User login
Hope for Hidradenitis Suppurativa
In September 2015, the US Food and Drug Administration approved adalimumab, the well-known injectable tumor necrosis factor (TNF)–α inhibitor indicated for psoriasis and other inflammatory conditions, for treatment of moderate to severe hidradenitis suppurativa (HS), classifying it as the first and only US Food and Drug Administration–approved therapy for adults with HS.
Pivotal studies (PIONEER/HS-I and -II, phase 3, double-blind) evaluated 633 patients (307 in HS-I and 326 in HS-II) with moderate to severe HS who were randomized to adalimumab versus placebo for 12 weeks. There was significant clinical response (at least 50% reduction in abscess and inflammatory nodule count, defined as hidradenitis suppurativa clinical response, HiSCR) in the adalimumab group (42% vs 26% in HS-I, 59% vs. 28% in HS-II, P<.001), reduction in pain (significant in the HS-II trial, 45.7% vs 20.7%, P<.001; HS-I 27.9% vs 24.8%, P>.05), and no new safety concerns when compared to other adalimumab dosages and indications. Some HS-II study patients (19.3%) were permitted to use oral antibiotics during the study.
For the indication of adult HS (moderate to severe disease), adalimumab should be administered subcutaneously at a dosing regimen of 160 mg/4 syringes on day 1 (or 80 mg/2 syringes on days 1 and 2), followed by 80 mg/2 syringes on day 15, then 40 mg/1 syringe on day 29, and every 7 days thereafter for an indefinite treatment period.
What’s the Issue?
It sits well with dermatologists when the indications for a medication with which we have great familiarity are broadened to include new disease entities; it is even better when the new entity is a condition for which every dermatologist pines for efficacious treatment options. Despite the disease burden of HS, which can include pain, scarring, disfigurement, social exclusion, and/or embarrassment, as well as the wasteful and burdensome effect that HS has on health care resources, such as injudicious use of antibiotics and unnecessary emergency department visits and inpatient hospital stays,1 there is nonetheless a wide-open and inviting playing field for effective therapies.
Although a much higher dosage of adalimumab is required in the treatment of HS compared to what is indicated for psoriasis patients, its safety concerns and side effect profile were unchanged in HS, and therefore its monitoring guidelines remain the same. Not all patients in these studies showed a notable reduction in lesion count, but given that HS classically is unresponsive to most medication regimens, will you embrace this therapy option for your patients with HS?
Reference
1. Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
In September 2015, the US Food and Drug Administration approved adalimumab, the well-known injectable tumor necrosis factor (TNF)–α inhibitor indicated for psoriasis and other inflammatory conditions, for treatment of moderate to severe hidradenitis suppurativa (HS), classifying it as the first and only US Food and Drug Administration–approved therapy for adults with HS.
Pivotal studies (PIONEER/HS-I and -II, phase 3, double-blind) evaluated 633 patients (307 in HS-I and 326 in HS-II) with moderate to severe HS who were randomized to adalimumab versus placebo for 12 weeks. There was significant clinical response (at least 50% reduction in abscess and inflammatory nodule count, defined as hidradenitis suppurativa clinical response, HiSCR) in the adalimumab group (42% vs 26% in HS-I, 59% vs. 28% in HS-II, P<.001), reduction in pain (significant in the HS-II trial, 45.7% vs 20.7%, P<.001; HS-I 27.9% vs 24.8%, P>.05), and no new safety concerns when compared to other adalimumab dosages and indications. Some HS-II study patients (19.3%) were permitted to use oral antibiotics during the study.
For the indication of adult HS (moderate to severe disease), adalimumab should be administered subcutaneously at a dosing regimen of 160 mg/4 syringes on day 1 (or 80 mg/2 syringes on days 1 and 2), followed by 80 mg/2 syringes on day 15, then 40 mg/1 syringe on day 29, and every 7 days thereafter for an indefinite treatment period.
What’s the Issue?
It sits well with dermatologists when the indications for a medication with which we have great familiarity are broadened to include new disease entities; it is even better when the new entity is a condition for which every dermatologist pines for efficacious treatment options. Despite the disease burden of HS, which can include pain, scarring, disfigurement, social exclusion, and/or embarrassment, as well as the wasteful and burdensome effect that HS has on health care resources, such as injudicious use of antibiotics and unnecessary emergency department visits and inpatient hospital stays,1 there is nonetheless a wide-open and inviting playing field for effective therapies.
Although a much higher dosage of adalimumab is required in the treatment of HS compared to what is indicated for psoriasis patients, its safety concerns and side effect profile were unchanged in HS, and therefore its monitoring guidelines remain the same. Not all patients in these studies showed a notable reduction in lesion count, but given that HS classically is unresponsive to most medication regimens, will you embrace this therapy option for your patients with HS?
In September 2015, the US Food and Drug Administration approved adalimumab, the well-known injectable tumor necrosis factor (TNF)–α inhibitor indicated for psoriasis and other inflammatory conditions, for treatment of moderate to severe hidradenitis suppurativa (HS), classifying it as the first and only US Food and Drug Administration–approved therapy for adults with HS.
Pivotal studies (PIONEER/HS-I and -II, phase 3, double-blind) evaluated 633 patients (307 in HS-I and 326 in HS-II) with moderate to severe HS who were randomized to adalimumab versus placebo for 12 weeks. There was significant clinical response (at least 50% reduction in abscess and inflammatory nodule count, defined as hidradenitis suppurativa clinical response, HiSCR) in the adalimumab group (42% vs 26% in HS-I, 59% vs. 28% in HS-II, P<.001), reduction in pain (significant in the HS-II trial, 45.7% vs 20.7%, P<.001; HS-I 27.9% vs 24.8%, P>.05), and no new safety concerns when compared to other adalimumab dosages and indications. Some HS-II study patients (19.3%) were permitted to use oral antibiotics during the study.
For the indication of adult HS (moderate to severe disease), adalimumab should be administered subcutaneously at a dosing regimen of 160 mg/4 syringes on day 1 (or 80 mg/2 syringes on days 1 and 2), followed by 80 mg/2 syringes on day 15, then 40 mg/1 syringe on day 29, and every 7 days thereafter for an indefinite treatment period.
What’s the Issue?
It sits well with dermatologists when the indications for a medication with which we have great familiarity are broadened to include new disease entities; it is even better when the new entity is a condition for which every dermatologist pines for efficacious treatment options. Despite the disease burden of HS, which can include pain, scarring, disfigurement, social exclusion, and/or embarrassment, as well as the wasteful and burdensome effect that HS has on health care resources, such as injudicious use of antibiotics and unnecessary emergency department visits and inpatient hospital stays,1 there is nonetheless a wide-open and inviting playing field for effective therapies.
Although a much higher dosage of adalimumab is required in the treatment of HS compared to what is indicated for psoriasis patients, its safety concerns and side effect profile were unchanged in HS, and therefore its monitoring guidelines remain the same. Not all patients in these studies showed a notable reduction in lesion count, but given that HS classically is unresponsive to most medication regimens, will you embrace this therapy option for your patients with HS?
Reference
1. Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
Reference
1. Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.

