User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Pink Papulonodular Eruption on the Trunk and Arms
Pink Papulonodular Eruption on the Trunk and Arms
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
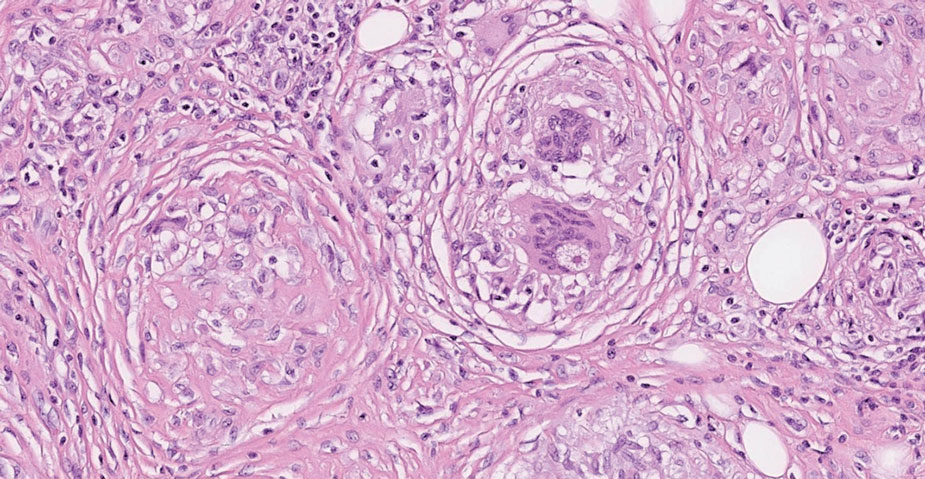
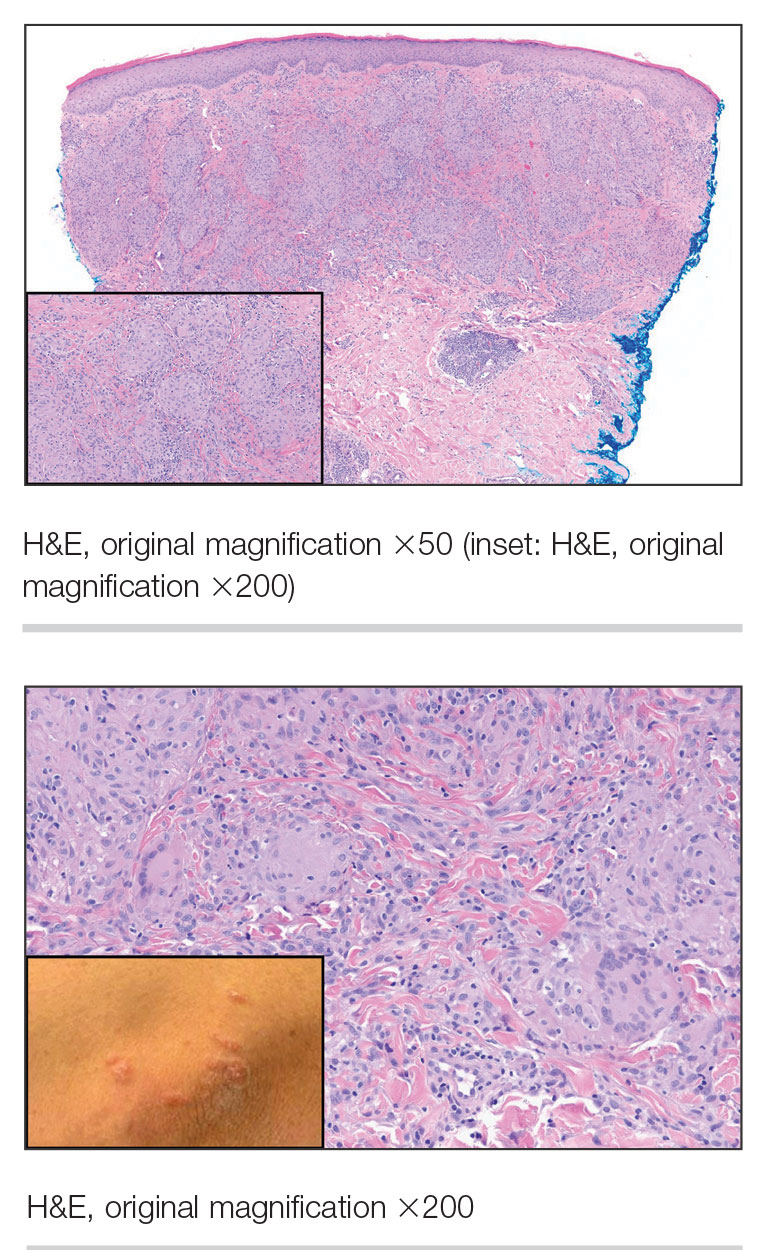
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
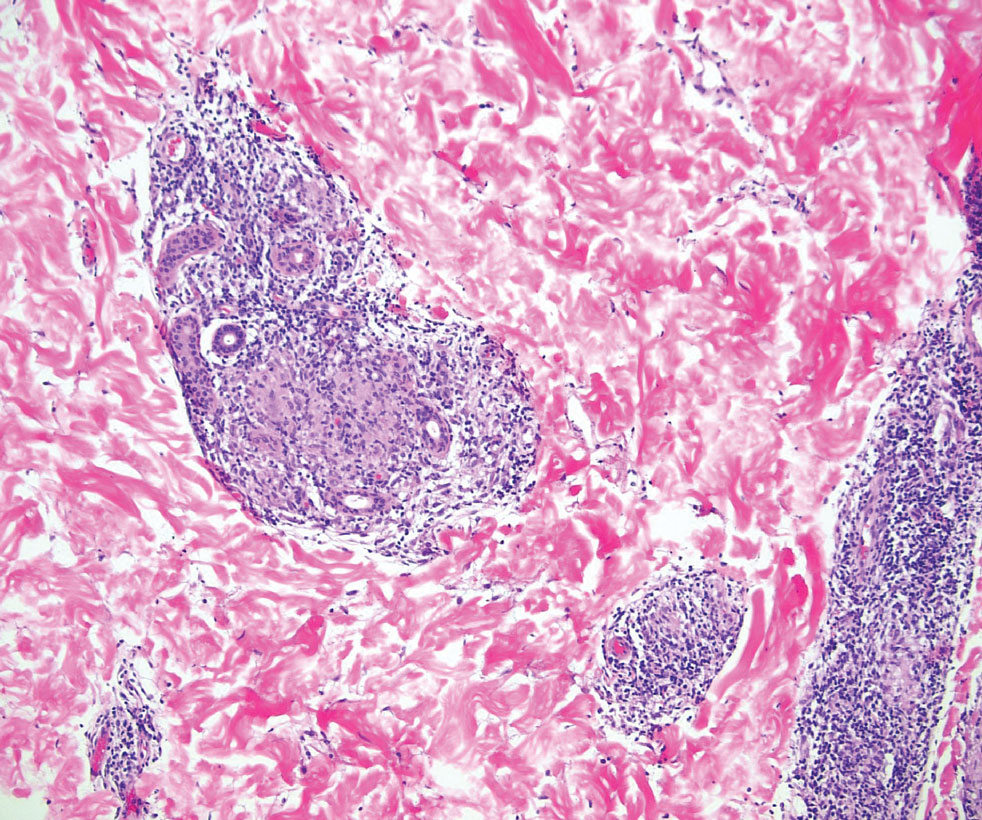
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
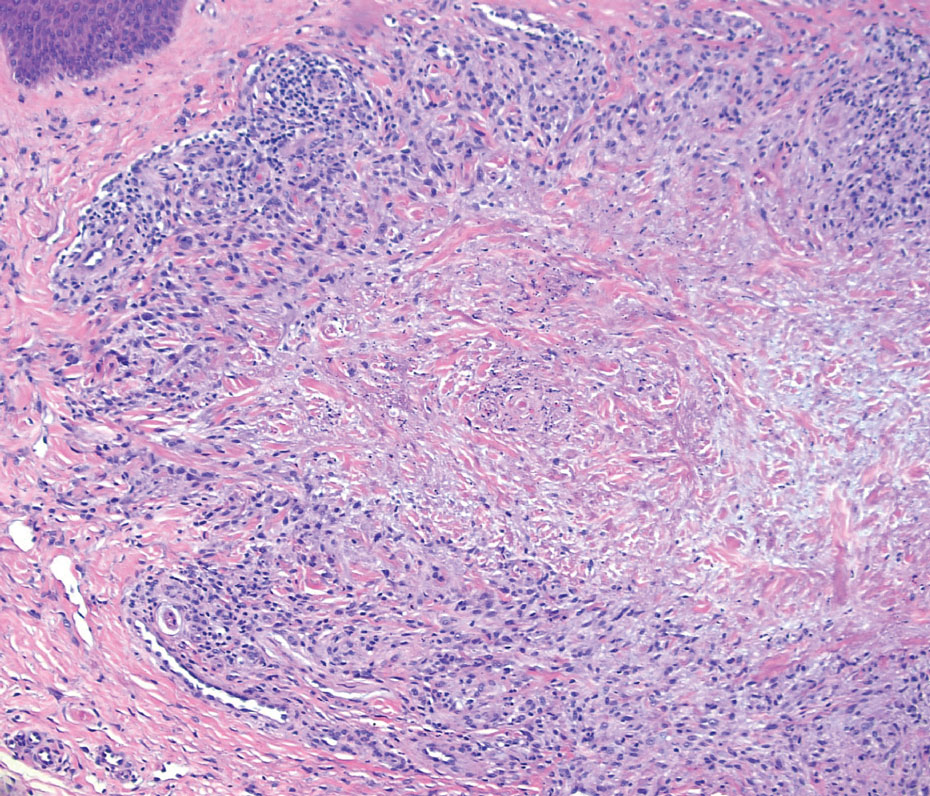
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
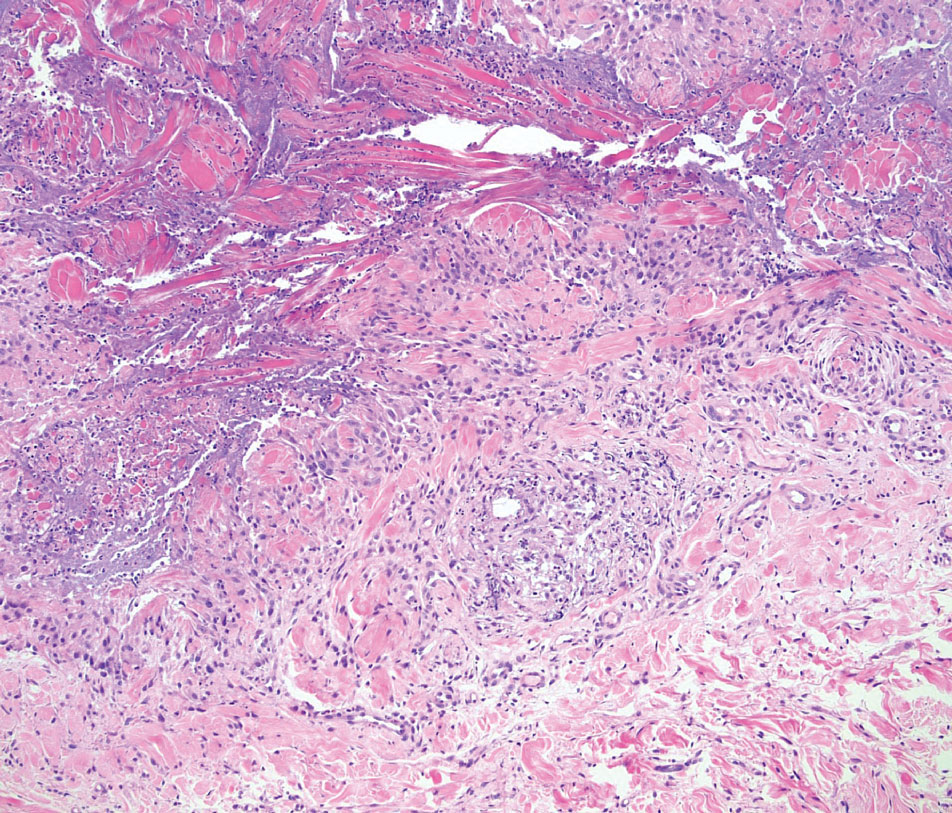
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
Pink Papulonodular Eruption on the Trunk and Arms
Pink Papulonodular Eruption on the Trunk and Arms
A 47-year-old man with a history of chronic kidney disease and bilateral clear cell renal cell carcinoma who was undergoing treatment with adjuvant pembrolizumab presented to the dermatology department with a scattered papulonodular eruption of several weeks’ duration. Physical examination revealed pink papules and nodules with coalescing erythema over the trunk and upper extremities, most pronounced on the right elbow (bottom [inset]). A 4-mm punch biopsy demonstrated dermal granulomatous inflammation. Special stains were negative for microorganisms. Computed tomography of the chest revealed a new subpleural nodule and new hilar lymphadenopathy.

