administer the first postoperative dose 6 to 10 hours after surgery, or when hemostasis is achieved (Table 1).
PT is recommended for rivaroxaban detection. Conventional assays are not sensitive at low concentrations, and degree of prolongation does not reliably predict amount of medication present.3,9 However, normal PT corresponds to a drug concentration of about 30 ng/mL and is considered safe for patients undergoing surgical intervention without increased risk for bleeding.6 This recommendation was extrapolated from data in the ROCKET AF study of patients who underwent elective surgeries while on rivaroxaban.6 Commercially available chromogenic anti–factor Xa assays, used with a rivaroxaban calibration curve, are sensitive and specific for rivaroxaban plasma concentrations.3,8 However, these assays are not widely available.
If a bleeding complication occurs in a patient who is being treated with rivaroxaban, the next rivaroxaban dose should be delayed, or treatment should be discontinued, as appropriate.8 Urgency of surgery should be weighed against risk for bleeding complications on a case-by-case basis. This decision is deferred to the clinical judgment of the surgeon. In the case of a patient with severe, life-threatening bleeding or a patient who requires emergent surgery, PCC 25-50 IU/kg is the recommended reversal agent.9 Recombinant factor VIIa and aPCC have been used in experimental settings, but there is concern about the greater prothrombotic potential of these agents compared with PCC8 (Table 2).
Apixaban
Apixaban is the second factor Xa inhibitor introduced in the United States and the first to show—in the ARISTOTLE (Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation) study—efficacy superior to that of warfarin for the prevention of stroke and systemic embolism, all-cause mortality, and major bleeding. Furthermore, in the AVERROES (Apixaban Versus Acetylsalicylic Acid to Prevent Stroke in Atrial Fibrillation Patients Who Have Failed or Are Unsuitable for Vitamin K Antagonist Treatment) study, apixaban used in AF patients who were deemed not suitable for warfarin proved to be more effective than aspirin for stroke prevention, and had a similar rate of major bleeding.10 Apixaban is administered in a 5-mg dose 2 times daily. It has a half-life of 10 to 14 hours, is highly protein-bound, and has predominantly fecal excretion (27% is renal). Apixaban can prolong PT, but the correlation is nonlinear. Barrett and colleagues11 found that chromogenic anti–factor Xa assays provided the most accurate readings of apixaban plasma concentrations. Normal anti–factor Xa activity in patients being treated with apixaban suggests low drug levels and an intact hemostatic function, which are indicators of low bleeding risk with surgical intervention3 (Table 1).
Similar to other NOACs, apixaban has no antidote. In vitro testing showed that PCC improved thrombin generation when added to the blood of healthy donors who had received apixaban. Despite the lack of clinical experience, use of PCC 50 IU/kg may be reasonable for apixaban patients with severe or life-threatening bleeding3 (Table 2). Unlike dabigatran, apixaban cannot be eliminated with dialysis because of its high degree of protein binding. In nonemergent circumstances, delaying surgery 24 to 48 hours is considered effective in reducing the concentration of apixaban to a range that does not cause additional risk for bleeding.
Conclusion
Compared with warfarin, the NOACs dabigatran, rivaroxaban, and apixaban are efficacious and safe. Because of their steady pharmacokinetics, they do not require regular coagulation testing, as is the case with warfarin. These NOACs have been approved for the prevention of stroke and thromboembolic events in patients with nonvalvular AF; rivaroxaban has also been approved for VTE prevention after total hip or knee arthroplasty, for DVT treatment, and for prevention of recurrent VTE after acute DVT. Other options for VTE prophylaxis after hip and knee surgery are addressed in the guidelines issued by the American Academy of Orthopaedic Surgeons in 2011.12 As the incidence of chronic anticoagulation continues to increase among patients undergoing TJA, orthopedic surgeons need to be aware of the mechanism of action of these NOACs, as well as their pharmacokinetics and available reversal agents. Aggarwal and colleagues1 found that AF patients undergoing TJA had longer hospital stays, increased transfusion requirements, and increased risk for periprosthetic joint infection and unplanned hospital readmission.
The anticoagulation tests recommended for evaluation of hemostasis and drug reversal are normalization of aPTT for dabigatran; PT for rivaroxaban; and chromogenic anti–factor Xa activity for apixaban3 (Table 2). Although several research projects are being planned to develop an antidote for these medications, no antidote has been approved for human trials. The coagulation agents currently being used for reversal of NOACs are nonactivated PCC (Kcentra, Bebulin) and aPCC. Kcentra is a 4-factor PCC (II, VII, IX, X), and Bebulin is a 3-factor PCC (II, IX, X). Most authors recommend using 4-factor PCC 25 to 50 IU/kg. In vivo studies and animal studies have shown that nanofiltered aPCC (Feiba NF) at doses of 30 to 50 IU/kg can to some extent reverse anticoagulation in patients receiving NOACs. The current, limited data support use of reversal agent PCC for rivaroxaban and apixaban (no human studies for apixaban) and use of aPCC for dabigatran.3,6,8 Activated charcoal can be used for patients who have taken dabigatran <6 hours before presentation.3 Hemodialysis is another option for dabigatran removal. Hemodialysis, however, takes 4 to 6 hours or longer to remove about 60% of the medication (Table 2).3,5
In major orthopedic surgeries, such as TJA, bleeding is a critical concern. Using reversal agents to overcome the anticoagulation effect adds to the potential concern for thromboembolism secondary to these agents. Therefore, in cases in which surgery cannot be delayed any longer, the decision to use reversal agents should be made on a case-by-case basis. For most patients on rivaroxaban or apixaban, it is sufficient to delay for 24 to 48 hours before proceeding safely with surgery; for dabigatran, a delay of 3 to 4 days is recommended. Delay before surgery may need to be extended for the elderly and for patients with renal failure. The pharmacokinetics of these medications is summarized in Table 1.
There are no guidelines for perioperative management of patients undergoing elective, urgent, or emergent surgeries while on NOACs. As discussed, Pernod and colleagues6 proposed better perioperative management of major bleeding risks in patients receiving rivaroxaban or dabigatran. Adapting their approach, and using the data available from the medical literature, we propose a perioperative algorithm that can guide practicing orthopedic surgeons performing urgent and emergent surgeries (Figure).
The population of patients receiving chronic anticoagulation therapy is growing, and anticoagulant and antiplatelet options are increasing in the United States and around the world. We propose a team approach for patient care, with orthopedic surgeon and cardiologist or vascular medicine specialist collaborating to ensure the safety and effectiveness of this treatment.
1. Aggarwal VK, Tischler EH, Post ZD, Kane I, Orozco FR, Ong A. Patients with atrial fibrillation undergoing total joint arthroplasty increase hospital burden. J Bone Joint Surg Am. 2013;95(17):1606-1611.
2. Curtis AB. Practice implications of the atrial fibrillation guidelines. Am J Cardiol. 2013;111(11):1660-1670.
3. Siegal DM, Crowther MA. Acute management of bleeding in patients on novel oral anticoagulants. Eur Heart J. 2013;34(7):489-498b.
4. van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost. 2010;103(6):1116-1127.
5. Lillo-Le Louët A, Wolf M, Soufir L, et al. Life-threatening bleeding in four patients with an unusual excessive response to dabigatran: implications for emergency surgery and resuscitation. Thromb Haemost. 2012;108(3):583-585.
6. Pernod G, Albaladejo P, Godier A, et al; Working Group on Perioperative Haemostasis. Management of major bleeding complications and emergency surgery in patients on long-term treatment with direct oral anticoagulants, thrombin or factor-Xa inhibitors: proposals of the Working Group on Perioperative Haemostasis (GIHP) - March 2013. Arch Cardiovasc Dis. 2013;106(6-7):382-393.
7. Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med. 2015;373(6):511-520.
8. Turpie AG, Kreutz R, Llau J, Norrving B, Haas S. Management consensus guidance for the use of rivaroxaban: an oral, direct factor Xa inhibitor. Thromb Haemost. 2012;108(5):876-886.
9. Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011;124(14):1573-1579.
10. Yates SW. Apixaban for stroke prevention in atrial fibrillation: a review of the clinical trial evidence. Hosp Pract. 2011;39(4):7-16.
11. Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost. 2010;104(6):1263-1271.
12. American Academy of Orthopaedic Surgeons. American Academy of Orthopaedic Surgeons clinical practice guideline on preventing venous thromboembolic disease in patients undergoing elective hip and knee arthroplasty. Agency for Healthcare Research and Quality website. http://www.guideline.gov/content.aspx?id=35173. Released 2007. Revised 2011. Accessed March 21, 2016.
1. Aggarwal VK, Tischler EH, Post ZD, Kane I, Orozco FR, Ong A. Patients with atrial fibrillation undergoing total joint arthroplasty increase hospital burden. J Bone Joint Surg Am. 2013;95(17):1606-1611.
2. Curtis AB. Practice implications of the atrial fibrillation guidelines. Am J Cardiol. 2013;111(11):1660-1670.
3. Siegal DM, Crowther MA. Acute management of bleeding in patients on novel oral anticoagulants. Eur Heart J. 2013;34(7):489-498b.
4. van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost. 2010;103(6):1116-1127.
5. Lillo-Le Louët A, Wolf M, Soufir L, et al. Life-threatening bleeding in four patients with an unusual excessive response to dabigatran: implications for emergency surgery and resuscitation. Thromb Haemost. 2012;108(3):583-585.