FDA okays prophylactic Pradaxa for VTE in hip replacement
The Food and Drug Administration has approved dabigatran for the prevention of deep venous thrombosis and pulmonary embolism for patients after hip replacement surgery.
The FDA’s approval was based on the results of two randomized, double-blind, phase III trials in patients undergoing total hip replacement, Boehringer Ingelheim, the manufacturer of the direct thrombin inhibitor, announced.
![]()
In RE-NOVATE I, the first trial, 3,494 patients were randomly assigned to three groups receiving prophylactic treatment with one of two doses of dabigatran (220 mg or 150 mg) once daily, or to the low-molecular-weight heparin enoxaparin at 40 mg once daily for 28-35 days. The first study drug arm was given 110 mg on the day of surgery and 220 mg daily thereafter; the second study drug arm received a dose of 75 mg on the day of surgery and 150 mg daily thereafter. Patients taking the dabigatran (Pradaxa) at 220 mg had a lower composite total of venous thromboembolism (VTE) and all-cause mortality (6.0%) than did those on enoxaparin 40 mg (6.7%), meeting the noninferiority mark (Lancet. 2007 Sep 15;370[9591]:949-56).
In RE-NOVATE II, 2,055 patients were randomly assigned prophylactic treatment for 28-35 days with the study drug dosed at 220 mg once daily, or enoxaparin 40 mg once daily. Patients receiving the study drug were treated with a dose of 110 mg on the day of surgery and 220 mg daily thereafter. The composite total of VTE and all-cause death occurred in 7.7% of patients in the study group vs. 8.8% of patients in the enoxaparin group, which was within the margin for noninferiority (Thromb Haemost. 2011 Apr;105[4]:721-9).
However, there were higher rates of major bleeding in RE-NOVATE I (2.0%, 1.6%) and II (1.4%, 0.9%) with 220 mg vs. enoxaparin. In both studies, the rate of major gastrointestinal bleeds in patients was the same (0.1%) for both the study and control drugs. The rate of any GI bleeds was 1.4% for the study drug and 0.9% for enoxaparin. The most common adverse events in both studies were GI disorders. The incidence rate was the same across all treatment groups (39.5%). Dyspepsia occurred more frequently in patients receiving the study drug (4.1%), compared with those taking enoxaparin (3.8%). Gastritislike symptoms were less common in patients receiving the study drug (0.6%), compared with enoxaparin (1.0%). Clinical myocardial infarction was reported in two (0.1%) study patients and six (0.3%) enoxaparin patients.
Pradaxa was initially indicated by the FDA in 2010 to reduce stroke and systemic embolism risk in patients with nonvalvular atrial fibrillation. In 2014, the FDA approved two additional indications for the drug for the treatment of VTE in patients treated with a parenteral anticoagulant for 5-10 day and to reduce the risk of recurrent VTE in patients who have been previously treated.
On Twitter @whitneymcknight
The Food and Drug Administration has approved dabigatran for the prevention of deep venous thrombosis and pulmonary embolism for patients after hip replacement surgery.
The FDA’s approval was based on the results of two randomized, double-blind, phase III trials in patients undergoing total hip replacement, Boehringer Ingelheim, the manufacturer of the direct thrombin inhibitor, announced.
![]()
In RE-NOVATE I, the first trial, 3,494 patients were randomly assigned to three groups receiving prophylactic treatment with one of two doses of dabigatran (220 mg or 150 mg) once daily, or to the low-molecular-weight heparin enoxaparin at 40 mg once daily for 28-35 days. The first study drug arm was given 110 mg on the day of surgery and 220 mg daily thereafter; the second study drug arm received a dose of 75 mg on the day of surgery and 150 mg daily thereafter. Patients taking the dabigatran (Pradaxa) at 220 mg had a lower composite total of venous thromboembolism (VTE) and all-cause mortality (6.0%) than did those on enoxaparin 40 mg (6.7%), meeting the noninferiority mark (Lancet. 2007 Sep 15;370[9591]:949-56).
In RE-NOVATE II, 2,055 patients were randomly assigned prophylactic treatment for 28-35 days with the study drug dosed at 220 mg once daily, or enoxaparin 40 mg once daily. Patients receiving the study drug were treated with a dose of 110 mg on the day of surgery and 220 mg daily thereafter. The composite total of VTE and all-cause death occurred in 7.7% of patients in the study group vs. 8.8% of patients in the enoxaparin group, which was within the margin for noninferiority (Thromb Haemost. 2011 Apr;105[4]:721-9).
However, there were higher rates of major bleeding in RE-NOVATE I (2.0%, 1.6%) and II (1.4%, 0.9%) with 220 mg vs. enoxaparin. In both studies, the rate of major gastrointestinal bleeds in patients was the same (0.1%) for both the study and control drugs. The rate of any GI bleeds was 1.4% for the study drug and 0.9% for enoxaparin. The most common adverse events in both studies were GI disorders. The incidence rate was the same across all treatment groups (39.5%). Dyspepsia occurred more frequently in patients receiving the study drug (4.1%), compared with those taking enoxaparin (3.8%). Gastritislike symptoms were less common in patients receiving the study drug (0.6%), compared with enoxaparin (1.0%). Clinical myocardial infarction was reported in two (0.1%) study patients and six (0.3%) enoxaparin patients.
Pradaxa was initially indicated by the FDA in 2010 to reduce stroke and systemic embolism risk in patients with nonvalvular atrial fibrillation. In 2014, the FDA approved two additional indications for the drug for the treatment of VTE in patients treated with a parenteral anticoagulant for 5-10 day and to reduce the risk of recurrent VTE in patients who have been previously treated.
On Twitter @whitneymcknight
The Food and Drug Administration has approved dabigatran for the prevention of deep venous thrombosis and pulmonary embolism for patients after hip replacement surgery.
The FDA’s approval was based on the results of two randomized, double-blind, phase III trials in patients undergoing total hip replacement, Boehringer Ingelheim, the manufacturer of the direct thrombin inhibitor, announced.
![]()
In RE-NOVATE I, the first trial, 3,494 patients were randomly assigned to three groups receiving prophylactic treatment with one of two doses of dabigatran (220 mg or 150 mg) once daily, or to the low-molecular-weight heparin enoxaparin at 40 mg once daily for 28-35 days. The first study drug arm was given 110 mg on the day of surgery and 220 mg daily thereafter; the second study drug arm received a dose of 75 mg on the day of surgery and 150 mg daily thereafter. Patients taking the dabigatran (Pradaxa) at 220 mg had a lower composite total of venous thromboembolism (VTE) and all-cause mortality (6.0%) than did those on enoxaparin 40 mg (6.7%), meeting the noninferiority mark (Lancet. 2007 Sep 15;370[9591]:949-56).
In RE-NOVATE II, 2,055 patients were randomly assigned prophylactic treatment for 28-35 days with the study drug dosed at 220 mg once daily, or enoxaparin 40 mg once daily. Patients receiving the study drug were treated with a dose of 110 mg on the day of surgery and 220 mg daily thereafter. The composite total of VTE and all-cause death occurred in 7.7% of patients in the study group vs. 8.8% of patients in the enoxaparin group, which was within the margin for noninferiority (Thromb Haemost. 2011 Apr;105[4]:721-9).
However, there were higher rates of major bleeding in RE-NOVATE I (2.0%, 1.6%) and II (1.4%, 0.9%) with 220 mg vs. enoxaparin. In both studies, the rate of major gastrointestinal bleeds in patients was the same (0.1%) for both the study and control drugs. The rate of any GI bleeds was 1.4% for the study drug and 0.9% for enoxaparin. The most common adverse events in both studies were GI disorders. The incidence rate was the same across all treatment groups (39.5%). Dyspepsia occurred more frequently in patients receiving the study drug (4.1%), compared with those taking enoxaparin (3.8%). Gastritislike symptoms were less common in patients receiving the study drug (0.6%), compared with enoxaparin (1.0%). Clinical myocardial infarction was reported in two (0.1%) study patients and six (0.3%) enoxaparin patients.
Pradaxa was initially indicated by the FDA in 2010 to reduce stroke and systemic embolism risk in patients with nonvalvular atrial fibrillation. In 2014, the FDA approved two additional indications for the drug for the treatment of VTE in patients treated with a parenteral anticoagulant for 5-10 day and to reduce the risk of recurrent VTE in patients who have been previously treated.
On Twitter @whitneymcknight
Stroke risk boosted by adult congenital heart disease
Adult congenital heart disease significantly increases the risk of both hemorrhagic and ischemic stroke, particularly in individuals under 55 years of age, new data suggests.
A retrospective study of 29,638 adults aged 18-64 years with adult congenital heart disease (ACHD) showed that women aged 15-54 years with the disease were more than 12 times as likely to experience an ischemic stroke compared to the general population, while men had a nine-fold increase in risk.
Women aged over 55 years with ACHD had a four-fold higher risk of ischemic stroke, and men had a two-fold increase in risk, compared to the general population, according to a study published Nov. 23 in Circulation.
In the case of hemorrhagic stroke, women aged under 55 had a five-fold greater risk and men had a more than six-fold greater risk of ischemic stroke, while the risk for those older than 55 years was 2-3 times higher (Circulation 2015, November 23 [doi: 10.1161/CIRCULATIONAHA.115.011241]).
The risk of ischemic stroke increased significantly with heart failure, diabetes, or a recent myocardial infarction, and overall, 8.9% of men and 6.8% of women with ACHD who reached the age of 18 years had at least one stroke before age 65.
“Whether subgroups of patients with heart failure and sinus rhythm could benefit from an antithrombotic treatment is a matter of ongoing research in the general population and based on our findings may warrant further investigation in ACHD-patients,” wrote Dr. Jonas Lanz, from the McGill Adult Unit for Congenital Heart Disease Excellence, and co-authors.
The study was funded by the Heart and Stroke Foundation of Québec, the Fonds de Recherche en Santé Québec and the Canadian Institute of Health Research. There were no conflicts of interest declared.
Adult congenital heart disease significantly increases the risk of both hemorrhagic and ischemic stroke, particularly in individuals under 55 years of age, new data suggests.
A retrospective study of 29,638 adults aged 18-64 years with adult congenital heart disease (ACHD) showed that women aged 15-54 years with the disease were more than 12 times as likely to experience an ischemic stroke compared to the general population, while men had a nine-fold increase in risk.
Women aged over 55 years with ACHD had a four-fold higher risk of ischemic stroke, and men had a two-fold increase in risk, compared to the general population, according to a study published Nov. 23 in Circulation.
In the case of hemorrhagic stroke, women aged under 55 had a five-fold greater risk and men had a more than six-fold greater risk of ischemic stroke, while the risk for those older than 55 years was 2-3 times higher (Circulation 2015, November 23 [doi: 10.1161/CIRCULATIONAHA.115.011241]).
The risk of ischemic stroke increased significantly with heart failure, diabetes, or a recent myocardial infarction, and overall, 8.9% of men and 6.8% of women with ACHD who reached the age of 18 years had at least one stroke before age 65.
“Whether subgroups of patients with heart failure and sinus rhythm could benefit from an antithrombotic treatment is a matter of ongoing research in the general population and based on our findings may warrant further investigation in ACHD-patients,” wrote Dr. Jonas Lanz, from the McGill Adult Unit for Congenital Heart Disease Excellence, and co-authors.
The study was funded by the Heart and Stroke Foundation of Québec, the Fonds de Recherche en Santé Québec and the Canadian Institute of Health Research. There were no conflicts of interest declared.
Adult congenital heart disease significantly increases the risk of both hemorrhagic and ischemic stroke, particularly in individuals under 55 years of age, new data suggests.
A retrospective study of 29,638 adults aged 18-64 years with adult congenital heart disease (ACHD) showed that women aged 15-54 years with the disease were more than 12 times as likely to experience an ischemic stroke compared to the general population, while men had a nine-fold increase in risk.
Women aged over 55 years with ACHD had a four-fold higher risk of ischemic stroke, and men had a two-fold increase in risk, compared to the general population, according to a study published Nov. 23 in Circulation.
In the case of hemorrhagic stroke, women aged under 55 had a five-fold greater risk and men had a more than six-fold greater risk of ischemic stroke, while the risk for those older than 55 years was 2-3 times higher (Circulation 2015, November 23 [doi: 10.1161/CIRCULATIONAHA.115.011241]).
The risk of ischemic stroke increased significantly with heart failure, diabetes, or a recent myocardial infarction, and overall, 8.9% of men and 6.8% of women with ACHD who reached the age of 18 years had at least one stroke before age 65.
“Whether subgroups of patients with heart failure and sinus rhythm could benefit from an antithrombotic treatment is a matter of ongoing research in the general population and based on our findings may warrant further investigation in ACHD-patients,” wrote Dr. Jonas Lanz, from the McGill Adult Unit for Congenital Heart Disease Excellence, and co-authors.
The study was funded by the Heart and Stroke Foundation of Québec, the Fonds de Recherche en Santé Québec and the Canadian Institute of Health Research. There were no conflicts of interest declared.
FROM CIRCULATION
Key clinical point:Adult congenital heart disease significantly increases the risk of both hemorrhagic and ischemic stroke, particularly in younger patients.
Major finding: Younger women with adult congenital heart disease have a 12-fold higher risk of ischemic stroke than the general population.
Data source: A retrospective study of 29,638 adults aged 18-64 years with adult congenital heart disease.
Disclosures: Authors were funded by the Heart and Stroke Foundation of Québec, the Fonds de Recherche en Santé Québec and the Canadian Institute of Health Research. There were no conflicts of interest declared.
Synovitis, effusion associated with increased pain sensitivity
Synovitis and effusion were associated with increases in pain sensitivity at the patella and wrist, respectively, in a study of 1,111 patients with or at risk of knee osteoarthritis (OA).
Radiographs and MRIs were taken of the patients’ knees, and the patients wrists and patellae were subjected to standardized quantitative sensory testing (QST) measures. The QST measures included temporal summation, which is a measure of central pain amplification based on “an augmented response to repetitive mechanical stimulation,” and pressure pain threshold (PPT), a measure of sensitivity to pain evoked by mechanical stimulation of nociceptors. (Lower PPTs represent a greater degree of sensitization or pain sensitivity.) All tests were conducted at baseline and 2 years later.