Association Between Hidradenitis Suppurativa and Polycystic Ovary Syndrome
Association Between Hidradenitis Suppurativa and Polycystic Ovary Syndrome
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful nodules, abscesses, scarring, and sinus tracts that commonly manifest in the axillary, inguinal, perianal, and inframammary regions.1 Hidradenitis suppurativa has been associated with several metabolic and cardiovascular comorbidities as well as polycystic ovary syndrome (PCOS)(recently renamed polyendocrine metabolic ovarian syndrome),2,3 a condition characterized by hyperandrogenism, chronic anovulation, and polycystic ovaries.2 Multiple comorbidities of PCOS overlap with those of HS, including type 2 diabetes, cardiovascular disease, and metabolic syndrome.1,3-5 While HS may be associated with PCOS, there is limited literature analyzing the association between these conditions. This study aimed to analyze the association between HS and PCOS using data from the National Institute of Health’s All of Us Research Program database (https://allofus.nih.gov/). While other studies have looked at the association between HS and PCOS, ours is among the first to analyze the relationship between multiple race/ ethnicity groups, which is especially important given racial disparities in HS and comorbid diseases.
Methods
A cross-sectional, population-based study of females included in the All of Us Research Program database was conducted. Patients with HS were identified using the Systematized Nomenclature of Medicine–Clinical Terms (SNOMED CT) code 59393003, while PCOS was identified with the code 237055002. Type 2 diabetes was identified with the following SNOMED CT codes: 44054006, 313436004, 237599002, 199230006, 359642000, and 81531005. Obesity was identified with the following codes: 414916001, 238136002, 190966007, 296526005, 294493008, 238134004, 83911000119104, and 415530009. Male patients and those who did not answer questions regarding sociodemographic variables were excluded from the final analysis. P values were calculated using Pearson χ2 tests. Multivariate logistic regression was used to calculate adjusted odds ratios and unadjusted odds ratios to analyze the association between HS and PCOS while controlling for age, race/ethnicity, smoking status, type 2 diabetes, and obesity. Statistical analyses were conducted using a 95% CI.