6. Pernod G, Albaladejo P, Godier A, et al; Working Group on Perioperative Haemostasis. Management of major bleeding complications and emergency surgery in patients on long-term treatment with direct oral anticoagulants, thrombin or factor-Xa inhibitors: proposals of the Working Group on Perioperative Haemostasis (GIHP) - March 2013. Arch Cardiovasc Dis. 2013;106(6-7):382-393.
7. Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med. 2015;373(6):511-520.
8. Turpie AG, Kreutz R, Llau J, Norrving B, Haas S. Management consensus guidance for the use of rivaroxaban: an oral, direct factor Xa inhibitor. Thromb Haemost. 2012;108(5):876-886.
9. Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011;124(14):1573-1579.
10. Yates SW. Apixaban for stroke prevention in atrial fibrillation: a review of the clinical trial evidence. Hosp Pract. 2011;39(4):7-16.
11. Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost. 2010;104(6):1263-1271.
12. American Academy of Orthopaedic Surgeons. American Academy of Orthopaedic Surgeons clinical practice guideline on preventing venous thromboembolic disease in patients undergoing elective hip and knee arthroplasty. Agency for Healthcare Research and Quality website. http://www.guideline.gov/content.aspx?id=35173. Released 2007. Revised 2011. Accessed March 21, 2016.
CMS: MACRA impact on small/solo practices not as dramatic as predicted in regs
MACRA will not be as hard on small and solo practices as it first appeared when draft implementing regulations were published, according to Andy Slavitt, administrator of the Centers for Medicare & Medicaid Services.
Mr. Slavitt testified May 11 before the House Ways & Means Health Subcommittee to address legislators’ concerns about how the government intends to implement the Medicare Access and CHIP Reauthorization Act of 2015.

Rep. Sam Johnson (R-Tex.) expressed concern that the draft regulations published April 27 project “the greatest negative impact on payments to practices with nine or fewer doctors and the least harm to large systems with 100 or more docs.”
The calculations in the draft regulation were based on data from 2014, a year in which few small and solo practices reported quality data.
“In 2015 and subsequent years, the reporting went up,” Mr. Slavitt testified. “So at best, this table would be very, very conservative. ... Reporting is going to be far easier going forward.”
Mr. Slavitt said that the CMS will do all it can to help ensure that small and solo practices have every opportunity to participate in the both the Merit-Based Incentive Payment System (MIPS) and in advanced alternative payment models.
“The question of making sure that small groups and solo practitioners can be successful is of utmost importance. Our data show that physicians who are in small and solo practices ... do just as well as physicians that are in practices that are larger than that,” he said, adding that technical assistance specific to solo and small practices is being developed to help them transition to these value-based payment models.
Other federal officials have been spreading the same message to physicians. Speaking May 7 at the annual meeting of the American College of Physicians, Dr. Thomas A. Mason, chief medical officer in the Office of the National Coordinator for Health Information Technology, pointed out that the MACRA legislation put aside $20 million a year for 5 years beginning in 2016 to help solo and small practices transition to MIPS and APMs.
“It is specifically to help with the shift and transforming practices to measuring quality and improving quality performance,” he said in an interview. “The MACRA statute specifically calls out what the dollars need to be used for and the two points are for assisting MIPS-eligible professionals and improving their MIPS composite score as well as the transition to advanced alternative payment models.”
The U.S. Department of Health & Human Services already has begun soliciting contractors to support small and solo practices, he added.
“Direct technical assistance through this program will target eligible clinicians in individual or small group practices of 15 or fewer, focusing on those practicing in historically under resourced areas,” according to a request for proposals. “Technical assistance is defined as provider outreach and education, practice readiness, practice facilitation, health information technology (HIT) optimization, practice workflow redesign, change management, strategic planning, assisting clinicians in fully transitioning to Alternative Payment Models, and enabling partnerships.”
The federal health IT office plans to provide more information on the availability of transition assistance soon, Dr. Mason said.
Dr. Michael E. Nelson, FCCP, comments: If you are unfamiliar with MACRA, or alternatively don’t feel concerned about it, you will very likely notice a reduction in your income over the next few years. As a punishment for advocating the demise of the Sustainable Growth Rate formula (SGR), the Federal Government has come up with the Quality Payment Program (QPP), which includes the Merit-Based Incentive Payment System (MIPS) and Alternative Payment Models (APM). The Physician Quality Reporting System (PQRS), Value-Based Payment Modifier (VM) and Medicare Electronic Health Record (EHR Meaningful Use) are being morphed into the Merit-based Incentive Payment System (MIPS). If you don’t like this, you may choose an Alternative Payment Model (APM). You may use either of these as an Eligible Professional (EP). These programs are being phased in between 2015 and 2021. If all of these eponyms have looked like gibberish to you, I would encourage you to go to the CMS website, Google, Facebook, or whatever information source you use and self-educate.
Dr. Michael E. Nelson, FCCP, comments: If you are unfamiliar with MACRA, or alternatively don’t feel concerned about it, you will very likely notice a reduction in your income over the next few years. As a punishment for advocating the demise of the Sustainable Growth Rate formula (SGR), the Federal Government has come up with the Quality Payment Program (QPP), which includes the Merit-Based Incentive Payment System (MIPS) and Alternative Payment Models (APM). The Physician Quality Reporting System (PQRS), Value-Based Payment Modifier (VM) and Medicare Electronic Health Record (EHR Meaningful Use) are being morphed into the Merit-based Incentive Payment System (MIPS). If you don’t like this, you may choose an Alternative Payment Model (APM). You may use either of these as an Eligible Professional (EP). These programs are being phased in between 2015 and 2021. If all of these eponyms have looked like gibberish to you, I would encourage you to go to the CMS website, Google, Facebook, or whatever information source you use and self-educate.
Dr. Michael E. Nelson, FCCP, comments: If you are unfamiliar with MACRA, or alternatively don’t feel concerned about it, you will very likely notice a reduction in your income over the next few years. As a punishment for advocating the demise of the Sustainable Growth Rate formula (SGR), the Federal Government has come up with the Quality Payment Program (QPP), which includes the Merit-Based Incentive Payment System (MIPS) and Alternative Payment Models (APM). The Physician Quality Reporting System (PQRS), Value-Based Payment Modifier (VM) and Medicare Electronic Health Record (EHR Meaningful Use) are being morphed into the Merit-based Incentive Payment System (MIPS). If you don’t like this, you may choose an Alternative Payment Model (APM). You may use either of these as an Eligible Professional (EP). These programs are being phased in between 2015 and 2021. If all of these eponyms have looked like gibberish to you, I would encourage you to go to the CMS website, Google, Facebook, or whatever information source you use and self-educate.
MACRA will not be as hard on small and solo practices as it first appeared when draft implementing regulations were published, according to Andy Slavitt, administrator of the Centers for Medicare & Medicaid Services.
Mr. Slavitt testified May 11 before the House Ways & Means Health Subcommittee to address legislators’ concerns about how the government intends to implement the Medicare Access and CHIP Reauthorization Act of 2015.
Rep. Sam Johnson (R-Tex.) expressed concern that the draft regulations published April 27 project “the greatest negative impact on payments to practices with nine or fewer doctors and the least harm to large systems with 100 or more docs.”
The calculations in the draft regulation were based on data from 2014, a year in which few small and solo practices reported quality data.
“In 2015 and subsequent years, the reporting went up,” Mr. Slavitt testified. “So at best, this table would be very, very conservative. ... Reporting is going to be far easier going forward.”
Mr. Slavitt said that the CMS will do all it can to help ensure that small and solo practices have every opportunity to participate in the both the Merit-Based Incentive Payment System (MIPS) and in advanced alternative payment models.
“The question of making sure that small groups and solo practitioners can be successful is of utmost importance. Our data show that physicians who are in small and solo practices ... do just as well as physicians that are in practices that are larger than that,” he said, adding that technical assistance specific to solo and small practices is being developed to help them transition to these value-based payment models.
Other federal officials have been spreading the same message to physicians. Speaking May 7 at the annual meeting of the American College of Physicians, Dr. Thomas A. Mason, chief medical officer in the Office of the National Coordinator for Health Information Technology, pointed out that the MACRA legislation put aside $20 million a year for 5 years beginning in 2016 to help solo and small practices transition to MIPS and APMs.
“It is specifically to help with the shift and transforming practices to measuring quality and improving quality performance,” he said in an interview. “The MACRA statute specifically calls out what the dollars need to be used for and the two points are for assisting MIPS-eligible professionals and improving their MIPS composite score as well as the transition to advanced alternative payment models.”
The U.S. Department of Health & Human Services already has begun soliciting contractors to support small and solo practices, he added.
“Direct technical assistance through this program will target eligible clinicians in individual or small group practices of 15 or fewer, focusing on those practicing in historically under resourced areas,” according to a request for proposals. “Technical assistance is defined as provider outreach and education, practice readiness, practice facilitation, health information technology (HIT) optimization, practice workflow redesign, change management, strategic planning, assisting clinicians in fully transitioning to Alternative Payment Models, and enabling partnerships.”
The federal health IT office plans to provide more information on the availability of transition assistance soon, Dr. Mason said.
MACRA will not be as hard on small and solo practices as it first appeared when draft implementing regulations were published, according to Andy Slavitt, administrator of the Centers for Medicare & Medicaid Services.