Synovitis was associated with a significant decrease in PPT at the patella, while effusion was associated with a decrease in PPT at the wrist. Effusion was additionally associated with risk of incident temporal summation. In contrast to synovitis and effusion, bone marrow lesions were not associated with either temporal summation or decreased pressure pain threshold.
“Our findings support the potential relevance of inflammation in the development and heightening of sensitization in knee osteoarthritis in humans. We found that synovitis was associated with lower PPT and a decrease in PPT at the patella over time, indicating increased pain sensitization or sensitivity. Effusion was associated with development of new temporal summation at the patella, and with a decrease in PPT at the wrist, a site distant to the pathology; both findings suggest the involvement of central sensitization. Thus inflammation appears to influence the development of and perhaps amplification of sensitization,” said Dr. Tuhina Neogi, of the department of medicine at Boston University and her colleagues.
Read the full study in Arthritis & Rheumatology (doi: 10.1002/art.39488).
Synovitis and effusion were associated with increases in pain sensitivity at the patella and wrist, respectively, in a study of 1,111 patients with or at risk of knee osteoarthritis (OA).
Radiographs and MRIs were taken of the patients’ knees, and the patients wrists and patellae were subjected to standardized quantitative sensory testing (QST) measures. The QST measures included temporal summation, which is a measure of central pain amplification based on “an augmented response to repetitive mechanical stimulation,” and pressure pain threshold (PPT), a measure of sensitivity to pain evoked by mechanical stimulation of nociceptors. (Lower PPTs represent a greater degree of sensitization or pain sensitivity.) All tests were conducted at baseline and 2 years later.
Synovitis was associated with a significant decrease in PPT at the patella, while effusion was associated with a decrease in PPT at the wrist. Effusion was additionally associated with risk of incident temporal summation. In contrast to synovitis and effusion, bone marrow lesions were not associated with either temporal summation or decreased pressure pain threshold.
“Our findings support the potential relevance of inflammation in the development and heightening of sensitization in knee osteoarthritis in humans. We found that synovitis was associated with lower PPT and a decrease in PPT at the patella over time, indicating increased pain sensitization or sensitivity. Effusion was associated with development of new temporal summation at the patella, and with a decrease in PPT at the wrist, a site distant to the pathology; both findings suggest the involvement of central sensitization. Thus inflammation appears to influence the development of and perhaps amplification of sensitization,” said Dr. Tuhina Neogi, of the department of medicine at Boston University and her colleagues.
Read the full study in Arthritis & Rheumatology (doi: 10.1002/art.39488).
Synovitis and effusion were associated with increases in pain sensitivity at the patella and wrist, respectively, in a study of 1,111 patients with or at risk of knee osteoarthritis (OA).
Radiographs and MRIs were taken of the patients’ knees, and the patients wrists and patellae were subjected to standardized quantitative sensory testing (QST) measures. The QST measures included temporal summation, which is a measure of central pain amplification based on “an augmented response to repetitive mechanical stimulation,” and pressure pain threshold (PPT), a measure of sensitivity to pain evoked by mechanical stimulation of nociceptors. (Lower PPTs represent a greater degree of sensitization or pain sensitivity.) All tests were conducted at baseline and 2 years later.
Synovitis was associated with a significant decrease in PPT at the patella, while effusion was associated with a decrease in PPT at the wrist. Effusion was additionally associated with risk of incident temporal summation. In contrast to synovitis and effusion, bone marrow lesions were not associated with either temporal summation or decreased pressure pain threshold.
“Our findings support the potential relevance of inflammation in the development and heightening of sensitization in knee osteoarthritis in humans. We found that synovitis was associated with lower PPT and a decrease in PPT at the patella over time, indicating increased pain sensitization or sensitivity. Effusion was associated with development of new temporal summation at the patella, and with a decrease in PPT at the wrist, a site distant to the pathology; both findings suggest the involvement of central sensitization. Thus inflammation appears to influence the development of and perhaps amplification of sensitization,” said Dr. Tuhina Neogi, of the department of medicine at Boston University and her colleagues.
Read the full study in Arthritis & Rheumatology (doi: 10.1002/art.39488).
FROM ARTHRITIS & RHEUMATOLOGY

Overweight, obese patients at greater risk for knee replacement surgery
Both overweight and obese patients with knee osteoarthritis (OA) are more likely to get knee replacement surgery, compared with normal-weight patients with knee OA, results of a population-based cohort study of people in Catalonia, Spain, suggest.
The study included 105,189 patients, who had been diagnosed with knee OA between 2006 and 2011. Patients with a history of knee OA or knee replacement in either knee before Jan. 1, 2006, and patients with a history inflammatory arthritis were not included in the study.

The patients were followed from the date of knee OA diagnosis until the date they underwent elective knee replacement surgery or until Dec. 31, 2011. (The researchers were unable to follow up with all individuals initially enrolled in the study.) The participants were broken up into the following categories based on their body mass index: normal (BMI was less than 25 kg/m2), overweight (BMI was 25 to less than 30 kg/m2), obese class I (BMI was 30 to less than 35 kg/m2), obese class II (BMI was 35 to less than 40 kg/m2), and obese class III (BMI was greater than or equal to 40 kg/m2).
The risk of knee replacement increased with BMI. For patients with a normal weight, the incidence rates of surgery were 1.35/100 person-years, compared with 3.49/100 person-years in patients in obese class III. Adjusted hazard ratios for knee replacement surgery were 1.41 for overweight, 1.97 for obese class I, 2.39 for obese class II, and 2.67 for obese class III, compared with normal-weight study participants.
An additional finding was a significant interaction between BMI and age on the risk of knee replacement (P is less than .001), with a higher relative hazard associated with obesity among patients aged less than 68 years.
“This research demonstrates that overweight and obesity are strong independent predictors of the clinical progression of knee OA, from disease onset/diagnosis to joint failure and subsequent [knee replacement]. Overweight subjects are at over 40% increased risk of surgery, and those who are obese have a more than doubled risk when compared to subjects with normal weight,” said Kristen M. Leyland, D.Phil., and her colleagues.
Read the full study in Arthritis & Rheumatology (doi: 10.1002/art.39486).
Both overweight and obese patients with knee osteoarthritis (OA) are more likely to get knee replacement surgery, compared with normal-weight patients with knee OA, results of a population-based cohort study of people in Catalonia, Spain, suggest.
The study included 105,189 patients, who had been diagnosed with knee OA between 2006 and 2011. Patients with a history of knee OA or knee replacement in either knee before Jan. 1, 2006, and patients with a history inflammatory arthritis were not included in the study.
The patients were followed from the date of knee OA diagnosis until the date they underwent elective knee replacement surgery or until Dec. 31, 2011. (The researchers were unable to follow up with all individuals initially enrolled in the study.) The participants were broken up into the following categories based on their body mass index: normal (BMI was less than 25 kg/m2), overweight (BMI was 25 to less than 30 kg/m2), obese class I (BMI was 30 to less than 35 kg/m2), obese class II (BMI was 35 to less than 40 kg/m2), and obese class III (BMI was greater than or equal to 40 kg/m2).
The risk of knee replacement increased with BMI. For patients with a normal weight, the incidence rates of surgery were 1.35/100 person-years, compared with 3.49/100 person-years in patients in obese class III. Adjusted hazard ratios for knee replacement surgery were 1.41 for overweight, 1.97 for obese class I, 2.39 for obese class II, and 2.67 for obese class III, compared with normal-weight study participants.
An additional finding was a significant interaction between BMI and age on the risk of knee replacement (P is less than .001), with a higher relative hazard associated with obesity among patients aged less than 68 years.
“This research demonstrates that overweight and obesity are strong independent predictors of the clinical progression of knee OA, from disease onset/diagnosis to joint failure and subsequent [knee replacement]. Overweight subjects are at over 40% increased risk of surgery, and those who are obese have a more than doubled risk when compared to subjects with normal weight,” said Kristen M. Leyland, D.Phil., and her colleagues.
Read the full study in Arthritis & Rheumatology (doi: 10.1002/art.39486).
Both overweight and obese patients with knee osteoarthritis (OA) are more likely to get knee replacement surgery, compared with normal-weight patients with knee OA, results of a population-based cohort study of people in Catalonia, Spain, suggest.
The study included 105,189 patients, who had been diagnosed with knee OA between 2006 and 2011. Patients with a history of knee OA or knee replacement in either knee before Jan. 1, 2006, and patients with a history inflammatory arthritis were not included in the study.
The patients were followed from the date of knee OA diagnosis until the date they underwent elective knee replacement surgery or until Dec. 31, 2011. (The researchers were unable to follow up with all individuals initially enrolled in the study.) The participants were broken up into the following categories based on their body mass index: normal (BMI was less than 25 kg/m2), overweight (BMI was 25 to less than 30 kg/m2), obese class I (BMI was 30 to less than 35 kg/m2), obese class II (BMI was 35 to less than 40 kg/m2), and obese class III (BMI was greater than or equal to 40 kg/m2).
The risk of knee replacement increased with BMI. For patients with a normal weight, the incidence rates of surgery were 1.35/100 person-years, compared with 3.49/100 person-years in patients in obese class III. Adjusted hazard ratios for knee replacement surgery were 1.41 for overweight, 1.97 for obese class I, 2.39 for obese class II, and 2.67 for obese class III, compared with normal-weight study participants.
An additional finding was a significant interaction between BMI and age on the risk of knee replacement (P is less than .001), with a higher relative hazard associated with obesity among patients aged less than 68 years.
“This research demonstrates that overweight and obesity are strong independent predictors of the clinical progression of knee OA, from disease onset/diagnosis to joint failure and subsequent [knee replacement]. Overweight subjects are at over 40% increased risk of surgery, and those who are obese have a more than doubled risk when compared to subjects with normal weight,” said Kristen M. Leyland, D.Phil., and her colleagues.
Read the full study in Arthritis & Rheumatology (doi: 10.1002/art.39486).
FROM ARTHRITIS & RHEUMATOLOGY

Prime-boost flu vaccination strategy effective in children
A prime-boost flu vaccination strategy proved effective in children and adolescents in an open-label phase III trial conducted by the vaccine manufacturer and reported online in the Pediatric Infectious Disease Journal.
This proof-of-concept result suggests that at the outbreak of the next flu pandemic, immediate priming with two doses of a subtype-matched flu vaccine, followed by boosting once a strain-matched vaccine becomes available, will offer the pediatric population better protection than simply waiting for adequate quantities of the strain-matched vaccine to be developed and marketed.