Results
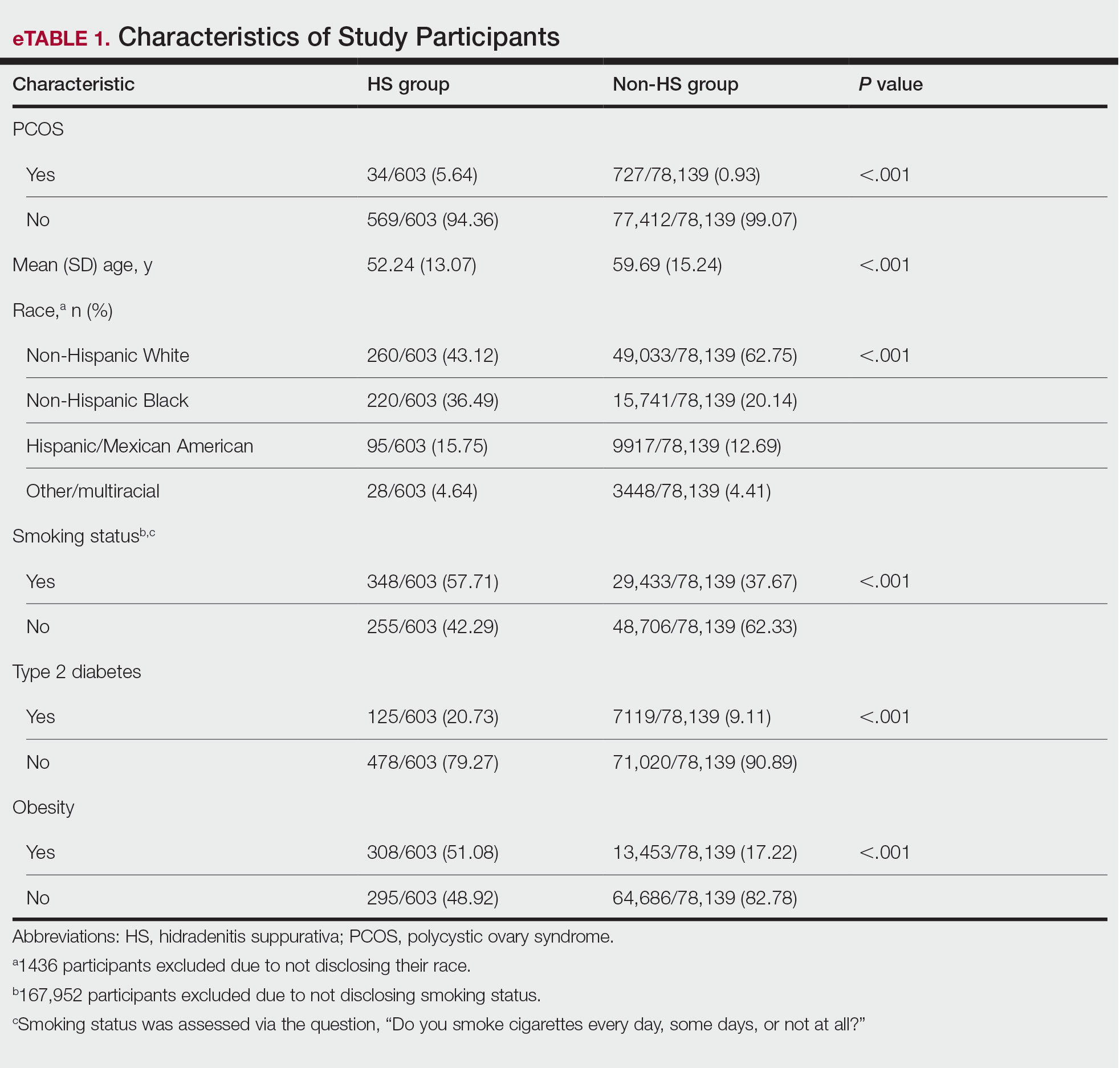
The final analysis included 78,742 patients. The prevalence of PCOS was 5.64% in the HS group vs 0.93% in the non-HS group (eTable 1). Individuals with HS had higher rates of smoking cigarettes (57.71% vs 37.67%), obesity (51.08% vs 17.22%), and type 2 diabetes (20.73% vs 9.11%) than individuals without HS, respectively.
Multivariate logistic regression analyses revealed that individuals with HS were 2.06 times more likely to have PCOS after adjusting for sociodemographic variables and comorbidities (95% CI, 1.41-3.02; P<.001). Adjusted subgroup analyses by race/ethnicity did not yield statistically significant results; however, unadjusted analyses revealed that individuals with HS had significantly increased odds of PCOS across all race/ethnicity groups (eTable 2). Interaction terms analysis to determine if the relationship between HS and PCOS differs by race/ ethnicity did not yield statistically significant results. However, independent of HS status, non-Hispanic Black and Hispanic patients were less likely to have PCOS compared to White individuals (adjusted odds ratio, 0.37 and 0.56, respectively; P<.001). Disparities in access to care could have led to underdiagnosis of PCOS among non-Hispanic Black and Hispanic patients. Lastly, individuals with type 2 diabetes were 10.43 times more likely to have PCOS than those without, while patients with obesity were 11.14 times more likely to have PCOS than those without.

Comment
This study demonstrated that females with HS are 2.06 times more likely to have PCOS than those without HS, even after controlling for important sociodemographic variables and comorbidities. While adjusted subgroup analyses did not yield statistically significant results, unadjusted analyses demonstrated increased odds of PCOS in patients with HS across all race/ethnicity groups, suggesting that sociodemographic variables and comorbidities substantially influence the relationship between HS and PCOS; for instance, patients with type 2 diabetes and obesity are approximately 10- to 11-fold more likely to have PCOS than patients without these conditions. Non-Hispanic Black and Hispanic patients were less likely to have PCOS compared with White patients, indicating possible underdiagnosis of PCOS in these populations and highlighting the need for increased PCOS screening. Limitations of this study include the reliance on SNOMED CT codes, which may have led to underdiagnosis of HS or PCOS, as well as the inability to differentiate between mild and severe HS in the database.
Hyperandrogenism is believed to contribute to the pathogenesis of both HS and PCOS, supporting the potential use of antiandrogen therapies, such as spironolactone, in managing both conditions.2,3 Furthermore, oral contraceptives may have a role in managing both conditions. In HS, oral contraceptives help to mitigate flares associated with hormonal changes during menstruation, while in PCOS, they are used to regulate the hormonal cycle and reduce hirsutism.2-4 However, not all women experience menstrual flares of HS, suggesting that variations in HS phenotypes may influence individual responses to hormonal changes.1 Additionally, the considerable overlap in metabolic and cardiovascular comorbidities between HS and PCOS indicates that shared pathomechanisms may contribute to the association between these conditions.1,2 For example, proinflammatory adipokines released in both HS and PCOS may contribute to inflammation, cardiovascular disease, and insulin resistance.3,5
Conclusion
Further research is needed to better understand the shared pathophysiology that links these 2 diseases and to identify targeted approaches for optimizing management and improving patient outcomes. The association between HS and PCOS highlights the importance of screening for metabolic and reproductive comorbidities in patients with HS. Early recognition and management of both HS and PCOS can improve long-term outcomes.