Mr. Slavitt testified May 11 before the House Ways & Means Health Subcommittee to address legislators’ concerns about how the government intends to implement the Medicare Access and CHIP Reauthorization Act of 2015.
Rep. Sam Johnson (R-Tex.) expressed concern that the draft regulations published April 27 project “the greatest negative impact on payments to practices with nine or fewer doctors and the least harm to large systems with 100 or more docs.”
The calculations in the draft regulation were based on data from 2014, a year in which few small and solo practices reported quality data.
“In 2015 and subsequent years, the reporting went up,” Mr. Slavitt testified. “So at best, this table would be very, very conservative. ... Reporting is going to be far easier going forward.”
Mr. Slavitt said that the CMS will do all it can to help ensure that small and solo practices have every opportunity to participate in the both the Merit-Based Incentive Payment System (MIPS) and in advanced alternative payment models.
“The question of making sure that small groups and solo practitioners can be successful is of utmost importance. Our data show that physicians who are in small and solo practices ... do just as well as physicians that are in practices that are larger than that,” he said, adding that technical assistance specific to solo and small practices is being developed to help them transition to these value-based payment models.
Other federal officials have been spreading the same message to physicians. Speaking May 7 at the annual meeting of the American College of Physicians, Dr. Thomas A. Mason, chief medical officer in the Office of the National Coordinator for Health Information Technology, pointed out that the MACRA legislation put aside $20 million a year for 5 years beginning in 2016 to help solo and small practices transition to MIPS and APMs.
“It is specifically to help with the shift and transforming practices to measuring quality and improving quality performance,” he said in an interview. “The MACRA statute specifically calls out what the dollars need to be used for and the two points are for assisting MIPS-eligible professionals and improving their MIPS composite score as well as the transition to advanced alternative payment models.”
The U.S. Department of Health & Human Services already has begun soliciting contractors to support small and solo practices, he added.
“Direct technical assistance through this program will target eligible clinicians in individual or small group practices of 15 or fewer, focusing on those practicing in historically under resourced areas,” according to a request for proposals. “Technical assistance is defined as provider outreach and education, practice readiness, practice facilitation, health information technology (HIT) optimization, practice workflow redesign, change management, strategic planning, assisting clinicians in fully transitioning to Alternative Payment Models, and enabling partnerships.”
The federal health IT office plans to provide more information on the availability of transition assistance soon, Dr. Mason said.

Death Rates for Brain Cancer Trend Downward
Brain cancer rates have declined slightly in recent years, according to the Annual Report to the Nation on the Status of Cancer, 1975-2012, which tracks trends in cancer incidence and deaths in the U.S.
Between 2003 and 2012, brain cancer was 1 of 7 common cancers for which incidence rates dropped among men. Death rates remained stable among men during that time for melanoma and cancers of the bladder, brain, oral cavity, and pharynx. Between 2003 and 2012, brain cancer ranked 11th of the top 17 cancers for whites, 15th for blacks, 13th for Asian/Pacific Islanders (API), 14th for American Indian/Alaska Natives (AI/AN), and 13th for Hispanics.
Related: Major Cancer Death Rates Are Down
When data from 1975 through 2012 were factored in, the long-term trend was a general decline in cancer deaths for adults. Overall, cancer deaths for both sexes decreased by 1.5% per year between 2003 and 2012. For men in all ethnic and racial groups, rates of brain cancer also trended downward. The annual percent change was 4.4% between 1975 and 1977; -0.4% between 1977 and 1982, 1.3% between 1982 and 1991, -1.0% between 1991 and 2007, and 0.7% between 2007 and 2012.
Among women, death rates declined slightly overall but remained stable for brain cancer. Between 2003 and 2012, brain cancer ranked 9th among the top 17 cancers for whites, 15th for blacks, 12th for API, 14th for AI/AN, and 12th for Hispanics.
Related: Predicting Tongue Cancer Recurrence
The annual updates are the joint production of The American Cancer Society, the CDC, National Cancer Institute, and the North American Association of Central Cancer Registries. This is the 18th year the report has been published.
Source:
Ryerson AB, Eheman CR, Altekruse SF, et al. Cancer. 2016;122(9):1312-137.
doi: 10.1002/cncr.29936.
Brain cancer rates have declined slightly in recent years, according to the Annual Report to the Nation on the Status of Cancer, 1975-2012, which tracks trends in cancer incidence and deaths in the U.S.
Between 2003 and 2012, brain cancer was 1 of 7 common cancers for which incidence rates dropped among men. Death rates remained stable among men during that time for melanoma and cancers of the bladder, brain, oral cavity, and pharynx. Between 2003 and 2012, brain cancer ranked 11th of the top 17 cancers for whites, 15th for blacks, 13th for Asian/Pacific Islanders (API), 14th for American Indian/Alaska Natives (AI/AN), and 13th for Hispanics.
Related: Major Cancer Death Rates Are Down
When data from 1975 through 2012 were factored in, the long-term trend was a general decline in cancer deaths for adults. Overall, cancer deaths for both sexes decreased by 1.5% per year between 2003 and 2012. For men in all ethnic and racial groups, rates of brain cancer also trended downward. The annual percent change was 4.4% between 1975 and 1977; -0.4% between 1977 and 1982, 1.3% between 1982 and 1991, -1.0% between 1991 and 2007, and 0.7% between 2007 and 2012.
Among women, death rates declined slightly overall but remained stable for brain cancer. Between 2003 and 2012, brain cancer ranked 9th among the top 17 cancers for whites, 15th for blacks, 12th for API, 14th for AI/AN, and 12th for Hispanics.
Related: Predicting Tongue Cancer Recurrence
The annual updates are the joint production of The American Cancer Society, the CDC, National Cancer Institute, and the North American Association of Central Cancer Registries. This is the 18th year the report has been published.
Source:
Ryerson AB, Eheman CR, Altekruse SF, et al. Cancer. 2016;122(9):1312-137.
doi: 10.1002/cncr.29936.
Brain cancer rates have declined slightly in recent years, according to the Annual Report to the Nation on the Status of Cancer, 1975-2012, which tracks trends in cancer incidence and deaths in the U.S.
Between 2003 and 2012, brain cancer was 1 of 7 common cancers for which incidence rates dropped among men. Death rates remained stable among men during that time for melanoma and cancers of the bladder, brain, oral cavity, and pharynx. Between 2003 and 2012, brain cancer ranked 11th of the top 17 cancers for whites, 15th for blacks, 13th for Asian/Pacific Islanders (API), 14th for American Indian/Alaska Natives (AI/AN), and 13th for Hispanics.
Related: Major Cancer Death Rates Are Down
When data from 1975 through 2012 were factored in, the long-term trend was a general decline in cancer deaths for adults. Overall, cancer deaths for both sexes decreased by 1.5% per year between 2003 and 2012. For men in all ethnic and racial groups, rates of brain cancer also trended downward. The annual percent change was 4.4% between 1975 and 1977; -0.4% between 1977 and 1982, 1.3% between 1982 and 1991, -1.0% between 1991 and 2007, and 0.7% between 2007 and 2012.
Among women, death rates declined slightly overall but remained stable for brain cancer. Between 2003 and 2012, brain cancer ranked 9th among the top 17 cancers for whites, 15th for blacks, 12th for API, 14th for AI/AN, and 12th for Hispanics.
Related: Predicting Tongue Cancer Recurrence
The annual updates are the joint production of The American Cancer Society, the CDC, National Cancer Institute, and the North American Association of Central Cancer Registries. This is the 18th year the report has been published.
Source:
Ryerson AB, Eheman CR, Altekruse SF, et al. Cancer. 2016;122(9):1312-137.
doi: 10.1002/cncr.29936.
Make informed treatment decisions about biosimilars
In recent years federal laws have been enacted to help provide more treatment options and possibly lower costs to patients for therapeutic biological products. You have likely heard or read about “biosimilars” on the U.S. drug market. But are you ready to make informed treatment decisions and answer patients’ questions about these new drugs?
Biosimilars are not “generic biologics.” Although the shared goal of making biosimilars and generic drugs available is to provide more treatment options, there are important differences between the two approval pathways. For instance, unlike generic drugs, which are “bioequivalent” to their reference listed drug, biosimilar products can be either “biosimilar” to or “interchangeable” with their reference product. The differences may seem subtle based on the terminology, but the differences are important, and health care professionals who prescribe, dispense, or administer these products will need to understand them – as well as other aspects of these new therapies.
To help busy health care professionals better understand biosimilar and interchangeable products, the FDA has created a free 1.5-hour continuing education program titled, FDA Overview of Biosimilar Products. The course describes the important characteristics of biological products, particularly their complexity. For instance, a single molecule of a biological product, such as a monoclonal antibody, can easily be many hundreds of times larger and much more complex than that of a “small molecule” drug such as aspirin. Their increased complexity is one reason they are regulated differently than generics – and why, in the case of biosimilars, the law allows some differences from the reference product in clinically inactive components. The CE course discusses the “inherent variability” in the production of all biological products – both reference and biosimilar – and how the FDA accounts for these differences in assessing safety and efficacy of the products. The course also provides detailed definitions of important terms – such as “biosimilar” and “interchangeable” – and an overview of the standards the FDA has established for reviewing and approving biosimilar and interchangeable products. It also outlines the FDA review and approval standards for biosimilars and interchangeable products to help practitioners be assured that these products have been demonstrated to be safe and effective treatment options for their patients.