It is impossible to predict which specific viral strain will cause the next flu pandemic, but H5N1 is considered a likely candidate virus, “to the extent that vaccine manufacturers and regulatory authorities are actively developing H5N1 vaccines” for all age groups, said Dr. Patricia Izurieta of GlaxoSmithKline Vaccines, Belgium, and her associates.
Waiting for a pandemic to begin and then producing strain-specific vaccines could take as long as 6 months, and even then supplies probably would be limited. Experts have theorized that preemptive “priming” with already stockpiled H5N1 vaccines immediately after a pandemic begins may provide broad cross-protection that could mitigate the intensity of the subsequent pandemic, potentially reducing morbidity, mortality, and viral transmission.
To test this theory, Dr. Izurieta and her associates mimicked a potential flu pandemic by priming children and adolescents with two doses of an H5N1-AS03 vaccine, giving a booster dose of a specific H5N1 strain at 6 months, and assessing antibody response to all the vaccinations through 12 months. (Assessing actual vaccine efficacy was impossible because it would be unethical to determine this by exposing children to a flu virus.)
A total of 520 participants at a single medical center in the Philippines who were aged 3-18 years (mean age, 9 years) were assigned to two intervention groups and two control groups. Approximately half of these children and adolescents received the intervention – two priming doses of A/Indonesia/05/2005/H5N1-AS03B vaccine – while the control subjects received a single dose of hepatitis A vaccine. At day 182, half of the primed and half of the unprimed participants then received a booster dose of A/turkey/Turkey/01/2005-H5N1-As03B, while the remainder received the hepatitis A vaccine.
Compared with no priming, priming afforded superior seroconversion and putative seroprotection against the second, specific strain of H5N1. This robust protective effect was seen within 10 days after vaccination, occurred across all ages, and persisted through 12 months of follow-up, the investigators said (Ped Infect Dis J. 2015 Nov 6. [doi: 10.1097/INF.0000000000000968]).
Adverse events within 7 days of the priming vaccinations developed in 68%-76% of the intervention group, compared with only 37%-64% of the control group. Adverse events within 7 days of the booster vaccinations developed in 69% of the intervention group, compared with only 40% of the control group. The most common adverse events were upper respiratory tract infection (20% vs 14%), nasopharyngitis (4% vs 3%), and rhinitis (3% in both study groups). The most serious adverse events affected two children and were classified as grade 3.
The study results suggest that a prime-boost strategy can induce broad and long-lasting immunity and could be effectively employed in this age group in the prepandemic setting, Dr. Izurieta and her associates reported.
This trial was funded by GlaxoSmithKline, which also was involved in the design and conduct of the study, the data analysis, and the writing and publishing of the report. Dr. Izurieta and two of her associates are employed by GSK.
A prime-boost flu vaccination strategy proved effective in children and adolescents in an open-label phase III trial conducted by the vaccine manufacturer and reported online in the Pediatric Infectious Disease Journal.
This proof-of-concept result suggests that at the outbreak of the next flu pandemic, immediate priming with two doses of a subtype-matched flu vaccine, followed by boosting once a strain-matched vaccine becomes available, will offer the pediatric population better protection than simply waiting for adequate quantities of the strain-matched vaccine to be developed and marketed.
It is impossible to predict which specific viral strain will cause the next flu pandemic, but H5N1 is considered a likely candidate virus, “to the extent that vaccine manufacturers and regulatory authorities are actively developing H5N1 vaccines” for all age groups, said Dr. Patricia Izurieta of GlaxoSmithKline Vaccines, Belgium, and her associates.
Waiting for a pandemic to begin and then producing strain-specific vaccines could take as long as 6 months, and even then supplies probably would be limited. Experts have theorized that preemptive “priming” with already stockpiled H5N1 vaccines immediately after a pandemic begins may provide broad cross-protection that could mitigate the intensity of the subsequent pandemic, potentially reducing morbidity, mortality, and viral transmission.
To test this theory, Dr. Izurieta and her associates mimicked a potential flu pandemic by priming children and adolescents with two doses of an H5N1-AS03 vaccine, giving a booster dose of a specific H5N1 strain at 6 months, and assessing antibody response to all the vaccinations through 12 months. (Assessing actual vaccine efficacy was impossible because it would be unethical to determine this by exposing children to a flu virus.)
A total of 520 participants at a single medical center in the Philippines who were aged 3-18 years (mean age, 9 years) were assigned to two intervention groups and two control groups. Approximately half of these children and adolescents received the intervention – two priming doses of A/Indonesia/05/2005/H5N1-AS03B vaccine – while the control subjects received a single dose of hepatitis A vaccine. At day 182, half of the primed and half of the unprimed participants then received a booster dose of A/turkey/Turkey/01/2005-H5N1-As03B, while the remainder received the hepatitis A vaccine.
Compared with no priming, priming afforded superior seroconversion and putative seroprotection against the second, specific strain of H5N1. This robust protective effect was seen within 10 days after vaccination, occurred across all ages, and persisted through 12 months of follow-up, the investigators said (Ped Infect Dis J. 2015 Nov 6. [doi: 10.1097/INF.0000000000000968]).
Adverse events within 7 days of the priming vaccinations developed in 68%-76% of the intervention group, compared with only 37%-64% of the control group. Adverse events within 7 days of the booster vaccinations developed in 69% of the intervention group, compared with only 40% of the control group. The most common adverse events were upper respiratory tract infection (20% vs 14%), nasopharyngitis (4% vs 3%), and rhinitis (3% in both study groups). The most serious adverse events affected two children and were classified as grade 3.
The study results suggest that a prime-boost strategy can induce broad and long-lasting immunity and could be effectively employed in this age group in the prepandemic setting, Dr. Izurieta and her associates reported.
This trial was funded by GlaxoSmithKline, which also was involved in the design and conduct of the study, the data analysis, and the writing and publishing of the report. Dr. Izurieta and two of her associates are employed by GSK.
A prime-boost flu vaccination strategy proved effective in children and adolescents in an open-label phase III trial conducted by the vaccine manufacturer and reported online in the Pediatric Infectious Disease Journal.
This proof-of-concept result suggests that at the outbreak of the next flu pandemic, immediate priming with two doses of a subtype-matched flu vaccine, followed by boosting once a strain-matched vaccine becomes available, will offer the pediatric population better protection than simply waiting for adequate quantities of the strain-matched vaccine to be developed and marketed.
It is impossible to predict which specific viral strain will cause the next flu pandemic, but H5N1 is considered a likely candidate virus, “to the extent that vaccine manufacturers and regulatory authorities are actively developing H5N1 vaccines” for all age groups, said Dr. Patricia Izurieta of GlaxoSmithKline Vaccines, Belgium, and her associates.
Waiting for a pandemic to begin and then producing strain-specific vaccines could take as long as 6 months, and even then supplies probably would be limited. Experts have theorized that preemptive “priming” with already stockpiled H5N1 vaccines immediately after a pandemic begins may provide broad cross-protection that could mitigate the intensity of the subsequent pandemic, potentially reducing morbidity, mortality, and viral transmission.
To test this theory, Dr. Izurieta and her associates mimicked a potential flu pandemic by priming children and adolescents with two doses of an H5N1-AS03 vaccine, giving a booster dose of a specific H5N1 strain at 6 months, and assessing antibody response to all the vaccinations through 12 months. (Assessing actual vaccine efficacy was impossible because it would be unethical to determine this by exposing children to a flu virus.)
A total of 520 participants at a single medical center in the Philippines who were aged 3-18 years (mean age, 9 years) were assigned to two intervention groups and two control groups. Approximately half of these children and adolescents received the intervention – two priming doses of A/Indonesia/05/2005/H5N1-AS03B vaccine – while the control subjects received a single dose of hepatitis A vaccine. At day 182, half of the primed and half of the unprimed participants then received a booster dose of A/turkey/Turkey/01/2005-H5N1-As03B, while the remainder received the hepatitis A vaccine.
Compared with no priming, priming afforded superior seroconversion and putative seroprotection against the second, specific strain of H5N1. This robust protective effect was seen within 10 days after vaccination, occurred across all ages, and persisted through 12 months of follow-up, the investigators said (Ped Infect Dis J. 2015 Nov 6. [doi: 10.1097/INF.0000000000000968]).
Adverse events within 7 days of the priming vaccinations developed in 68%-76% of the intervention group, compared with only 37%-64% of the control group. Adverse events within 7 days of the booster vaccinations developed in 69% of the intervention group, compared with only 40% of the control group. The most common adverse events were upper respiratory tract infection (20% vs 14%), nasopharyngitis (4% vs 3%), and rhinitis (3% in both study groups). The most serious adverse events affected two children and were classified as grade 3.
The study results suggest that a prime-boost strategy can induce broad and long-lasting immunity and could be effectively employed in this age group in the prepandemic setting, Dr. Izurieta and her associates reported.
This trial was funded by GlaxoSmithKline, which also was involved in the design and conduct of the study, the data analysis, and the writing and publishing of the report. Dr. Izurieta and two of her associates are employed by GSK.
FROM THE PEDIATRIC INFECTIOUS DISEASE JOURNAL
Key clinical point: A prime-boost flu vaccination strategy was found effective in children and adolescents.
Major finding: Compared with no priming, priming afforded superior seroconversion and putative seroprotection against the second, specific strain of H5N1.
Data source: An industry-sponsored, single-center, open-label phase III trial involving 520 participants aged 3-18 years followed for 1 year.
Disclosures: This trial was funded by GlaxoSmithKline, which also was involved in the design and conduct of the study, the data analysis, and the writing and publishing of the report. Dr. Izurieta and two of her associates are employed by GSK.

AHA: DAPT score helps decide whether DAPT continues
ORLANDO – The common challenge faced by clinicians in deciding whether or not to continue dual antiplatelet therapy beyond a year in a patient who underwent percutaneous coronary intervention has gotten easier.
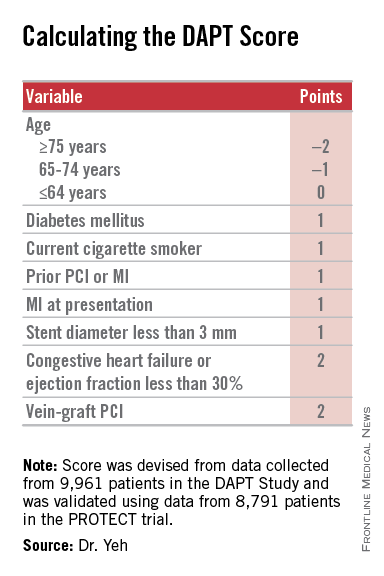
Researchers have devised a simple, eight-element scoring system using information already available in a patient’s records to help determine whether an individual patient will be more likely to benefit from continuing or stopping dual antiplatelet therapy (DAPT).

“The DAPT score may help clinicians decide who should and who should not be treated with extended DAPT,” Dr. Robert W. Yeh said at the American Heart Association Scientific Sessions.
“This is a step forward for an issue we deal with daily, balancing an individual patient’s risk from ischemia and bleeding,” commented Dr. Alice Jacobs, professor of medicine at Boston University and director of the cardiac catheterization laboratory and interventional cardiology at Boston Medical Center.
Dr. Yeh and his associates devised the DAPT score from the data collected in the DAPT study, which enrolled more than 25,000 patients and randomized about 10,000 to test whether patients fared better by stopping or continuing DAPT after completing their initial year of DAPT following percutaneous coronary intervention (PCI). The DAPT study results showed that after 18 additional months, continuing DAPT cut the rate of definite or probable stent thrombosis by 1 percentage point and the combined rate of death, MI, or stroke by 1.6 percentage points, both statistically significant differences, compared with patients randomized to treatment with aspirin plus placebo. The results also showed that continued DAPT increased GUSTO moderate or severe bleeding events by 1 percentage point, compared with the control patients (N Engl J Med. 2014 Dec 4;371[23]:2155-66).