- van Straalen KR, Prens EP, Gudjonsson JE. Insights into hidradenitis suppurativa. J Allergy Clin Immunol. 2022;149:1150-1161. doi:10.1016 /j.jaci.2022.02.003
- Choudhari R, Tayade S, Tiwari A, et al. Diagnosis, management, and associated comorbidities of polycystic ovary syndrome: a narrative review. Cureus. 2024;16:e58733. doi:10.7759/cureus.58733
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Montero-Vilchez T, Valenzuela-Amigo A, Cuenca-Barrales C, et al. The role of oral contraceptive pills in hidradenitis suppurativa: a cohort study. Life (Basel). 2021;11:697. doi:10.3390/life11070697
- Randeva HS, Tan BK, Weickert MO, et al. Cardiometabolic aspects of the polycystic ovary syndrome. Endocr Rev. 2012;33:812-841. doi:10.1210/er.2012-1003
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful nodules, abscesses, scarring, and sinus tracts that commonly manifest in the axillary, inguinal, perianal, and inframammary regions.1 Hidradenitis suppurativa has been associated with several metabolic and cardiovascular comorbidities as well as polycystic ovary syndrome (PCOS)(recently renamed polyendocrine metabolic ovarian syndrome),2,3 a condition characterized by hyperandrogenism, chronic anovulation, and polycystic ovaries.2 Multiple comorbidities of PCOS overlap with those of HS, including type 2 diabetes, cardiovascular disease, and metabolic syndrome.1,3-5 While HS may be associated with PCOS, there is limited literature analyzing the association between these conditions. This study aimed to analyze the association between HS and PCOS using data from the National Institute of Health’s All of Us Research Program database (https://allofus.nih.gov/). While other studies have looked at the association between HS and PCOS, ours is among the first to analyze the relationship between multiple race/ ethnicity groups, which is especially important given racial disparities in HS and comorbid diseases.
Methods
A cross-sectional, population-based study of females included in the All of Us Research Program database was conducted. Patients with HS were identified using the Systematized Nomenclature of Medicine–Clinical Terms (SNOMED CT) code 59393003, while PCOS was identified with the code 237055002. Type 2 diabetes was identified with the following SNOMED CT codes: 44054006, 313436004, 237599002, 199230006, 359642000, and 81531005. Obesity was identified with the following codes: 414916001, 238136002, 190966007, 296526005, 294493008, 238134004, 83911000119104, and 415530009. Male patients and those who did not answer questions regarding sociodemographic variables were excluded from the final analysis. P values were calculated using Pearson χ2 tests. Multivariate logistic regression was used to calculate adjusted odds ratios and unadjusted odds ratios to analyze the association between HS and PCOS while controlling for age, race/ethnicity, smoking status, type 2 diabetes, and obesity. Statistical analyses were conducted using a 95% CI.
Results
The final analysis included 78,742 patients. The prevalence of PCOS was 5.64% in the HS group vs 0.93% in the non-HS group (eTable 1). Individuals with HS had higher rates of smoking cigarettes (57.71% vs 37.67%), obesity (51.08% vs 17.22%), and type 2 diabetes (20.73% vs 9.11%) than individuals without HS, respectively.
Multivariate logistic regression analyses revealed that individuals with HS were 2.06 times more likely to have PCOS after adjusting for sociodemographic variables and comorbidities (95% CI, 1.41-3.02; P<.001). Adjusted subgroup analyses by race/ethnicity did not yield statistically significant results; however, unadjusted analyses revealed that individuals with HS had significantly increased odds of PCOS across all race/ethnicity groups (eTable 2). Interaction terms analysis to determine if the relationship between HS and PCOS differs by race/ ethnicity did not yield statistically significant results. However, independent of HS status, non-Hispanic Black and Hispanic patients were less likely to have PCOS compared to White individuals (adjusted odds ratio, 0.37 and 0.56, respectively; P<.001). Disparities in access to care could have led to underdiagnosis of PCOS among non-Hispanic Black and Hispanic patients. Lastly, individuals with type 2 diabetes were 10.43 times more likely to have PCOS than those without, while patients with obesity were 11.14 times more likely to have PCOS than those without.
Comment
This study demonstrated that females with HS are 2.06 times more likely to have PCOS than those without HS, even after controlling for important sociodemographic variables and comorbidities. While adjusted subgroup analyses did not yield statistically significant results, unadjusted analyses demonstrated increased odds of PCOS in patients with HS across all race/ethnicity groups, suggesting that sociodemographic variables and comorbidities substantially influence the relationship between HS and PCOS; for instance, patients with type 2 diabetes and obesity are approximately 10- to 11-fold more likely to have PCOS than patients without these conditions. Non-Hispanic Black and Hispanic patients were less likely to have PCOS compared with White patients, indicating possible underdiagnosis of PCOS in these populations and highlighting the need for increased PCOS screening. Limitations of this study include the reliance on SNOMED CT codes, which may have led to underdiagnosis of HS or PCOS, as well as the inability to differentiate between mild and severe HS in the database.