Some of our most important and expensive drugs are biological products used to treat patients who have serious medical conditions that are often life threatening and usually life altering. In April 2016, the Food and Drug Administration approved Inflectra (infliximab-dyyb) to help treat certain patients with certain gastrointestinal disorders, such as Crohn’s disease, and for other indications such as treatment of psoriasis and rheumatoid arthritis. The product is approved but not yet marketed in the United States. However, last year, Zarxio (filgrastim-sndz) became the first biosimilar actually available in the U.S. marketplace – approved to help boost white blood cell production for patients with severe neutropenia as well as for patients receiving various cancer therapies. These products are “biosimilar” to already-approved biological products called “reference products.” Inflectra is biosimilar to the reference product, Remicade (infliximab), and Zarxio is biosimilar to Neupogen (filgrastim).
Use of safe and effective biosimilar and interchangeable biological products can potentially make treatment more affordable for patients in need and improve public health. As patients begin to ask about the use of biosimilar and interchangeable products, the FDA hopes this course will help prepare health care practitioners to make informed decisions on behalf of their patients.
Dr. Christl is Associate Director for Therapeutic Biologics at the Office of New Drugs, Center for Drug Evaluation and Research, FDA.
The Food and Drug Administration’s recent approval of Inflectra (infliximab-dyyb) signals the advent of new therapeutic options in the treatment of patients with digestive diseases. However, there is still much to learn about biosimilars, their safety, and their efficacy. In the absence of sufficient clinical data, the American Gastroenterological Association has deferred taking a position on biosimilars. However, we recognize a critical need to educate gastroenterologists and their patients on this issue based on the information we have to date.
 |
Dr. Gary R. Lichtenstein |
In April 2016, the AGA Biosimilars Advisory Panel was created to determine key knowledge gaps regarding biosimilars, anticipate emerging issues around which to prepare AGA members, and recommend educational activities that address these gaps and issues. Upon review and approval by the AGA Institute Governing Board, the panel’s recommendations will be implemented by the AGA Center for Diagnostics and Therapeutics (CDT) in conjunction with other AGA committees. I am honored to chair this panel and look forward to working with my esteemed colleagues, all of whom are thought leaders in the field:
• Jean-Frederic Colombel, M.D., Mount Sinai Hospital
• Stephen B. Hanauer, M.D., AGAF, Northwestern University
• Sunanda V. Kane, M.D., MSPH, AGAF, Mayo Clinic
• Garrett Lawlor, M.D., Columbia University Medical Center
• James D. Lewis, M.D., MSCE, AGAF, University of Pennsylvania
• Loren A. Laine, M.D., AGAF, Yale University (CDT scientific advisory board liaison)
We hope to develop our initial recommendations in the next few months, recognizing that the field will continue to evolve quickly. AGA will also continue to work with the FDA and industry to ensure that the concerns of gastroenterologists are appropriately communicated and ensure that all of us are working together to improve the lives of patients with digestive diseases.
Dr. Gary R. Lichtenstein, AGAF, is chair, AGA Biosimilars Advisory Panel, and professor of medicine, Hospital of the University of Pennsylvania, Philadelphia.
The Food and Drug Administration’s recent approval of Inflectra (infliximab-dyyb) signals the advent of new therapeutic options in the treatment of patients with digestive diseases. However, there is still much to learn about biosimilars, their safety, and their efficacy. In the absence of sufficient clinical data, the American Gastroenterological Association has deferred taking a position on biosimilars. However, we recognize a critical need to educate gastroenterologists and their patients on this issue based on the information we have to date.
|
Dr. Gary R. Lichtenstein |
In April 2016, the AGA Biosimilars Advisory Panel was created to determine key knowledge gaps regarding biosimilars, anticipate emerging issues around which to prepare AGA members, and recommend educational activities that address these gaps and issues. Upon review and approval by the AGA Institute Governing Board, the panel’s recommendations will be implemented by the AGA Center for Diagnostics and Therapeutics (CDT) in conjunction with other AGA committees. I am honored to chair this panel and look forward to working with my esteemed colleagues, all of whom are thought leaders in the field:
• Jean-Frederic Colombel, M.D., Mount Sinai Hospital
• Stephen B. Hanauer, M.D., AGAF, Northwestern University
• Sunanda V. Kane, M.D., MSPH, AGAF, Mayo Clinic
• Garrett Lawlor, M.D., Columbia University Medical Center
• James D. Lewis, M.D., MSCE, AGAF, University of Pennsylvania
• Loren A. Laine, M.D., AGAF, Yale University (CDT scientific advisory board liaison)
We hope to develop our initial recommendations in the next few months, recognizing that the field will continue to evolve quickly. AGA will also continue to work with the FDA and industry to ensure that the concerns of gastroenterologists are appropriately communicated and ensure that all of us are working together to improve the lives of patients with digestive diseases.
Dr. Gary R. Lichtenstein, AGAF, is chair, AGA Biosimilars Advisory Panel, and professor of medicine, Hospital of the University of Pennsylvania, Philadelphia.
The Food and Drug Administration’s recent approval of Inflectra (infliximab-dyyb) signals the advent of new therapeutic options in the treatment of patients with digestive diseases. However, there is still much to learn about biosimilars, their safety, and their efficacy. In the absence of sufficient clinical data, the American Gastroenterological Association has deferred taking a position on biosimilars. However, we recognize a critical need to educate gastroenterologists and their patients on this issue based on the information we have to date.
|
Dr. Gary R. Lichtenstein |
In April 2016, the AGA Biosimilars Advisory Panel was created to determine key knowledge gaps regarding biosimilars, anticipate emerging issues around which to prepare AGA members, and recommend educational activities that address these gaps and issues. Upon review and approval by the AGA Institute Governing Board, the panel’s recommendations will be implemented by the AGA Center for Diagnostics and Therapeutics (CDT) in conjunction with other AGA committees. I am honored to chair this panel and look forward to working with my esteemed colleagues, all of whom are thought leaders in the field:
• Jean-Frederic Colombel, M.D., Mount Sinai Hospital
• Stephen B. Hanauer, M.D., AGAF, Northwestern University
• Sunanda V. Kane, M.D., MSPH, AGAF, Mayo Clinic
• Garrett Lawlor, M.D., Columbia University Medical Center
• James D. Lewis, M.D., MSCE, AGAF, University of Pennsylvania
• Loren A. Laine, M.D., AGAF, Yale University (CDT scientific advisory board liaison)
We hope to develop our initial recommendations in the next few months, recognizing that the field will continue to evolve quickly. AGA will also continue to work with the FDA and industry to ensure that the concerns of gastroenterologists are appropriately communicated and ensure that all of us are working together to improve the lives of patients with digestive diseases.
Dr. Gary R. Lichtenstein, AGAF, is chair, AGA Biosimilars Advisory Panel, and professor of medicine, Hospital of the University of Pennsylvania, Philadelphia.
In recent years federal laws have been enacted to help provide more treatment options and possibly lower costs to patients for therapeutic biological products. You have likely heard or read about “biosimilars” on the U.S. drug market. But are you ready to make informed treatment decisions and answer patients’ questions about these new drugs?
Biosimilars are not “generic biologics.” Although the shared goal of making biosimilars and generic drugs available is to provide more treatment options, there are important differences between the two approval pathways. For instance, unlike generic drugs, which are “bioequivalent” to their reference listed drug, biosimilar products can be either “biosimilar” to or “interchangeable” with their reference product. The differences may seem subtle based on the terminology, but the differences are important, and health care professionals who prescribe, dispense, or administer these products will need to understand them – as well as other aspects of these new therapies.
To help busy health care professionals better understand biosimilar and interchangeable products, the FDA has created a free 1.5-hour continuing education program titled, FDA Overview of Biosimilar Products. The course describes the important characteristics of biological products, particularly their complexity. For instance, a single molecule of a biological product, such as a monoclonal antibody, can easily be many hundreds of times larger and much more complex than that of a “small molecule” drug such as aspirin. Their increased complexity is one reason they are regulated differently than generics – and why, in the case of biosimilars, the law allows some differences from the reference product in clinically inactive components. The CE course discusses the “inherent variability” in the production of all biological products – both reference and biosimilar – and how the FDA accounts for these differences in assessing safety and efficacy of the products. The course also provides detailed definitions of important terms – such as “biosimilar” and “interchangeable” – and an overview of the standards the FDA has established for reviewing and approving biosimilar and interchangeable products. It also outlines the FDA review and approval standards for biosimilars and interchangeable products to help practitioners be assured that these products have been demonstrated to be safe and effective treatment options for their patients.
Some of our most important and expensive drugs are biological products used to treat patients who have serious medical conditions that are often life threatening and usually life altering. In April 2016, the Food and Drug Administration approved Inflectra (infliximab-dyyb) to help treat certain patients with certain gastrointestinal disorders, such as Crohn’s disease, and for other indications such as treatment of psoriasis and rheumatoid arthritis. The product is approved but not yet marketed in the United States. However, last year, Zarxio (filgrastim-sndz) became the first biosimilar actually available in the U.S. marketplace – approved to help boost white blood cell production for patients with severe neutropenia as well as for patients receiving various cancer therapies. These products are “biosimilar” to already-approved biological products called “reference products.” Inflectra is biosimilar to the reference product, Remicade (infliximab), and Zarxio is biosimilar to Neupogen (filgrastim).