The researchers used data collected in the DAPT study to build risk models using patient- and procedure-specific variables that predicted the ischemic and bleeding outcomes, and then combined the two into a single model. That meant abandoning some variables that had significant impact on both outcomes.
The result was a scoring system that includes eight variables that result in a score that ranges from –2 to 9. The analysis showed that a score of 1 or less identified patients for whom the risk for bleeding outweighs their potential gain by avoiding an ischemic event by about 2.5-fold, and hence likely would fare better by stopping DAPT. A score of 2 or higher flagged patients who benefited about eightfold more from avoided ischemic events, compared with their risk for a moderate or severe bleed.
Patient scores showed a classic bell-shaped curve, with roughly a quarter of the DAPT study patients having a score of 1 and about a quarter with a score of 2, about 16% had a score of 0 and about 16% had a score of 3, and about 8% had a score of –1 or –2, while about 9% had a score of 4 or more.
The investigators validated the scoring system using data collected in the PROTECT trial, which included 8,791 patients who underwent PCI during 2007-2008. Dr. Yeh acknowledged that the discrimination strength of the models he and his associated developed was “modest,” but added that its efficacy was greater than what has been shown in validation cohorts for the commonly used CH2ADS2-VASc and HAS-BLED scoring systems.
Dr. Yeh stressed that using the DAPT score “cannot trump clinical judgment.” He suggested that a clinician use the score to help facilitate a conversation with a PCI patient when the time comes to decide whether or not to continue DAPT beyond 1 year.
Other factors that could influence the decision include the length of the stented coronary lesions or prior radiation exposure to the patient’s coronary arteries, said Dr. Laura Mauri, who led the DAPT study and collaborated on developing the DAPT score. “It requires judgment to decide [on whether to continue DAPT] for patients who are on the borderline” for risk and benefit. “This gives patients a way to better understand what they might gain or lose” by continuing treatment. Without the quantification that the DAPT score provides, the balance of risk and benefit “is somewhat nebulous,” said Dr. Mauri, an interventional cardiologist and director of the Center for Clinical Biometrics in the division of cardiovascular medicine at Brigham and Women’s Hospital in Boston.