Hyperandrogenism is believed to contribute to the pathogenesis of both HS and PCOS, supporting the potential use of antiandrogen therapies, such as spironolactone, in managing both conditions.2,3 Furthermore, oral contraceptives may have a role in managing both conditions. In HS, oral contraceptives help to mitigate flares associated with hormonal changes during menstruation, while in PCOS, they are used to regulate the hormonal cycle and reduce hirsutism.2-4 However, not all women experience menstrual flares of HS, suggesting that variations in HS phenotypes may influence individual responses to hormonal changes.1 Additionally, the considerable overlap in metabolic and cardiovascular comorbidities between HS and PCOS indicates that shared pathomechanisms may contribute to the association between these conditions.1,2 For example, proinflammatory adipokines released in both HS and PCOS may contribute to inflammation, cardiovascular disease, and insulin resistance.3,5
Conclusion
Further research is needed to better understand the shared pathophysiology that links these 2 diseases and to identify targeted approaches for optimizing management and improving patient outcomes. The association between HS and PCOS highlights the importance of screening for metabolic and reproductive comorbidities in patients with HS. Early recognition and management of both HS and PCOS can improve long-term outcomes.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful nodules, abscesses, scarring, and sinus tracts that commonly manifest in the axillary, inguinal, perianal, and inframammary regions.1 Hidradenitis suppurativa has been associated with several metabolic and cardiovascular comorbidities as well as polycystic ovary syndrome (PCOS)(recently renamed polyendocrine metabolic ovarian syndrome),2,3 a condition characterized by hyperandrogenism, chronic anovulation, and polycystic ovaries.2 Multiple comorbidities of PCOS overlap with those of HS, including type 2 diabetes, cardiovascular disease, and metabolic syndrome.1,3-5 While HS may be associated with PCOS, there is limited literature analyzing the association between these conditions. This study aimed to analyze the association between HS and PCOS using data from the National Institute of Health’s All of Us Research Program database (https://allofus.nih.gov/). While other studies have looked at the association between HS and PCOS, ours is among the first to analyze the relationship between multiple race/ ethnicity groups, which is especially important given racial disparities in HS and comorbid diseases.
Methods
A cross-sectional, population-based study of females included in the All of Us Research Program database was conducted. Patients with HS were identified using the Systematized Nomenclature of Medicine–Clinical Terms (SNOMED CT) code 59393003, while PCOS was identified with the code 237055002. Type 2 diabetes was identified with the following SNOMED CT codes: 44054006, 313436004, 237599002, 199230006, 359642000, and 81531005. Obesity was identified with the following codes: 414916001, 238136002, 190966007, 296526005, 294493008, 238134004, 83911000119104, and 415530009. Male patients and those who did not answer questions regarding sociodemographic variables were excluded from the final analysis. P values were calculated using Pearson χ2 tests. Multivariate logistic regression was used to calculate adjusted odds ratios and unadjusted odds ratios to analyze the association between HS and PCOS while controlling for age, race/ethnicity, smoking status, type 2 diabetes, and obesity. Statistical analyses were conducted using a 95% CI.
Results
The final analysis included 78,742 patients. The prevalence of PCOS was 5.64% in the HS group vs 0.93% in the non-HS group (eTable 1). Individuals with HS had higher rates of smoking cigarettes (57.71% vs 37.67%), obesity (51.08% vs 17.22%), and type 2 diabetes (20.73% vs 9.11%) than individuals without HS, respectively.
Multivariate logistic regression analyses revealed that individuals with HS were 2.06 times more likely to have PCOS after adjusting for sociodemographic variables and comorbidities (95% CI, 1.41-3.02; P<.001). Adjusted subgroup analyses by race/ethnicity did not yield statistically significant results; however, unadjusted analyses revealed that individuals with HS had significantly increased odds of PCOS across all race/ethnicity groups (eTable 2). Interaction terms analysis to determine if the relationship between HS and PCOS differs by race/ ethnicity did not yield statistically significant results. However, independent of HS status, non-Hispanic Black and Hispanic patients were less likely to have PCOS compared to White individuals (adjusted odds ratio, 0.37 and 0.56, respectively; P<.001). Disparities in access to care could have led to underdiagnosis of PCOS among non-Hispanic Black and Hispanic patients. Lastly, individuals with type 2 diabetes were 10.43 times more likely to have PCOS than those without, while patients with obesity were 11.14 times more likely to have PCOS than those without.
Comment
This study demonstrated that females with HS are 2.06 times more likely to have PCOS than those without HS, even after controlling for important sociodemographic variables and comorbidities. While adjusted subgroup analyses did not yield statistically significant results, unadjusted analyses demonstrated increased odds of PCOS in patients with HS across all race/ethnicity groups, suggesting that sociodemographic variables and comorbidities substantially influence the relationship between HS and PCOS; for instance, patients with type 2 diabetes and obesity are approximately 10- to 11-fold more likely to have PCOS than patients without these conditions. Non-Hispanic Black and Hispanic patients were less likely to have PCOS compared with White patients, indicating possible underdiagnosis of PCOS in these populations and highlighting the need for increased PCOS screening. Limitations of this study include the reliance on SNOMED CT codes, which may have led to underdiagnosis of HS or PCOS, as well as the inability to differentiate between mild and severe HS in the database.