Use of safe and effective biosimilar and interchangeable biological products can potentially make treatment more affordable for patients in need and improve public health. As patients begin to ask about the use of biosimilar and interchangeable products, the FDA hopes this course will help prepare health care practitioners to make informed decisions on behalf of their patients.
Dr. Christl is Associate Director for Therapeutic Biologics at the Office of New Drugs, Center for Drug Evaluation and Research, FDA.
In recent years federal laws have been enacted to help provide more treatment options and possibly lower costs to patients for therapeutic biological products. You have likely heard or read about “biosimilars” on the U.S. drug market. But are you ready to make informed treatment decisions and answer patients’ questions about these new drugs?
Biosimilars are not “generic biologics.” Although the shared goal of making biosimilars and generic drugs available is to provide more treatment options, there are important differences between the two approval pathways. For instance, unlike generic drugs, which are “bioequivalent” to their reference listed drug, biosimilar products can be either “biosimilar” to or “interchangeable” with their reference product. The differences may seem subtle based on the terminology, but the differences are important, and health care professionals who prescribe, dispense, or administer these products will need to understand them – as well as other aspects of these new therapies.
To help busy health care professionals better understand biosimilar and interchangeable products, the FDA has created a free 1.5-hour continuing education program titled, FDA Overview of Biosimilar Products. The course describes the important characteristics of biological products, particularly their complexity. For instance, a single molecule of a biological product, such as a monoclonal antibody, can easily be many hundreds of times larger and much more complex than that of a “small molecule” drug such as aspirin. Their increased complexity is one reason they are regulated differently than generics – and why, in the case of biosimilars, the law allows some differences from the reference product in clinically inactive components. The CE course discusses the “inherent variability” in the production of all biological products – both reference and biosimilar – and how the FDA accounts for these differences in assessing safety and efficacy of the products. The course also provides detailed definitions of important terms – such as “biosimilar” and “interchangeable” – and an overview of the standards the FDA has established for reviewing and approving biosimilar and interchangeable products. It also outlines the FDA review and approval standards for biosimilars and interchangeable products to help practitioners be assured that these products have been demonstrated to be safe and effective treatment options for their patients.
Some of our most important and expensive drugs are biological products used to treat patients who have serious medical conditions that are often life threatening and usually life altering. In April 2016, the Food and Drug Administration approved Inflectra (infliximab-dyyb) to help treat certain patients with certain gastrointestinal disorders, such as Crohn’s disease, and for other indications such as treatment of psoriasis and rheumatoid arthritis. The product is approved but not yet marketed in the United States. However, last year, Zarxio (filgrastim-sndz) became the first biosimilar actually available in the U.S. marketplace – approved to help boost white blood cell production for patients with severe neutropenia as well as for patients receiving various cancer therapies. These products are “biosimilar” to already-approved biological products called “reference products.” Inflectra is biosimilar to the reference product, Remicade (infliximab), and Zarxio is biosimilar to Neupogen (filgrastim).
Use of safe and effective biosimilar and interchangeable biological products can potentially make treatment more affordable for patients in need and improve public health. As patients begin to ask about the use of biosimilar and interchangeable products, the FDA hopes this course will help prepare health care practitioners to make informed decisions on behalf of their patients.
Dr. Christl is Associate Director for Therapeutic Biologics at the Office of New Drugs, Center for Drug Evaluation and Research, FDA.

The Treatment of Obstructive Hypertrophic Cardiomyopathy is best in Higher-Volume Hospitals
NEW YORK (Reuters Health) - Higher-volume hospitals do better in treatment of obstructive hypertrophic cardiomyopathy (HCM), but more efforts are needed to direct patients to these centers, according to New York-based researchers.
In an April 27 online paper in JAMA Cardiology, they note that recommendations are that the treatments, septal myectomy (SM) and alcohol septal ablation (ASA), be performed only by experienced operators with dedicated HCM clinical programs.
"Our study demonstrates that a significant number of cases of septal myectomy and alcohol septal ablation are not being performed at centers of excellence despite the guideline," Dr. Luke K. Kim, of Weill Cornell Medical College, told Reuters Health by email.
Dr. Kim and colleagues investigated compliance and its influence on outcome by examining nationwide data from 2003 to 2009 on 6,386 patients who underwent SM and 4,862 who had ASA. During this period almost 60% of institutions performed 10 or fewer SM procedures. The corresponding proportion for ASAs was 67%.
The incidence of in-hospital death after SM was significantly lower in hospitals in the highest volume tertile (3.8%) than those in the middle (9.6%) and lowest tertiles (15.6%). Corresponding proportions for ASA were 0.6%, 0.8% and 2.3%. There was a similar pattern for acute renal failure after ASA (2.4%, 7.6% and 6.2%).
After adjustment, being in the lowest tertile of SM volume was an independent predictor of in-hospital all-cause mortality (odds ratio, 3.11) and bleeding (OR, 3.77). However, being in the lowest volume for ASA was not independently associated with an increased risk of adverse post-procedural events.
In addition, hospitalization at a high-volume center was associated with a shorter stay and lower costs for both procedures.
However, over the study period, wrote the investigators, "Most centers that provide septal reduction therapy performed few SM and ASA procedures" and were "below the threshold recommended."
In particular, they concluded "Low SM volume was associated with worse outcomes, including higher mortality, longer length of stay, and higher costs. More efforts are needed to encourage referral of patients to centers of excellence for septal reduction therapy."
Commenting on the findings by email, Dr. Steve R. Ommen, coauthor of an accompanying invited opinion, told Reuters Health that "patients deserve to be offered the best care and outcomes possible and that appears to be possible only at centers with focused expertise in the management of HCM. Simply being a high-volume facility does not translate into achieving the safety nor the success observed at expert centers."
Dr. Ommen of the Mayo Clinic, Rochester, Minnesota, added that "Success takes a comprehensive understanding of the underlying HCM disease process. That is really the main take-home message from my point of view."
The other really astonishing finding," he concluded, "was that the median number of procedures performed was only one per year per hospital in the study."
The Michael Wolk Heart Foundation and the New York Cardiac Center supported this research. Two coauthors reported disclosures.
NEW YORK (Reuters Health) - Higher-volume hospitals do better in treatment of obstructive hypertrophic cardiomyopathy (HCM), but more efforts are needed to direct patients to these centers, according to New York-based researchers.
In an April 27 online paper in JAMA Cardiology, they note that recommendations are that the treatments, septal myectomy (SM) and alcohol septal ablation (ASA), be performed only by experienced operators with dedicated HCM clinical programs.
"Our study demonstrates that a significant number of cases of septal myectomy and alcohol septal ablation are not being performed at centers of excellence despite the guideline," Dr. Luke K. Kim, of Weill Cornell Medical College, told Reuters Health by email.
Dr. Kim and colleagues investigated compliance and its influence on outcome by examining nationwide data from 2003 to 2009 on 6,386 patients who underwent SM and 4,862 who had ASA. During this period almost 60% of institutions performed 10 or fewer SM procedures. The corresponding proportion for ASAs was 67%.
The incidence of in-hospital death after SM was significantly lower in hospitals in the highest volume tertile (3.8%) than those in the middle (9.6%) and lowest tertiles (15.6%). Corresponding proportions for ASA were 0.6%, 0.8% and 2.3%. There was a similar pattern for acute renal failure after ASA (2.4%, 7.6% and 6.2%).
After adjustment, being in the lowest tertile of SM volume was an independent predictor of in-hospital all-cause mortality (odds ratio, 3.11) and bleeding (OR, 3.77). However, being in the lowest volume for ASA was not independently associated with an increased risk of adverse post-procedural events.
In addition, hospitalization at a high-volume center was associated with a shorter stay and lower costs for both procedures.
However, over the study period, wrote the investigators, "Most centers that provide septal reduction therapy performed few SM and ASA procedures" and were "below the threshold recommended."
In particular, they concluded "Low SM volume was associated with worse outcomes, including higher mortality, longer length of stay, and higher costs. More efforts are needed to encourage referral of patients to centers of excellence for septal reduction therapy."
Commenting on the findings by email, Dr. Steve R. Ommen, coauthor of an accompanying invited opinion, told Reuters Health that "patients deserve to be offered the best care and outcomes possible and that appears to be possible only at centers with focused expertise in the management of HCM. Simply being a high-volume facility does not translate into achieving the safety nor the success observed at expert centers."
Dr. Ommen of the Mayo Clinic, Rochester, Minnesota, added that "Success takes a comprehensive understanding of the underlying HCM disease process. That is really the main take-home message from my point of view."
The other really astonishing finding," he concluded, "was that the median number of procedures performed was only one per year per hospital in the study."
The Michael Wolk Heart Foundation and the New York Cardiac Center supported this research. Two coauthors reported disclosures.
NEW YORK (Reuters Health) - Higher-volume hospitals do better in treatment of obstructive hypertrophic cardiomyopathy (HCM), but more efforts are needed to direct patients to these centers, according to New York-based researchers.
In an April 27 online paper in JAMA Cardiology, they note that recommendations are that the treatments, septal myectomy (SM) and alcohol septal ablation (ASA), be performed only by experienced operators with dedicated HCM clinical programs.