The investigators who ran the DAPT study realized several years before the study finished that development of the DAPT score was a critical part of applying the findings from the study into clinical practice, she said in an interview.
Dr. Yeh cautioned that the score is only appropriate for patients who match those enrolled into the DAPT study: patients who went through their first year post PCI on DAPT without having any ischemic or bleeding complications. For these patients, “we feel the DAPT score is incredibly valuable,” said Dr. Yeh, an interventional cardiologist and director of the Center for Outcomes Research in Cardiology at Beth Israel Deaconess Medical Center in Boston. He and his associates are now using data from the DAPT study to model bleeding and ischemia risks during the first year following PCI to try to come up with risk models that can address DAPT use during this period.
Dr. Yeh and Dr. Mauri have placed a link to the electronic DAPT score calculator on the website for the DAPT study (www.daptstudy.org), and Dr. Yeh said that an app version will soon become available. Interventionalists in the programs that Dr. Yeh and Mauri are affiliated with have recently begun using the DAPT score calculator in their routine practice, Dr. Yeh said.
The DAPT study received funding from Abbott, Boston Scientific, Cordis, Medtronic, Bristol-Myers Squib-Sanofi, Eli Lilly, and Daiichi Sankyo. Derivation of the DAPT score was funded by the National Institutes of Health. Dr. Yeh has received honoraria from Abbott, Boston Scientific, and Merck. Dr. Jacobs had no disclosures. Dr. Mauri has been a consultant to Biotronik, Medtronic, and St. Jude and has received research funding from several companies.
On Twitter @mitchelzoler
The DAPT score is a clever and innovative idea. It is a major step forward in helping clinicians decide which patients should continue dual antiplatelet therapy after safely completing a year on this therapy following percutaneous coronary intervention. The DAPT score was data driven and provides a tool to help personalize decision making with a simple, practical solution to a common clinical dilemma. It’s a welcome addition to our decision-making process.
The competing risks from bleeding events caused by continued dual antiplatelet therapy (DAPT) beyond 1 year and ischemic events caused by stopping DAPT creates difficulty in determining whether or not to continue or stop DAPT for an individual patient. The DAPT score helps make that decision.
 |
| Mitchel L. Zoler/Frontline Medical News Dr. James de Lemos |
The analysis performed by Dr. Yeh and his associates produced a clear and convincing result. The primary caveat is that it is only applicable to patients who entered the randomized phase of the DAPT study, specifically patients who underwent a full first year of DAPT treatment following PCI without an ischemic or major bleeding event. I would like to see replication of the score’s validation in an additional data set, although few data sets exist that are suitable for such replication. Although the discrimination produced by the DAPT score is moderate, it compares favorably with other widely used clinical decision scores such as the CHA2ADS2-VASc.
The added decision-making ability facilitated by this score revises my interpretation of the results from the DAPT study. When the results of the trial appeared in 2014, I considered the outcome null because of the problem it highlighted in balancing the competing risks of ischemic and bleeding events when deciding about continuing DAPT beyond 1 year. The DAPT score helps produce a much clearer risk versus benefit decision for a sizable subset of patients who undergo percutaneous coronary intervention.
Dr. James de Lemos is a professor of medicine at UT Southwestern Medical Center, Dallas, and chief of the cardiology service at Parkland Memorial Hospital in Dallas. He has received honoraria from Novo Nordisk and St. Jude and research funding from Roche Diagnostics and Abbott Diagnostics. He made these comments as the designated discussant for Dr. Yeh’s report.
The DAPT score is a clever and innovative idea. It is a major step forward in helping clinicians decide which patients should continue dual antiplatelet therapy after safely completing a year on this therapy following percutaneous coronary intervention. The DAPT score was data driven and provides a tool to help personalize decision making with a simple, practical solution to a common clinical dilemma. It’s a welcome addition to our decision-making process.
The competing risks from bleeding events caused by continued dual antiplatelet therapy (DAPT) beyond 1 year and ischemic events caused by stopping DAPT creates difficulty in determining whether or not to continue or stop DAPT for an individual patient. The DAPT score helps make that decision.
|
| Mitchel L. Zoler/Frontline Medical News Dr. James de Lemos |
The analysis performed by Dr. Yeh and his associates produced a clear and convincing result. The primary caveat is that it is only applicable to patients who entered the randomized phase of the DAPT study, specifically patients who underwent a full first year of DAPT treatment following PCI without an ischemic or major bleeding event. I would like to see replication of the score’s validation in an additional data set, although few data sets exist that are suitable for such replication. Although the discrimination produced by the DAPT score is moderate, it compares favorably with other widely used clinical decision scores such as the CHA2ADS2-VASc.
The added decision-making ability facilitated by this score revises my interpretation of the results from the DAPT study. When the results of the trial appeared in 2014, I considered the outcome null because of the problem it highlighted in balancing the competing risks of ischemic and bleeding events when deciding about continuing DAPT beyond 1 year. The DAPT score helps produce a much clearer risk versus benefit decision for a sizable subset of patients who undergo percutaneous coronary intervention.
Dr. James de Lemos is a professor of medicine at UT Southwestern Medical Center, Dallas, and chief of the cardiology service at Parkland Memorial Hospital in Dallas. He has received honoraria from Novo Nordisk and St. Jude and research funding from Roche Diagnostics and Abbott Diagnostics. He made these comments as the designated discussant for Dr. Yeh’s report.
The DAPT score is a clever and innovative idea. It is a major step forward in helping clinicians decide which patients should continue dual antiplatelet therapy after safely completing a year on this therapy following percutaneous coronary intervention. The DAPT score was data driven and provides a tool to help personalize decision making with a simple, practical solution to a common clinical dilemma. It’s a welcome addition to our decision-making process.
The competing risks from bleeding events caused by continued dual antiplatelet therapy (DAPT) beyond 1 year and ischemic events caused by stopping DAPT creates difficulty in determining whether or not to continue or stop DAPT for an individual patient. The DAPT score helps make that decision.
|
| Mitchel L. Zoler/Frontline Medical News Dr. James de Lemos |
The analysis performed by Dr. Yeh and his associates produced a clear and convincing result. The primary caveat is that it is only applicable to patients who entered the randomized phase of the DAPT study, specifically patients who underwent a full first year of DAPT treatment following PCI without an ischemic or major bleeding event. I would like to see replication of the score’s validation in an additional data set, although few data sets exist that are suitable for such replication. Although the discrimination produced by the DAPT score is moderate, it compares favorably with other widely used clinical decision scores such as the CHA2ADS2-VASc.
The added decision-making ability facilitated by this score revises my interpretation of the results from the DAPT study. When the results of the trial appeared in 2014, I considered the outcome null because of the problem it highlighted in balancing the competing risks of ischemic and bleeding events when deciding about continuing DAPT beyond 1 year. The DAPT score helps produce a much clearer risk versus benefit decision for a sizable subset of patients who undergo percutaneous coronary intervention.
Dr. James de Lemos is a professor of medicine at UT Southwestern Medical Center, Dallas, and chief of the cardiology service at Parkland Memorial Hospital in Dallas. He has received honoraria from Novo Nordisk and St. Jude and research funding from Roche Diagnostics and Abbott Diagnostics. He made these comments as the designated discussant for Dr. Yeh’s report.
ORLANDO – The common challenge faced by clinicians in deciding whether or not to continue dual antiplatelet therapy beyond a year in a patient who underwent percutaneous coronary intervention has gotten easier.
Researchers have devised a simple, eight-element scoring system using information already available in a patient’s records to help determine whether an individual patient will be more likely to benefit from continuing or stopping dual antiplatelet therapy (DAPT).
“The DAPT score may help clinicians decide who should and who should not be treated with extended DAPT,” Dr. Robert W. Yeh said at the American Heart Association Scientific Sessions.
“This is a step forward for an issue we deal with daily, balancing an individual patient’s risk from ischemia and bleeding,” commented Dr. Alice Jacobs, professor of medicine at Boston University and director of the cardiac catheterization laboratory and interventional cardiology at Boston Medical Center.
Dr. Yeh and his associates devised the DAPT score from the data collected in the DAPT study, which enrolled more than 25,000 patients and randomized about 10,000 to test whether patients fared better by stopping or continuing DAPT after completing their initial year of DAPT following percutaneous coronary intervention (PCI). The DAPT study results showed that after 18 additional months, continuing DAPT cut the rate of definite or probable stent thrombosis by 1 percentage point and the combined rate of death, MI, or stroke by 1.6 percentage points, both statistically significant differences, compared with patients randomized to treatment with aspirin plus placebo. The results also showed that continued DAPT increased GUSTO moderate or severe bleeding events by 1 percentage point, compared with the control patients (N Engl J Med. 2014 Dec 4;371[23]:2155-66).
The researchers used data collected in the DAPT study to build risk models using patient- and procedure-specific variables that predicted the ischemic and bleeding outcomes, and then combined the two into a single model. That meant abandoning some variables that had significant impact on both outcomes.
The result was a scoring system that includes eight variables that result in a score that ranges from –2 to 9. The analysis showed that a score of 1 or less identified patients for whom the risk for bleeding outweighs their potential gain by avoiding an ischemic event by about 2.5-fold, and hence likely would fare better by stopping DAPT. A score of 2 or higher flagged patients who benefited about eightfold more from avoided ischemic events, compared with their risk for a moderate or severe bleed.
Patient scores showed a classic bell-shaped curve, with roughly a quarter of the DAPT study patients having a score of 1 and about a quarter with a score of 2, about 16% had a score of 0 and about 16% had a score of 3, and about 8% had a score of –1 or –2, while about 9% had a score of 4 or more.
The investigators validated the scoring system using data collected in the PROTECT trial, which included 8,791 patients who underwent PCI during 2007-2008. Dr. Yeh acknowledged that the discrimination strength of the models he and his associated developed was “modest,” but added that its efficacy was greater than what has been shown in validation cohorts for the commonly used CH2ADS2-VASc and HAS-BLED scoring systems.
Dr. Yeh stressed that using the DAPT score “cannot trump clinical judgment.” He suggested that a clinician use the score to help facilitate a conversation with a PCI patient when the time comes to decide whether or not to continue DAPT beyond 1 year.
Other factors that could influence the decision include the length of the stented coronary lesions or prior radiation exposure to the patient’s coronary arteries, said Dr. Laura Mauri, who led the DAPT study and collaborated on developing the DAPT score. “It requires judgment to decide [on whether to continue DAPT] for patients who are on the borderline” for risk and benefit. “This gives patients a way to better understand what they might gain or lose” by continuing treatment. Without the quantification that the DAPT score provides, the balance of risk and benefit “is somewhat nebulous,” said Dr. Mauri, an interventional cardiologist and director of the Center for Clinical Biometrics in the division of cardiovascular medicine at Brigham and Women’s Hospital in Boston.
The investigators who ran the DAPT study realized several years before the study finished that development of the DAPT score was a critical part of applying the findings from the study into clinical practice, she said in an interview.
Dr. Yeh cautioned that the score is only appropriate for patients who match those enrolled into the DAPT study: patients who went through their first year post PCI on DAPT without having any ischemic or bleeding complications. For these patients, “we feel the DAPT score is incredibly valuable,” said Dr. Yeh, an interventional cardiologist and director of the Center for Outcomes Research in Cardiology at Beth Israel Deaconess Medical Center in Boston. He and his associates are now using data from the DAPT study to model bleeding and ischemia risks during the first year following PCI to try to come up with risk models that can address DAPT use during this period.
Dr. Yeh and Dr. Mauri have placed a link to the electronic DAPT score calculator on the website for the DAPT study (www.daptstudy.org), and Dr. Yeh said that an app version will soon become available. Interventionalists in the programs that Dr. Yeh and Mauri are affiliated with have recently begun using the DAPT score calculator in their routine practice, Dr. Yeh said.
The DAPT study received funding from Abbott, Boston Scientific, Cordis, Medtronic, Bristol-Myers Squib-Sanofi, Eli Lilly, and Daiichi Sankyo. Derivation of the DAPT score was funded by the National Institutes of Health. Dr. Yeh has received honoraria from Abbott, Boston Scientific, and Merck. Dr. Jacobs had no disclosures. Dr. Mauri has been a consultant to Biotronik, Medtronic, and St. Jude and has received research funding from several companies.
On Twitter @mitchelzoler
ORLANDO – The common challenge faced by clinicians in deciding whether or not to continue dual antiplatelet therapy beyond a year in a patient who underwent percutaneous coronary intervention has gotten easier.
Researchers have devised a simple, eight-element scoring system using information already available in a patient’s records to help determine whether an individual patient will be more likely to benefit from continuing or stopping dual antiplatelet therapy (DAPT).
“The DAPT score may help clinicians decide who should and who should not be treated with extended DAPT,” Dr. Robert W. Yeh said at the American Heart Association Scientific Sessions.
“This is a step forward for an issue we deal with daily, balancing an individual patient’s risk from ischemia and bleeding,” commented Dr. Alice Jacobs, professor of medicine at Boston University and director of the cardiac catheterization laboratory and interventional cardiology at Boston Medical Center.
Dr. Yeh and his associates devised the DAPT score from the data collected in the DAPT study, which enrolled more than 25,000 patients and randomized about 10,000 to test whether patients fared better by stopping or continuing DAPT after completing their initial year of DAPT following percutaneous coronary intervention (PCI). The DAPT study results showed that after 18 additional months, continuing DAPT cut the rate of definite or probable stent thrombosis by 1 percentage point and the combined rate of death, MI, or stroke by 1.6 percentage points, both statistically significant differences, compared with patients randomized to treatment with aspirin plus placebo. The results also showed that continued DAPT increased GUSTO moderate or severe bleeding events by 1 percentage point, compared with the control patients (N Engl J Med. 2014 Dec 4;371[23]:2155-66).
The researchers used data collected in the DAPT study to build risk models using patient- and procedure-specific variables that predicted the ischemic and bleeding outcomes, and then combined the two into a single model. That meant abandoning some variables that had significant impact on both outcomes.
The result was a scoring system that includes eight variables that result in a score that ranges from –2 to 9. The analysis showed that a score of 1 or less identified patients for whom the risk for bleeding outweighs their potential gain by avoiding an ischemic event by about 2.5-fold, and hence likely would fare better by stopping DAPT. A score of 2 or higher flagged patients who benefited about eightfold more from avoided ischemic events, compared with their risk for a moderate or severe bleed.
Patient scores showed a classic bell-shaped curve, with roughly a quarter of the DAPT study patients having a score of 1 and about a quarter with a score of 2, about 16% had a score of 0 and about 16% had a score of 3, and about 8% had a score of –1 or –2, while about 9% had a score of 4 or more.
The investigators validated the scoring system using data collected in the PROTECT trial, which included 8,791 patients who underwent PCI during 2007-2008. Dr. Yeh acknowledged that the discrimination strength of the models he and his associated developed was “modest,” but added that its efficacy was greater than what has been shown in validation cohorts for the commonly used CH2ADS2-VASc and HAS-BLED scoring systems.
Dr. Yeh stressed that using the DAPT score “cannot trump clinical judgment.” He suggested that a clinician use the score to help facilitate a conversation with a PCI patient when the time comes to decide whether or not to continue DAPT beyond 1 year.
Other factors that could influence the decision include the length of the stented coronary lesions or prior radiation exposure to the patient’s coronary arteries, said Dr. Laura Mauri, who led the DAPT study and collaborated on developing the DAPT score. “It requires judgment to decide [on whether to continue DAPT] for patients who are on the borderline” for risk and benefit. “This gives patients a way to better understand what they might gain or lose” by continuing treatment. Without the quantification that the DAPT score provides, the balance of risk and benefit “is somewhat nebulous,” said Dr. Mauri, an interventional cardiologist and director of the Center for Clinical Biometrics in the division of cardiovascular medicine at Brigham and Women’s Hospital in Boston.
The investigators who ran the DAPT study realized several years before the study finished that development of the DAPT score was a critical part of applying the findings from the study into clinical practice, she said in an interview.
Dr. Yeh cautioned that the score is only appropriate for patients who match those enrolled into the DAPT study: patients who went through their first year post PCI on DAPT without having any ischemic or bleeding complications. For these patients, “we feel the DAPT score is incredibly valuable,” said Dr. Yeh, an interventional cardiologist and director of the Center for Outcomes Research in Cardiology at Beth Israel Deaconess Medical Center in Boston. He and his associates are now using data from the DAPT study to model bleeding and ischemia risks during the first year following PCI to try to come up with risk models that can address DAPT use during this period.
Dr. Yeh and Dr. Mauri have placed a link to the electronic DAPT score calculator on the website for the DAPT study (www.daptstudy.org), and Dr. Yeh said that an app version will soon become available. Interventionalists in the programs that Dr. Yeh and Mauri are affiliated with have recently begun using the DAPT score calculator in their routine practice, Dr. Yeh said.
The DAPT study received funding from Abbott, Boston Scientific, Cordis, Medtronic, Bristol-Myers Squib-Sanofi, Eli Lilly, and Daiichi Sankyo. Derivation of the DAPT score was funded by the National Institutes of Health. Dr. Yeh has received honoraria from Abbott, Boston Scientific, and Merck. Dr. Jacobs had no disclosures. Dr. Mauri has been a consultant to Biotronik, Medtronic, and St. Jude and has received research funding from several companies.
On Twitter @mitchelzoler
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: A year after percutaneous coronary intervention, the DAPT score helps clinicians decide whether to continue dual antiplatelet therapy.
Major finding: DAPT scores of 1 or less were linked with a 2.5-fold higher risk of bleeding than ischemic events prevented by continued DAPT; scores of 2 or more were linked with an eightfold increased rate of ischemic events prevented, compared with bleeding events triggered.
Data source: The DAPT study, a multicenter, international, randomized trial that enrolled 25,682 patients.
Disclosures: The DAPT study received funding from Abbott, Boston Scientific, Cordis, Medtronic, Bristol-Myers Squibb-Sanofi, Eli Lilly, and Daiichi Sankyo. Derivation of the DAPT score was funded by the National Institutes of Health. Dr. Yeh has received honoraria from Abbott, Boston Scientific, and Merck.

ACC/AHA guidelines upgrade multivessel PCI, downgrade thrombectomy
ORLANDO – Two guideline panels jointly upgraded percutaneous coronary interventions in noninfarct coronary arteries in patients with a recent MI, and downgraded manual aspiration thrombectomy in acute MI patients.
The panels, organized by the American College of Cardiology and the American Heart Association, cover, respectively, percutaneous coronary intervention (PCI) and ST-elevation MI (STEMI).
The revised guidance on percutaneous coronary intervention for multivessel coronary disease affects roughly half the patients with an acute ST-elevation MI, those who have significant stenoses in noninfarct coronaries. The 2011 guidelines from the PCI panel (J Am Coll Cardiol. 2011 Dec;58[24]:e44-122) and the 2013 guidelines from the STEMI committee (J Am Coll Cardiol. 2013 Jan;61[4]:e78-140) only endorsed revascularization of stenotic, noninfarct arteries in patients with cardiogenic shock or severe heart failure. Multivessel PCI for patients without hemodynamic compromise at the time of primary PCI was deemed a class III indication, “thought to confer harm,” based on the results of several observational studies, Dr. Patrick T. O’Gara said at the American Heart Association scientific sessions.