Hyperandrogenism is believed to contribute to the pathogenesis of both HS and PCOS, supporting the potential use of antiandrogen therapies, such as spironolactone, in managing both conditions.2,3 Furthermore, oral contraceptives may have a role in managing both conditions. In HS, oral contraceptives help to mitigate flares associated with hormonal changes during menstruation, while in PCOS, they are used to regulate the hormonal cycle and reduce hirsutism.2-4 However, not all women experience menstrual flares of HS, suggesting that variations in HS phenotypes may influence individual responses to hormonal changes.1 Additionally, the considerable overlap in metabolic and cardiovascular comorbidities between HS and PCOS indicates that shared pathomechanisms may contribute to the association between these conditions.1,2 For example, proinflammatory adipokines released in both HS and PCOS may contribute to inflammation, cardiovascular disease, and insulin resistance.3,5
Conclusion
Further research is needed to better understand the shared pathophysiology that links these 2 diseases and to identify targeted approaches for optimizing management and improving patient outcomes. The association between HS and PCOS highlights the importance of screening for metabolic and reproductive comorbidities in patients with HS. Early recognition and management of both HS and PCOS can improve long-term outcomes.
- van Straalen KR, Prens EP, Gudjonsson JE. Insights into hidradenitis suppurativa. J Allergy Clin Immunol. 2022;149:1150-1161. doi:10.1016 /j.jaci.2022.02.003
- Choudhari R, Tayade S, Tiwari A, et al. Diagnosis, management, and associated comorbidities of polycystic ovary syndrome: a narrative review. Cureus. 2024;16:e58733. doi:10.7759/cureus.58733
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Montero-Vilchez T, Valenzuela-Amigo A, Cuenca-Barrales C, et al. The role of oral contraceptive pills in hidradenitis suppurativa: a cohort study. Life (Basel). 2021;11:697. doi:10.3390/life11070697
- Randeva HS, Tan BK, Weickert MO, et al. Cardiometabolic aspects of the polycystic ovary syndrome. Endocr Rev. 2012;33:812-841. doi:10.1210/er.2012-1003
- van Straalen KR, Prens EP, Gudjonsson JE. Insights into hidradenitis suppurativa. J Allergy Clin Immunol. 2022;149:1150-1161. doi:10.1016 /j.jaci.2022.02.003
- Choudhari R, Tayade S, Tiwari A, et al. Diagnosis, management, and associated comorbidities of polycystic ovary syndrome: a narrative review. Cureus. 2024;16:e58733. doi:10.7759/cureus.58733
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Montero-Vilchez T, Valenzuela-Amigo A, Cuenca-Barrales C, et al. The role of oral contraceptive pills in hidradenitis suppurativa: a cohort study. Life (Basel). 2021;11:697. doi:10.3390/life11070697
- Randeva HS, Tan BK, Weickert MO, et al. Cardiometabolic aspects of the polycystic ovary syndrome. Endocr Rev. 2012;33:812-841. doi:10.1210/er.2012-1003
Association Between Hidradenitis Suppurativa and Polycystic Ovary Syndrome
Association Between Hidradenitis Suppurativa and Polycystic Ovary Syndrome
PRACTICE POINTS
- Patients with hidradenitis suppurativa were 2.06 times more likely to have polycystic ovary syndrome (PCOS) than patients without HS after controlling for age, race/ ethnicity, tobacco use, type 2 diabetes, and obesity.
- Non-Hispanic Black and Hispanic patients were less likely than White patients to have a diagnosis of PCOS, potentially reflecting underdiagnosis in these populations.
- Individuals with type 2 diabetes and obesity were 10.43 and 11.14 times more likely, respectively, to have PCOS.

Atopic Dermatitis: New Insights and Expanded Treatment Options
Atopic Dermatitis: New Insights and Expanded Treatment Options
Atopic dermatitis (AD) is a chronic skin condition generally characterized by pruritic and erythematous papules and plaques.1 While AD commonly manifests in childhood, 1 in 4 patients living with AD report adult onset of the disease.2 The clinical presentation and prevalence of AD vary across age groups, skin tones, and racial and ethnic groups. Globally, AD is estimated to have a prevalence of 2.6%; however, rates vary widely by region.1 Morphology and distribution of AD lesions also vary by population; therefore, defining one classic presentation of AD is not sufficient in diverse patient populations.3
Epidemiology
The prevalence of AD ranges from 0.2% to 24.6% worldwide, with higher rates in Africa and Oceania and lower rates in India and Northern and Eastern Europe.1 In the United States, AD affects all racial and ethnic groups; however, prevalence and severity are increased in Black children compared with White children.4 In one prospective cohort study, Hispanic children and non-Hispanic Black children aged 3 years and younger had greater odds of AD persisting into mid childhood (approximately age 7 years) compared with non-Hispanic White children.5,6
Key Clinical Features
Clinical features of AD are heterogeneous and may include differences in color, morphology, and distribution. Brown, hyperpigmented, gray, and/or violaceous plaques may predominate in patients with skin of color (SOC) compared with the erythematous plaques commonly described in lighter skin tones.1,3 Established scoring systems for AD rely on erythema as a key diagnostic feature, but because erythema can be difficult to detect in darker skin tones, disease severity may be underestimated and diagnosis may be delayed in this population.4
Atopic dermatitis in SOC may manifest as lichenoid plaques,7 prurigo nodules,7,8 lichenification,1 and follicular accentuation.9 Lichen planus–like AD is a distinct variant characterized by lichenoid plaques with a predilection for the extensor surfaces and face in patients with darker skin tones1,8 occurring in approximately 9% of patients in one study.10