"Our study demonstrates that a significant number of cases of septal myectomy and alcohol septal ablation are not being performed at centers of excellence despite the guideline," Dr. Luke K. Kim, of Weill Cornell Medical College, told Reuters Health by email.
Dr. Kim and colleagues investigated compliance and its influence on outcome by examining nationwide data from 2003 to 2009 on 6,386 patients who underwent SM and 4,862 who had ASA. During this period almost 60% of institutions performed 10 or fewer SM procedures. The corresponding proportion for ASAs was 67%.
The incidence of in-hospital death after SM was significantly lower in hospitals in the highest volume tertile (3.8%) than those in the middle (9.6%) and lowest tertiles (15.6%). Corresponding proportions for ASA were 0.6%, 0.8% and 2.3%. There was a similar pattern for acute renal failure after ASA (2.4%, 7.6% and 6.2%).
After adjustment, being in the lowest tertile of SM volume was an independent predictor of in-hospital all-cause mortality (odds ratio, 3.11) and bleeding (OR, 3.77). However, being in the lowest volume for ASA was not independently associated with an increased risk of adverse post-procedural events.
In addition, hospitalization at a high-volume center was associated with a shorter stay and lower costs for both procedures.
However, over the study period, wrote the investigators, "Most centers that provide septal reduction therapy performed few SM and ASA procedures" and were "below the threshold recommended."
In particular, they concluded "Low SM volume was associated with worse outcomes, including higher mortality, longer length of stay, and higher costs. More efforts are needed to encourage referral of patients to centers of excellence for septal reduction therapy."
Commenting on the findings by email, Dr. Steve R. Ommen, coauthor of an accompanying invited opinion, told Reuters Health that "patients deserve to be offered the best care and outcomes possible and that appears to be possible only at centers with focused expertise in the management of HCM. Simply being a high-volume facility does not translate into achieving the safety nor the success observed at expert centers."
Dr. Ommen of the Mayo Clinic, Rochester, Minnesota, added that "Success takes a comprehensive understanding of the underlying HCM disease process. That is really the main take-home message from my point of view."
The other really astonishing finding," he concluded, "was that the median number of procedures performed was only one per year per hospital in the study."
The Michael Wolk Heart Foundation and the New York Cardiac Center supported this research. Two coauthors reported disclosures.
EC approves first immunostimulatory antibody to treat MM
Photo courtesy of
Bristol-Myers Squibb
The European Commission (EC) has approved elotuzumab (Empliciti) for use in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received at least one prior therapy.
Elotuzumab is an immunostimulatory antibody that specifically targets signaling lymphocyte activation molecule family member 7 (SLAMF7), a cell-surface glycoprotein expressed on myeloma cells, natural killer (NK) cells, plasma cells, and specific immune cell subsets of differentiated cells in the hematopoietic lineage.
Elotuzumab has a dual mechanism of action. It directly activates the immune system through NK cells via the SLAMF7 pathway, and it targets SLAMF7 on myeloma cells, tagging them for NK-cell-mediated destruction via antibody-dependent cellular toxicity.
Elotuzumab is the first immunostimulatory antibody approved to treat MM in the European Union.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Phase 3 trial
The EC approved elotuzumab based on data from the phase 3 ELOQUENT-2 trial, which were presented at ASCO 2015 and published in NEJM.
The trial included 646 MM patients who had received 1 to 3 prior therapies.
The patients were randomized 1:1 to receive either elotuzumab at 10 mg/kg in combination with lenalidomide and dexamethasone (len-dex) or len-dex alone in 4-week cycles until disease progression or unacceptable toxicity.
Baseline patient demographics and disease characteristics were well balanced between treatment arms.
The minimum follow-up for all study subjects was 24 months. The co-primary endpoints were progression-free survival (PFS) and overall response rate.
The overall response rate was 78.5% in the elotuzumab arm and 65.5% in the len-dex arm (P=0.0002).
The median PFS was 19.4 months in the elotuzumab arm and 14.9 months in the len-dex arm (P=0.0004). At 1 year, the PFS was 68% in the elotuzumab arm and 57% in the len-dex arm. At 2 years, the PFS was 41% and 27%, respectively.
The most common adverse events in the elotuzumab arm and len-dex arm, respectively, were fatigue (61.6% vs 51.7%), diarrhea (46.9% vs 36.0%), pyrexia (37.4% vs 24.6%), constipation (35.5% vs 27.1%), cough (34.3% vs 18.9%), peripheral neuropathy (26.7% vs 20.8%), nasopharyngitis (24.5% vs 19.2%), upper respiratory tract infection (22.6% vs 17.4%), decreased appetite (20.8% vs 12.6%), and pneumonia (20.1% vs 14.2%).
Serious adverse events occurred in 65.4% of patients in the elotuzumab arm and 56.5% in the len-dex arm. The most frequent events were pneumonia, pyrexia, respiratory tract infection, anemia, pulmonary embolism, and acute renal failure. ![]()
Photo courtesy of
Bristol-Myers Squibb
The European Commission (EC) has approved elotuzumab (Empliciti) for use in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received at least one prior therapy.
Elotuzumab is an immunostimulatory antibody that specifically targets signaling lymphocyte activation molecule family member 7 (SLAMF7), a cell-surface glycoprotein expressed on myeloma cells, natural killer (NK) cells, plasma cells, and specific immune cell subsets of differentiated cells in the hematopoietic lineage.
Elotuzumab has a dual mechanism of action. It directly activates the immune system through NK cells via the SLAMF7 pathway, and it targets SLAMF7 on myeloma cells, tagging them for NK-cell-mediated destruction via antibody-dependent cellular toxicity.
Elotuzumab is the first immunostimulatory antibody approved to treat MM in the European Union.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Phase 3 trial
The EC approved elotuzumab based on data from the phase 3 ELOQUENT-2 trial, which were presented at ASCO 2015 and published in NEJM.
The trial included 646 MM patients who had received 1 to 3 prior therapies.
The patients were randomized 1:1 to receive either elotuzumab at 10 mg/kg in combination with lenalidomide and dexamethasone (len-dex) or len-dex alone in 4-week cycles until disease progression or unacceptable toxicity.
Baseline patient demographics and disease characteristics were well balanced between treatment arms.
The minimum follow-up for all study subjects was 24 months. The co-primary endpoints were progression-free survival (PFS) and overall response rate.
The overall response rate was 78.5% in the elotuzumab arm and 65.5% in the len-dex arm (P=0.0002).
The median PFS was 19.4 months in the elotuzumab arm and 14.9 months in the len-dex arm (P=0.0004). At 1 year, the PFS was 68% in the elotuzumab arm and 57% in the len-dex arm. At 2 years, the PFS was 41% and 27%, respectively.
The most common adverse events in the elotuzumab arm and len-dex arm, respectively, were fatigue (61.6% vs 51.7%), diarrhea (46.9% vs 36.0%), pyrexia (37.4% vs 24.6%), constipation (35.5% vs 27.1%), cough (34.3% vs 18.9%), peripheral neuropathy (26.7% vs 20.8%), nasopharyngitis (24.5% vs 19.2%), upper respiratory tract infection (22.6% vs 17.4%), decreased appetite (20.8% vs 12.6%), and pneumonia (20.1% vs 14.2%).
Serious adverse events occurred in 65.4% of patients in the elotuzumab arm and 56.5% in the len-dex arm. The most frequent events were pneumonia, pyrexia, respiratory tract infection, anemia, pulmonary embolism, and acute renal failure. ![]()
Photo courtesy of
Bristol-Myers Squibb
The European Commission (EC) has approved elotuzumab (Empliciti) for use in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received at least one prior therapy.
Elotuzumab is an immunostimulatory antibody that specifically targets signaling lymphocyte activation molecule family member 7 (SLAMF7), a cell-surface glycoprotein expressed on myeloma cells, natural killer (NK) cells, plasma cells, and specific immune cell subsets of differentiated cells in the hematopoietic lineage.
Elotuzumab has a dual mechanism of action. It directly activates the immune system through NK cells via the SLAMF7 pathway, and it targets SLAMF7 on myeloma cells, tagging them for NK-cell-mediated destruction via antibody-dependent cellular toxicity.
Elotuzumab is the first immunostimulatory antibody approved to treat MM in the European Union.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Phase 3 trial
The EC approved elotuzumab based on data from the phase 3 ELOQUENT-2 trial, which were presented at ASCO 2015 and published in NEJM.
The trial included 646 MM patients who had received 1 to 3 prior therapies.
The patients were randomized 1:1 to receive either elotuzumab at 10 mg/kg in combination with lenalidomide and dexamethasone (len-dex) or len-dex alone in 4-week cycles until disease progression or unacceptable toxicity.
Baseline patient demographics and disease characteristics were well balanced between treatment arms.
The minimum follow-up for all study subjects was 24 months. The co-primary endpoints were progression-free survival (PFS) and overall response rate.
The overall response rate was 78.5% in the elotuzumab arm and 65.5% in the len-dex arm (P=0.0002).
The median PFS was 19.4 months in the elotuzumab arm and 14.9 months in the len-dex arm (P=0.0004). At 1 year, the PFS was 68% in the elotuzumab arm and 57% in the len-dex arm. At 2 years, the PFS was 41% and 27%, respectively.