After the 2013 STEMI guidelines came out, results from four studies showed either benefit or no harm from multivessel PCI in patients with appropriate anatomy performed either at the time of primary PCI or as a scheduled, staged procedure soon after primary PCI, said Dr. O’Gara, professor and director of clinical cardiology at Brigham and Women’s Hospital in Boston. Three of the four studies, PRAMI (N Engl J Med. 2013 Sept 19;369[12]:1115-23), CvLPRIT (J Am Coll Cardiol. 2015 March;65[10]:963-72), and DANAMI-3–PRIMULTI (Lancet. 2015 Aug 15;386[9994]:665-71), are published, while results from the fourth, PRAGUE-13, came out in a meeting report this year but as of mid-November had not been published.
Based on these data the two committees reset PCI of noninfarct arteries as a class IIb recommendation that “could be considered” in selected STEMI patients with multivessel disease who are hemodynamically stable.
The guidelines further said that “insufficient data exist to inform a recommendation” about the optimal time for this multivessel PCI, which could be coincident with primary PCI or 2 days, 3 days, a week, or even later after initial PCI, said Dr. O’Gara. “The writing committees do not advocate for routine use of multivessel PCI; clinical judgment is obviously needed,” he cautioned.
The two committees also reassessed manual aspiration thrombectomy during primary PCI for STEMI, which until now has been a class IIa recommendation, “reasonable” for STEMI patients undergoing primary PCI. But based on the neutral results for thrombectomy in the recent findings from the two largest randomized trials of thrombectomy, TOTAL (N Engl J Med. 2015 April 9;372[15]:1389-98) and TASTE (N Engl J Med. 2013 Oct 24;369[17]:1587-97), as well as a recent, 17-trial meta-analysis, the revised guidelines rate routine aspiration thrombectomy as class III, with “no benefit” and “not useful,” and selective and bailout thrombectomy as a class IIb recommendation, with usefulness that is “not well established.”
Selective use of thrombectomy will require good judgment, noted Dr. O’Gara, who added “it will be important to see what effect, if any, these observations, this evidence, and this distillation of the recommendations from the writing committees have on a change in practice” – using thrombectomy to treat STEMI patients.
Dr. O’Gara chairs the STEMI writing panel of the American College of Cardiology and American Heart Association.
On Twitter @mitchelzoler
ORLANDO – Two guideline panels jointly upgraded percutaneous coronary interventions in noninfarct coronary arteries in patients with a recent MI, and downgraded manual aspiration thrombectomy in acute MI patients.
The panels, organized by the American College of Cardiology and the American Heart Association, cover, respectively, percutaneous coronary intervention (PCI) and ST-elevation MI (STEMI).
The revised guidance on percutaneous coronary intervention for multivessel coronary disease affects roughly half the patients with an acute ST-elevation MI, those who have significant stenoses in noninfarct coronaries. The 2011 guidelines from the PCI panel (J Am Coll Cardiol. 2011 Dec;58[24]:e44-122) and the 2013 guidelines from the STEMI committee (J Am Coll Cardiol. 2013 Jan;61[4]:e78-140) only endorsed revascularization of stenotic, noninfarct arteries in patients with cardiogenic shock or severe heart failure. Multivessel PCI for patients without hemodynamic compromise at the time of primary PCI was deemed a class III indication, “thought to confer harm,” based on the results of several observational studies, Dr. Patrick T. O’Gara said at the American Heart Association scientific sessions.
After the 2013 STEMI guidelines came out, results from four studies showed either benefit or no harm from multivessel PCI in patients with appropriate anatomy performed either at the time of primary PCI or as a scheduled, staged procedure soon after primary PCI, said Dr. O’Gara, professor and director of clinical cardiology at Brigham and Women’s Hospital in Boston. Three of the four studies, PRAMI (N Engl J Med. 2013 Sept 19;369[12]:1115-23), CvLPRIT (J Am Coll Cardiol. 2015 March;65[10]:963-72), and DANAMI-3–PRIMULTI (Lancet. 2015 Aug 15;386[9994]:665-71), are published, while results from the fourth, PRAGUE-13, came out in a meeting report this year but as of mid-November had not been published.
Based on these data the two committees reset PCI of noninfarct arteries as a class IIb recommendation that “could be considered” in selected STEMI patients with multivessel disease who are hemodynamically stable.
The guidelines further said that “insufficient data exist to inform a recommendation” about the optimal time for this multivessel PCI, which could be coincident with primary PCI or 2 days, 3 days, a week, or even later after initial PCI, said Dr. O’Gara. “The writing committees do not advocate for routine use of multivessel PCI; clinical judgment is obviously needed,” he cautioned.
The two committees also reassessed manual aspiration thrombectomy during primary PCI for STEMI, which until now has been a class IIa recommendation, “reasonable” for STEMI patients undergoing primary PCI. But based on the neutral results for thrombectomy in the recent findings from the two largest randomized trials of thrombectomy, TOTAL (N Engl J Med. 2015 April 9;372[15]:1389-98) and TASTE (N Engl J Med. 2013 Oct 24;369[17]:1587-97), as well as a recent, 17-trial meta-analysis, the revised guidelines rate routine aspiration thrombectomy as class III, with “no benefit” and “not useful,” and selective and bailout thrombectomy as a class IIb recommendation, with usefulness that is “not well established.”
Selective use of thrombectomy will require good judgment, noted Dr. O’Gara, who added “it will be important to see what effect, if any, these observations, this evidence, and this distillation of the recommendations from the writing committees have on a change in practice” – using thrombectomy to treat STEMI patients.
Dr. O’Gara chairs the STEMI writing panel of the American College of Cardiology and American Heart Association.
On Twitter @mitchelzoler
ORLANDO – Two guideline panels jointly upgraded percutaneous coronary interventions in noninfarct coronary arteries in patients with a recent MI, and downgraded manual aspiration thrombectomy in acute MI patients.
The panels, organized by the American College of Cardiology and the American Heart Association, cover, respectively, percutaneous coronary intervention (PCI) and ST-elevation MI (STEMI).
The revised guidance on percutaneous coronary intervention for multivessel coronary disease affects roughly half the patients with an acute ST-elevation MI, those who have significant stenoses in noninfarct coronaries. The 2011 guidelines from the PCI panel (J Am Coll Cardiol. 2011 Dec;58[24]:e44-122) and the 2013 guidelines from the STEMI committee (J Am Coll Cardiol. 2013 Jan;61[4]:e78-140) only endorsed revascularization of stenotic, noninfarct arteries in patients with cardiogenic shock or severe heart failure. Multivessel PCI for patients without hemodynamic compromise at the time of primary PCI was deemed a class III indication, “thought to confer harm,” based on the results of several observational studies, Dr. Patrick T. O’Gara said at the American Heart Association scientific sessions.
After the 2013 STEMI guidelines came out, results from four studies showed either benefit or no harm from multivessel PCI in patients with appropriate anatomy performed either at the time of primary PCI or as a scheduled, staged procedure soon after primary PCI, said Dr. O’Gara, professor and director of clinical cardiology at Brigham and Women’s Hospital in Boston. Three of the four studies, PRAMI (N Engl J Med. 2013 Sept 19;369[12]:1115-23), CvLPRIT (J Am Coll Cardiol. 2015 March;65[10]:963-72), and DANAMI-3–PRIMULTI (Lancet. 2015 Aug 15;386[9994]:665-71), are published, while results from the fourth, PRAGUE-13, came out in a meeting report this year but as of mid-November had not been published.
Based on these data the two committees reset PCI of noninfarct arteries as a class IIb recommendation that “could be considered” in selected STEMI patients with multivessel disease who are hemodynamically stable.
The guidelines further said that “insufficient data exist to inform a recommendation” about the optimal time for this multivessel PCI, which could be coincident with primary PCI or 2 days, 3 days, a week, or even later after initial PCI, said Dr. O’Gara. “The writing committees do not advocate for routine use of multivessel PCI; clinical judgment is obviously needed,” he cautioned.
The two committees also reassessed manual aspiration thrombectomy during primary PCI for STEMI, which until now has been a class IIa recommendation, “reasonable” for STEMI patients undergoing primary PCI. But based on the neutral results for thrombectomy in the recent findings from the two largest randomized trials of thrombectomy, TOTAL (N Engl J Med. 2015 April 9;372[15]:1389-98) and TASTE (N Engl J Med. 2013 Oct 24;369[17]:1587-97), as well as a recent, 17-trial meta-analysis, the revised guidelines rate routine aspiration thrombectomy as class III, with “no benefit” and “not useful,” and selective and bailout thrombectomy as a class IIb recommendation, with usefulness that is “not well established.”
Selective use of thrombectomy will require good judgment, noted Dr. O’Gara, who added “it will be important to see what effect, if any, these observations, this evidence, and this distillation of the recommendations from the writing committees have on a change in practice” – using thrombectomy to treat STEMI patients.
Dr. O’Gara chairs the STEMI writing panel of the American College of Cardiology and American Heart Association.
On Twitter @mitchelzoler
AT THE AHA SCIENTIFIC SESSIONS

AHA: It’s best to have a cardiac arrest in Midwest
ORLANDO – Considerable regional variation exists across the United States in outcomes, including survival and hospital charges following out-of-hospital cardiac arrest, Dr. Aiham Albaeni reported at the American Heart Association scientific sessions.
He presented an analysis of 155,592 adults who survived at least until hospital admission following non–trauma-related out-of-hospital cardiac arrest (OHCA) during 2002-2012. The data came from the Agency for Healthcare Research and Quality’s Nationwide Inpatient Sample, the largest all-payer U.S. inpatient database.