The most common adverse events in the elotuzumab arm and len-dex arm, respectively, were fatigue (61.6% vs 51.7%), diarrhea (46.9% vs 36.0%), pyrexia (37.4% vs 24.6%), constipation (35.5% vs 27.1%), cough (34.3% vs 18.9%), peripheral neuropathy (26.7% vs 20.8%), nasopharyngitis (24.5% vs 19.2%), upper respiratory tract infection (22.6% vs 17.4%), decreased appetite (20.8% vs 12.6%), and pneumonia (20.1% vs 14.2%).
Serious adverse events occurred in 65.4% of patients in the elotuzumab arm and 56.5% in the len-dex arm. The most frequent events were pneumonia, pyrexia, respiratory tract infection, anemia, pulmonary embolism, and acute renal failure. ![]()
Study reveals potential treatment avenue for DBA, MDS

The production of two components of hemoglobin may be out of sync in Diamond Blackfan anemia (DBA) and myelodysplastic syndromes (MDS), according to a new study.
Researchers found that, in samples from patients with DBA or MDS, ribosome dysfunction delayed globin production, while heme synthesis proceeded normally.
This disruption in heme-globin coordination led to a buildup of toxic heme that killed red blood cell (RBC) precursors.
However, treating patient samples with a compound that blocks heme synthesis increased RBC production in both DBA and MDS.
Zhantao Yang, MD, of the University of Washington in Seattle, and his colleagues reported these findings in Science Translational Medicine.
Both DBA and MDS have been linked to defects in ribosome assembly, which is critical to protein production, but how this leads to anemia remains unknown.
To find out, Dr Yang and his colleagues analyzed bone marrow cells from patients with DBA (n=3) or MDS with del(5q) (n=6).
The researchers found that globin translation proceeded slowly in these samples, but heme synthesis proceeded normally.
This resulted in insufficient globin, excess heme, and excess reactive oxygen species in early erythroid precursors and, ultimately, the death of colony-forming unit–erythroid/proerythroblast cells.
The cells that were able to rapidly export heme or slow its synthesis survived and matured into RBCs, but the other colony-forming unit–erythroid cells/early proerythroblasts died.
The researchers noted that it is not clear how excess heme induces cell death in RBC precursors, but they said it likely involves both ferroptosis and apoptosis.
Regardless of the mechanism of cell death, the team found that treating the patients’ cells with succinylacetone (10 mM), a compound that blocks heme synthesis, improved RBC production.
The treatment improved RBC production in DBA and del(5q) MDS marrow cultures by 68% to 95% (P=0.03 to 0.05). In comparison, RBC production in control marrow cultures decreased by 4% to 13%.
The researchers said their experiments revealed additional important findings. First, they found that erythroid differentiation in the marrow cultures “excellently” phenocopied erythroid differentiation in vivo. This suggests these cultures can serve as a reliable platform in preclinical studies.
Second, the team said the fact that epigenetic differences between RBC precursors can lead to their preferential death or survival has broad implications. And querying the cells that preferentially survive could provide important insights. ![]()
The production of two components of hemoglobin may be out of sync in Diamond Blackfan anemia (DBA) and myelodysplastic syndromes (MDS), according to a new study.
Researchers found that, in samples from patients with DBA or MDS, ribosome dysfunction delayed globin production, while heme synthesis proceeded normally.
This disruption in heme-globin coordination led to a buildup of toxic heme that killed red blood cell (RBC) precursors.
However, treating patient samples with a compound that blocks heme synthesis increased RBC production in both DBA and MDS.
Zhantao Yang, MD, of the University of Washington in Seattle, and his colleagues reported these findings in Science Translational Medicine.
Both DBA and MDS have been linked to defects in ribosome assembly, which is critical to protein production, but how this leads to anemia remains unknown.
To find out, Dr Yang and his colleagues analyzed bone marrow cells from patients with DBA (n=3) or MDS with del(5q) (n=6).
The researchers found that globin translation proceeded slowly in these samples, but heme synthesis proceeded normally.
This resulted in insufficient globin, excess heme, and excess reactive oxygen species in early erythroid precursors and, ultimately, the death of colony-forming unit–erythroid/proerythroblast cells.
The cells that were able to rapidly export heme or slow its synthesis survived and matured into RBCs, but the other colony-forming unit–erythroid cells/early proerythroblasts died.
The researchers noted that it is not clear how excess heme induces cell death in RBC precursors, but they said it likely involves both ferroptosis and apoptosis.
Regardless of the mechanism of cell death, the team found that treating the patients’ cells with succinylacetone (10 mM), a compound that blocks heme synthesis, improved RBC production.
The treatment improved RBC production in DBA and del(5q) MDS marrow cultures by 68% to 95% (P=0.03 to 0.05). In comparison, RBC production in control marrow cultures decreased by 4% to 13%.
The researchers said their experiments revealed additional important findings. First, they found that erythroid differentiation in the marrow cultures “excellently” phenocopied erythroid differentiation in vivo. This suggests these cultures can serve as a reliable platform in preclinical studies.
Second, the team said the fact that epigenetic differences between RBC precursors can lead to their preferential death or survival has broad implications. And querying the cells that preferentially survive could provide important insights. ![]()
The production of two components of hemoglobin may be out of sync in Diamond Blackfan anemia (DBA) and myelodysplastic syndromes (MDS), according to a new study.
Researchers found that, in samples from patients with DBA or MDS, ribosome dysfunction delayed globin production, while heme synthesis proceeded normally.
This disruption in heme-globin coordination led to a buildup of toxic heme that killed red blood cell (RBC) precursors.
However, treating patient samples with a compound that blocks heme synthesis increased RBC production in both DBA and MDS.
Zhantao Yang, MD, of the University of Washington in Seattle, and his colleagues reported these findings in Science Translational Medicine.
Both DBA and MDS have been linked to defects in ribosome assembly, which is critical to protein production, but how this leads to anemia remains unknown.
To find out, Dr Yang and his colleagues analyzed bone marrow cells from patients with DBA (n=3) or MDS with del(5q) (n=6).
The researchers found that globin translation proceeded slowly in these samples, but heme synthesis proceeded normally.
This resulted in insufficient globin, excess heme, and excess reactive oxygen species in early erythroid precursors and, ultimately, the death of colony-forming unit–erythroid/proerythroblast cells.
The cells that were able to rapidly export heme or slow its synthesis survived and matured into RBCs, but the other colony-forming unit–erythroid cells/early proerythroblasts died.
The researchers noted that it is not clear how excess heme induces cell death in RBC precursors, but they said it likely involves both ferroptosis and apoptosis.
Regardless of the mechanism of cell death, the team found that treating the patients’ cells with succinylacetone (10 mM), a compound that blocks heme synthesis, improved RBC production.
The treatment improved RBC production in DBA and del(5q) MDS marrow cultures by 68% to 95% (P=0.03 to 0.05). In comparison, RBC production in control marrow cultures decreased by 4% to 13%.
The researchers said their experiments revealed additional important findings. First, they found that erythroid differentiation in the marrow cultures “excellently” phenocopied erythroid differentiation in vivo. This suggests these cultures can serve as a reliable platform in preclinical studies.
Second, the team said the fact that epigenetic differences between RBC precursors can lead to their preferential death or survival has broad implications. And querying the cells that preferentially survive could provide important insights. ![]()
Long-term PPI use damages endothelial cells
Image courtesy of NIH
Chronic exposure to proton pump inhibitors (PPIs) accelerates biological aging in human endothelial cells, according to preclinical research.
Investigators said this finding, published in Circulation Research, supports recent epidemiological and retrospective studies that revealed associations between the long-term use of PPIs and an increased risk of heart attack, renal failure, and dementia.
“When we exposed human endothelial cells over a period of time to these PPIs, we observed accelerated aging of the cells,” said John P. Cooke, MD, PhD, of the Houston Methodist Research Institute in Texas.
“The PPIs also reduce acidity in lysosomes of the endothelial cell. The lysosomes are like cellular garbage disposals and need acid to work properly. We observed cellular garbage accumulating in the endothelial cells, which sped up the aging process.”
Dr Cooke suspects this may be the unifying mechanism that explains the increased risk of heart attack, renal failure, and dementia observed in long-term PPI users.
“These drugs do not seem to adversely affect the heart and blood vessels when taken for a few weeks,” he said. “However, we urgently need studies to assess the impact of long-term use of these drugs on vascular health in a broad patient population. We also need to consider if these drugs should be so accessible without medical supervision.”
Dr Cooke and his colleagues noted that, while PPIs like esomeprazole (Nexium) were shown to affect vascular aging, H2 blockers like ranitidine (Zantac) did not adversely affect the endothelium.
Therefore, other approaches to long-term treatment that should be considered for gastroesophageal reflux disease include H2 antagonists, lifestyle modifications, and, in severe cases, surgical approaches.
The investigators also pointed out that this study had some limitations. Although two different PPIs were tested, only one of these, esomeprazole, is commercially available.
In addition, the study did not show how PPIs actually impair the lysosome’s ability to produce enough acid to clear waste.
Finally, since this study was conducted in a laboratory setting, it did not show whether PPIs act in the same way within the human body as they do in vitro. ![]()
Image courtesy of NIH
Chronic exposure to proton pump inhibitors (PPIs) accelerates biological aging in human endothelial cells, according to preclinical research.
Investigators said this finding, published in Circulation Research, supports recent epidemiological and retrospective studies that revealed associations between the long-term use of PPIs and an increased risk of heart attack, renal failure, and dementia.
“When we exposed human endothelial cells over a period of time to these PPIs, we observed accelerated aging of the cells,” said John P. Cooke, MD, PhD, of the Houston Methodist Research Institute in Texas.
“The PPIs also reduce acidity in lysosomes of the endothelial cell. The lysosomes are like cellular garbage disposals and need acid to work properly. We observed cellular garbage accumulating in the endothelial cells, which sped up the aging process.”
Dr Cooke suspects this may be the unifying mechanism that explains the increased risk of heart attack, renal failure, and dementia observed in long-term PPI users.
“These drugs do not seem to adversely affect the heart and blood vessels when taken for a few weeks,” he said. “However, we urgently need studies to assess the impact of long-term use of these drugs on vascular health in a broad patient population. We also need to consider if these drugs should be so accessible without medical supervision.”
Dr Cooke and his colleagues noted that, while PPIs like esomeprazole (Nexium) were shown to affect vascular aging, H2 blockers like ranitidine (Zantac) did not adversely affect the endothelium.
Therefore, other approaches to long-term treatment that should be considered for gastroesophageal reflux disease include H2 antagonists, lifestyle modifications, and, in severe cases, surgical approaches.
The investigators also pointed out that this study had some limitations. Although two different PPIs were tested, only one of these, esomeprazole, is commercially available.
In addition, the study did not show how PPIs actually impair the lysosome’s ability to produce enough acid to clear waste.
Finally, since this study was conducted in a laboratory setting, it did not show whether PPIs act in the same way within the human body as they do in vitro. ![]()
Image courtesy of NIH
Chronic exposure to proton pump inhibitors (PPIs) accelerates biological aging in human endothelial cells, according to preclinical research.
Investigators said this finding, published in Circulation Research, supports recent epidemiological and retrospective studies that revealed associations between the long-term use of PPIs and an increased risk of heart attack, renal failure, and dementia.
“When we exposed human endothelial cells over a period of time to these PPIs, we observed accelerated aging of the cells,” said John P. Cooke, MD, PhD, of the Houston Methodist Research Institute in Texas.
“The PPIs also reduce acidity in lysosomes of the endothelial cell. The lysosomes are like cellular garbage disposals and need acid to work properly. We observed cellular garbage accumulating in the endothelial cells, which sped up the aging process.”
Dr Cooke suspects this may be the unifying mechanism that explains the increased risk of heart attack, renal failure, and dementia observed in long-term PPI users.
“These drugs do not seem to adversely affect the heart and blood vessels when taken for a few weeks,” he said. “However, we urgently need studies to assess the impact of long-term use of these drugs on vascular health in a broad patient population. We also need to consider if these drugs should be so accessible without medical supervision.”
Dr Cooke and his colleagues noted that, while PPIs like esomeprazole (Nexium) were shown to affect vascular aging, H2 blockers like ranitidine (Zantac) did not adversely affect the endothelium.
Therefore, other approaches to long-term treatment that should be considered for gastroesophageal reflux disease include H2 antagonists, lifestyle modifications, and, in severe cases, surgical approaches.
The investigators also pointed out that this study had some limitations. Although two different PPIs were tested, only one of these, esomeprazole, is commercially available.
In addition, the study did not show how PPIs actually impair the lysosome’s ability to produce enough acid to clear waste.
Finally, since this study was conducted in a laboratory setting, it did not show whether PPIs act in the same way within the human body as they do in vitro. ![]()
EC approves long-acting hemophilia B therapy

The European Commission (EC) has approved albutrepenonacog alfa (Idelvion) to treat and prevent bleeding in children and adults with hemophilia B.
Albutrepenonacog alfa is a long-acting albumin fusion protein linking recombinant coagulation factor IX with recombinant albumin.
The product is now approved for use as routine prophylaxis, for on-demand control of bleeding, and for the perioperative management of bleeding.
Appropriate patients age 12 and older can go up to 14 days between albutrepenonacog alfa infusions. This dosing interval has been achieved while maintaining high levels of factor IX activity—above 5% over 14 days at 75 IU/kg.
“Offering 14-day dosing, Idelvion helps patients maintain higher factor IX levels over a long period of time, providing them with greater freedom from frequent infusions,” said Elena Santagostino, MD, PhD, of the University of Milan/IRCCS Maggiore Hospital in Italy.
“This is an important attribute for my patients who require a prophylactic regimen but don’t want treatment to disrupt their active lives.”
Albutrepenonacog alfa is being developed by CSL Behring. The company said the product will be launched in European markets in the coming months, as market access and pricing are obtained.
Phase 3 trial
The EC approved albutrepenonacog alfa based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks,
followed by a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache. ![]()
The European Commission (EC) has approved albutrepenonacog alfa (Idelvion) to treat and prevent bleeding in children and adults with hemophilia B.
Albutrepenonacog alfa is a long-acting albumin fusion protein linking recombinant coagulation factor IX with recombinant albumin.
The product is now approved for use as routine prophylaxis, for on-demand control of bleeding, and for the perioperative management of bleeding.
Appropriate patients age 12 and older can go up to 14 days between albutrepenonacog alfa infusions. This dosing interval has been achieved while maintaining high levels of factor IX activity—above 5% over 14 days at 75 IU/kg.
“Offering 14-day dosing, Idelvion helps patients maintain higher factor IX levels over a long period of time, providing them with greater freedom from frequent infusions,” said Elena Santagostino, MD, PhD, of the University of Milan/IRCCS Maggiore Hospital in Italy.
“This is an important attribute for my patients who require a prophylactic regimen but don’t want treatment to disrupt their active lives.”
Albutrepenonacog alfa is being developed by CSL Behring. The company said the product will be launched in European markets in the coming months, as market access and pricing are obtained.
Phase 3 trial
The EC approved albutrepenonacog alfa based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks,
followed by a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache. ![]()
The European Commission (EC) has approved albutrepenonacog alfa (Idelvion) to treat and prevent bleeding in children and adults with hemophilia B.
Albutrepenonacog alfa is a long-acting albumin fusion protein linking recombinant coagulation factor IX with recombinant albumin.
The product is now approved for use as routine prophylaxis, for on-demand control of bleeding, and for the perioperative management of bleeding.
Appropriate patients age 12 and older can go up to 14 days between albutrepenonacog alfa infusions. This dosing interval has been achieved while maintaining high levels of factor IX activity—above 5% over 14 days at 75 IU/kg.
“Offering 14-day dosing, Idelvion helps patients maintain higher factor IX levels over a long period of time, providing them with greater freedom from frequent infusions,” said Elena Santagostino, MD, PhD, of the University of Milan/IRCCS Maggiore Hospital in Italy.
“This is an important attribute for my patients who require a prophylactic regimen but don’t want treatment to disrupt their active lives.”
Albutrepenonacog alfa is being developed by CSL Behring. The company said the product will be launched in European markets in the coming months, as market access and pricing are obtained.
Phase 3 trial
The EC approved albutrepenonacog alfa based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks,
followed by a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache. ![]()
Clinical Trials Update
Are you a clinical trials investigator with unused capacity? Would you like to refer patients to participate in groundbreaking clinical trials?
The CHEST Clinical Trials Registry is a free service that connects physicians to information about clinical trials in respiratory disease conducted by participating pharmaceutical companies.
Ongoing groundbreaking research could have a measurable impact on patient care, but a lack of clinical trial participants is significantly slowing research and threatening the development of new treatments. Recruiting and retaining trial participants are the greatest challenges to developing the next generation of treatment options.
Participation in clinical trials provides an opportunity to advance and accelerate medical research and contribute to an improved health outlook for future generations. Use our registry to get immediate information on how you can be involved in a clinical trial.
Access to learn more: chestnet.org/Guidelines-and-Resources/Clinical-Trials/Clinical-Trials-Registry.
Are you a clinical trials investigator with unused capacity? Would you like to refer patients to participate in groundbreaking clinical trials?
The CHEST Clinical Trials Registry is a free service that connects physicians to information about clinical trials in respiratory disease conducted by participating pharmaceutical companies.
Ongoing groundbreaking research could have a measurable impact on patient care, but a lack of clinical trial participants is significantly slowing research and threatening the development of new treatments. Recruiting and retaining trial participants are the greatest challenges to developing the next generation of treatment options.
Participation in clinical trials provides an opportunity to advance and accelerate medical research and contribute to an improved health outlook for future generations. Use our registry to get immediate information on how you can be involved in a clinical trial.
Access to learn more: chestnet.org/Guidelines-and-Resources/Clinical-Trials/Clinical-Trials-Registry.
Are you a clinical trials investigator with unused capacity? Would you like to refer patients to participate in groundbreaking clinical trials?
The CHEST Clinical Trials Registry is a free service that connects physicians to information about clinical trials in respiratory disease conducted by participating pharmaceutical companies.
Ongoing groundbreaking research could have a measurable impact on patient care, but a lack of clinical trial participants is significantly slowing research and threatening the development of new treatments. Recruiting and retaining trial participants are the greatest challenges to developing the next generation of treatment options.
Participation in clinical trials provides an opportunity to advance and accelerate medical research and contribute to an improved health outlook for future generations. Use our registry to get immediate information on how you can be involved in a clinical trial.
Access to learn more: chestnet.org/Guidelines-and-Resources/Clinical-Trials/Clinical-Trials-Registry.