Mortality was lowest among patients whose OHCA occurred in the Midwest. Indeed, taking the Northeast region as the reference point in a multivariate analysis, the adjusted mortality risk was 14% lower in the Midwest and 9% lower in the South. There was no difference in survival rates between the West and Northeast in this analysis adjusted for age, gender, race, primary diagnosis, income, Charlson Comorbidity Index, primary payer, and hospital size and teaching status, reported Dr. Albaeni of Johns Hopkins University, Baltimore.
Total hospital charges for patients admitted following OHCA were far and away highest in the West, and this increased expenditure didn’t pay off in terms of a survival advantage. The Consumer Price Index–adjusted mean total hospital charges averaged $85,592 per patient in the West, $66,290 in the Northeast, $55,257 in the Midwest, and $54,878 in the South.
Outliers in terms of cost of care – that is, patients admitted with OHCA whose total hospital charges exceeded $109,000 per admission – were 85% more common in the West than the other three regions, he noted.
Hospital length of stay greater than 8 days occurred most often in the Northeast. These lengthier stays were 10%-12% less common in the other regions.
The explanation for the marked regional differences observed in this study remains unknown.
“These findings call for more efforts to identify a high-quality model of excellence that standardizes health care delivery and improves quality of care in low-performing regions,” said Dr. Albaeni.
He reported having no financial conflicts of interest regarding his study.
ORLANDO – Considerable regional variation exists across the United States in outcomes, including survival and hospital charges following out-of-hospital cardiac arrest, Dr. Aiham Albaeni reported at the American Heart Association scientific sessions.
He presented an analysis of 155,592 adults who survived at least until hospital admission following non–trauma-related out-of-hospital cardiac arrest (OHCA) during 2002-2012. The data came from the Agency for Healthcare Research and Quality’s Nationwide Inpatient Sample, the largest all-payer U.S. inpatient database.
Mortality was lowest among patients whose OHCA occurred in the Midwest. Indeed, taking the Northeast region as the reference point in a multivariate analysis, the adjusted mortality risk was 14% lower in the Midwest and 9% lower in the South. There was no difference in survival rates between the West and Northeast in this analysis adjusted for age, gender, race, primary diagnosis, income, Charlson Comorbidity Index, primary payer, and hospital size and teaching status, reported Dr. Albaeni of Johns Hopkins University, Baltimore.
Total hospital charges for patients admitted following OHCA were far and away highest in the West, and this increased expenditure didn’t pay off in terms of a survival advantage. The Consumer Price Index–adjusted mean total hospital charges averaged $85,592 per patient in the West, $66,290 in the Northeast, $55,257 in the Midwest, and $54,878 in the South.
Outliers in terms of cost of care – that is, patients admitted with OHCA whose total hospital charges exceeded $109,000 per admission – were 85% more common in the West than the other three regions, he noted.
Hospital length of stay greater than 8 days occurred most often in the Northeast. These lengthier stays were 10%-12% less common in the other regions.
The explanation for the marked regional differences observed in this study remains unknown.
“These findings call for more efforts to identify a high-quality model of excellence that standardizes health care delivery and improves quality of care in low-performing regions,” said Dr. Albaeni.
He reported having no financial conflicts of interest regarding his study.
ORLANDO – Considerable regional variation exists across the United States in outcomes, including survival and hospital charges following out-of-hospital cardiac arrest, Dr. Aiham Albaeni reported at the American Heart Association scientific sessions.
He presented an analysis of 155,592 adults who survived at least until hospital admission following non–trauma-related out-of-hospital cardiac arrest (OHCA) during 2002-2012. The data came from the Agency for Healthcare Research and Quality’s Nationwide Inpatient Sample, the largest all-payer U.S. inpatient database.
Mortality was lowest among patients whose OHCA occurred in the Midwest. Indeed, taking the Northeast region as the reference point in a multivariate analysis, the adjusted mortality risk was 14% lower in the Midwest and 9% lower in the South. There was no difference in survival rates between the West and Northeast in this analysis adjusted for age, gender, race, primary diagnosis, income, Charlson Comorbidity Index, primary payer, and hospital size and teaching status, reported Dr. Albaeni of Johns Hopkins University, Baltimore.
Total hospital charges for patients admitted following OHCA were far and away highest in the West, and this increased expenditure didn’t pay off in terms of a survival advantage. The Consumer Price Index–adjusted mean total hospital charges averaged $85,592 per patient in the West, $66,290 in the Northeast, $55,257 in the Midwest, and $54,878 in the South.
Outliers in terms of cost of care – that is, patients admitted with OHCA whose total hospital charges exceeded $109,000 per admission – were 85% more common in the West than the other three regions, he noted.
Hospital length of stay greater than 8 days occurred most often in the Northeast. These lengthier stays were 10%-12% less common in the other regions.
The explanation for the marked regional differences observed in this study remains unknown.
“These findings call for more efforts to identify a high-quality model of excellence that standardizes health care delivery and improves quality of care in low-performing regions,” said Dr. Albaeni.
He reported having no financial conflicts of interest regarding his study.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Substantial and as-yet unexplained regional differences in survival and total hospital charges following out-of-hospital cardiac arrest exist across the United States.
Major finding: Mortality among adults hospitalized after experiencing out-of-hospital cardiac arrest was 14% lower in the Midwest than in the Northeast.
Data source: A retrospective analysis of data from the Nationwide Inpatient Sample for 2002-2012 that included 155,592 adults with out-of-hospital cardiac arrest who survived to hospital admission.
Disclosures: The presenter reported having no financial conflicts of interest.

Postsurgical Analgesic Found to Decrease Opioid Use, Hospital Stay, and Readmission Rates After Knee Replacement Surgery
DALLAS—Positive data about the use of Exparel (bupivacaine liposome injectable suspension) as a postsurgical analgesic following total knee replacement surgery was presented at the 25th Annual Meeting of the American Association of Hip and Knee Surgeons.
The study, which compared the use of bupivacaine liposome injectable suspension infiltration to the standard of care in 1,110 patients, found that bupivacaine liposome injectable suspension was associated with significant improvements in a variety of patient and health economic outcomes, including opioid use, hospital stay, and readmission rate.

Patients who underwent total knee arthroplasty (TKA) received identical pre-, intra-, and postoperative pain management protocols, with the exception of 527 patients who received bupivacaine liposome injectable suspension infiltration in place of a femoral nerve block.
The study authors compared several patient and cost-related outcomes. Opioid use during hospitalization was statistically significantly reduced in the bupivacaine liposome injectable suspension group. Other key findings included:
• Shorter hospital length of stay (2.93 days for the bupivacaine liposome injectable suspension group vs 3.19 days for the femoral nerve block group, P<0.001)
• Increased rate of discharge to home (77.8% for the bupivacaine liposome injectable suspension group vs 72.21% for the femoral nerve block group, P=0.032)
• Reduced inpatient fall rate (0.56% for the bupivacaine liposome injectable suspension group vs 2.11% for the femoral nerve block group, P=0.03)
• Lower 30-day all-cause readmission rate (0.95% for the bupivacaine liposome injectable suspension group vs 2.57% for the femoral nerve block group, P=0.041)
“Based on our analysis, incorporating liposomal bupivacaine into the postsurgical analgesic protocol following total knee arthroplasty has significant and quantifiable benefits to both the patient and the institution,” said Richard Iorio, MD, Professor of Orthopaedic Surgery at NYU School of Medicine in New York. “The measurable opioid-sparing effect of this new regimen has enabled us to virtually eliminate intravenous patient-controlled analgesia, or PCA, devices from the standard of care in total joint arthroplasty patients, without compromising patient comfort. In addition, we found that the incremental cost of adding this new modality was offset by meaningful savings from shorter anesthesia induction time in the operating room, shorter hospital stays and lower rates of 30-day readmission.”
DALLAS—Positive data about the use of Exparel (bupivacaine liposome injectable suspension) as a postsurgical analgesic following total knee replacement surgery was presented at the 25th Annual Meeting of the American Association of Hip and Knee Surgeons.
The study, which compared the use of bupivacaine liposome injectable suspension infiltration to the standard of care in 1,110 patients, found that bupivacaine liposome injectable suspension was associated with significant improvements in a variety of patient and health economic outcomes, including opioid use, hospital stay, and readmission rate.
Patients who underwent total knee arthroplasty (TKA) received identical pre-, intra-, and postoperative pain management protocols, with the exception of 527 patients who received bupivacaine liposome injectable suspension infiltration in place of a femoral nerve block.
The study authors compared several patient and cost-related outcomes. Opioid use during hospitalization was statistically significantly reduced in the bupivacaine liposome injectable suspension group. Other key findings included:
• Shorter hospital length of stay (2.93 days for the bupivacaine liposome injectable suspension group vs 3.19 days for the femoral nerve block group, P<0.001)
• Increased rate of discharge to home (77.8% for the bupivacaine liposome injectable suspension group vs 72.21% for the femoral nerve block group, P=0.032)
• Reduced inpatient fall rate (0.56% for the bupivacaine liposome injectable suspension group vs 2.11% for the femoral nerve block group, P=0.03)
• Lower 30-day all-cause readmission rate (0.95% for the bupivacaine liposome injectable suspension group vs 2.57% for the femoral nerve block group, P=0.041)
“Based on our analysis, incorporating liposomal bupivacaine into the postsurgical analgesic protocol following total knee arthroplasty has significant and quantifiable benefits to both the patient and the institution,” said Richard Iorio, MD, Professor of Orthopaedic Surgery at NYU School of Medicine in New York. “The measurable opioid-sparing effect of this new regimen has enabled us to virtually eliminate intravenous patient-controlled analgesia, or PCA, devices from the standard of care in total joint arthroplasty patients, without compromising patient comfort. In addition, we found that the incremental cost of adding this new modality was offset by meaningful savings from shorter anesthesia induction time in the operating room, shorter hospital stays and lower rates of 30-day readmission.”
DALLAS—Positive data about the use of Exparel (bupivacaine liposome injectable suspension) as a postsurgical analgesic following total knee replacement surgery was presented at the 25th Annual Meeting of the American Association of Hip and Knee Surgeons.
The study, which compared the use of bupivacaine liposome injectable suspension infiltration to the standard of care in 1,110 patients, found that bupivacaine liposome injectable suspension was associated with significant improvements in a variety of patient and health economic outcomes, including opioid use, hospital stay, and readmission rate.
Patients who underwent total knee arthroplasty (TKA) received identical pre-, intra-, and postoperative pain management protocols, with the exception of 527 patients who received bupivacaine liposome injectable suspension infiltration in place of a femoral nerve block.
The study authors compared several patient and cost-related outcomes. Opioid use during hospitalization was statistically significantly reduced in the bupivacaine liposome injectable suspension group. Other key findings included:
• Shorter hospital length of stay (2.93 days for the bupivacaine liposome injectable suspension group vs 3.19 days for the femoral nerve block group, P<0.001)
• Increased rate of discharge to home (77.8% for the bupivacaine liposome injectable suspension group vs 72.21% for the femoral nerve block group, P=0.032)
• Reduced inpatient fall rate (0.56% for the bupivacaine liposome injectable suspension group vs 2.11% for the femoral nerve block group, P=0.03)
• Lower 30-day all-cause readmission rate (0.95% for the bupivacaine liposome injectable suspension group vs 2.57% for the femoral nerve block group, P=0.041)
“Based on our analysis, incorporating liposomal bupivacaine into the postsurgical analgesic protocol following total knee arthroplasty has significant and quantifiable benefits to both the patient and the institution,” said Richard Iorio, MD, Professor of Orthopaedic Surgery at NYU School of Medicine in New York. “The measurable opioid-sparing effect of this new regimen has enabled us to virtually eliminate intravenous patient-controlled analgesia, or PCA, devices from the standard of care in total joint arthroplasty patients, without compromising patient comfort. In addition, we found that the incremental cost of adding this new modality was offset by meaningful savings from shorter anesthesia induction time in the operating room, shorter hospital stays and lower rates of 30-day readmission.”