Other key clinical features of AD in patients with SOC include pityriasis alba,10 increased risk for postinflammatory pigment alteration (including hyperpigmentation and/or hypopigmentation),1 and greater trunk and extensor involvement.1,11
Worth Noting
The scientific landscape for AD has grown rapidly, increasing our understanding of its pathophysiology, treatment, and social impact. Nonsteroidal treatments available for pediatric and adult patients with AD have increased in recent years, including crisaborole (approved for use in those ages ≥3 months), tacrolimus (≥2 years), and pimecrolimus (≥2 years). Injectable options include dupilumab (≥6 months), lebrikizumab (≥12 years), nemolizumab (≥12 years), and tralokinumab (≥12 years). Oral options include abrocitinib (≥12 years) and upadacitinib (≥12 years).12 Topical options include roflumilast 0.15% cream (≥6 years)12 and 0.05% cream (≥2-5 years),13 ruxolitinib 1.5% cream (≥2 years),14 and tapinarof 1% cream (≥2 years).12
For some patients, postinflammatory pigment alteration associated with AD has a higher impact on quality of life than the AD itself.7 In a study of 260 US adults with AD, the emotional impact of pigmentary changes was greatest in Black patients, with 53.3% reporting that pigment changes bothered them “a lot” or “very much.”15
Genome-wide association studies have not identified a single determinant that explains racial and ethnic differences in susceptibility to AD.4 Instead, social determinants of health are thought to play a role in the difference in AD prevalence and severity across groups in the United States.16
Health Disparity Highlight
In an analysis of 20 US metropolitan cities, urban and inner-city residence was associated with approximately 1.7-fold increased odds of AD.4 Among pediatric patients with moderate to severe AD, Black children were more likely to be exposed to tobacco smoke17 and traffic-related air pollution.18 Low socioeconomic status and low income also have been associated with moderate16 and severe19 AD. At the same education level, Black individuals in the United States receive less income than their White counterparts and have markedly less wealth at equivalent incomes.20
In utero exposure to maternal stress is associated with AD.4 Increased IgE levels have been recorded in children who develop AD, with Black children having the highest IgE levels overall compared to other children.18
An analysis of medical records from an urban medical center in Baltimore, Maryland, from 2013 through 2018 showed that Black patients with AD were less likely to receive topical corticosteroids, topical calcineurin inhibitors, a topical phosphodiesterase 4 inhibitor, and a biologic compared to White patients with AD.21
Since the disproportionate burden experienced by patients with AD is not physiologic, it is imperative to address these systemic complexities and address the barriers impacting treatment availability to improve health outcomes for all patients living with AD.
- Kaufman BP, Guttman-Yassky E, Alexis AF. Atopic dermatitis in diverse racial and ethnic groups—variations in epidemiology, genetics, clinical presentation and treatment. Exp Dermatol. 2018;27:340-357.
- Lee HH, Patel KR, Singam V, et al. A systematic review and meta-analysis of the prevalence and phenotype of adult-onset atopic dermatitis. J Am Acad Dermatol. 2019;80:1526-1532.E7.
- Adawi W, Cornman H, Kambala A, et al. Diagnosing atopic dermatitis in skin of color. Dermatol Clin. 2023;41:417-429.
- Narla S, Silverberg JI. Current updates in the epidemiology and comorbidities of atopic dermatitis. Ann Allergy Asthma Immunol. 2025;135:511-520.
- Croce EA, Levy ML, Adamson AS, et al. Reframing racial and ethnic disparities in atopic dermatitis in Black and Latinx populations. J Allergy Clin Immunol. 2021;148:1104-1111.
- Kim Y, Blomberg M, Rifas-Shiman SL, et al. Racial/ethnic differences in incidence and persistence of childhood atopic dermatitis. J Invest Dermatol. 2019;139:827-834.
- Nomura T, Wu J, Kabashima K, et al. Endophenotypic variations of atopic dermatitis by age, race, and ethnicity. J Allergy Clin Immunol. 2020;8:1840-1852.
- McColl M, Boozalis E, Aguh C, et al. Pruritus in Black skin: unique molecular characteristics and clinical features. J Natl Med Assoc. 2021;114:30-38.
- Silverberg JI, Margolis DJ, Boguniewicz M, et al. Distribution of atopic dermatitis lesions in United States adults. J Eur Acad Dermatol Venereol. 2019;33:1341-1348.
- Summey BT, Bowen SE, Allen HB. Lichen planus-like atopic dermatitis: expanding the differential diagnosis of spongiotic dermatitis. J Cutan Pathol. 2008;35:311-314.
- Odhiambo JA, Williams HC, Clayton TO, et al; ISAAC Phase Three Study Group. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124:1251-1258.E23.
- Gallagher K, Halperin-Goldstein S, Paller AS. New treatments in atopic dermatitis update. Ann Allergy Asthma Immunol. 2025;135:498-510.E10.
- Shaw ML. FDA expands roflumilast use for atopic dermatitis to children aged 2 to 5 years. Am J Managed Care. October 6, 2025. Accessed April 30, 2026. https://www.ajmc.com/view/fda-expands -roflumilast-use-for-atopic-dermatitis-to-children-aged-2-to-5-years
- Eichenfield LF, Stein Gold LF, Simpson EL, et al. Efficacy and safety of ruxolitinib cream in children aged 2 to 11 years with atopic dermatitis: results from TRuE-AD3, a phase 3, randomized double-blind study. J Am Acad of Dermatol. 2025;93:689-698.
- Heath CR, Dosono B, Shi VY, et al. Variability in skin tone changes by race and ethnicity among US adults with atopic dermatitis. Presented at: Skin of Color Update 2024, September 13-15, 2024, New York, NY.
- Tackett KJ, Jenkins F, Morrell DS, et al. Structural racism and its influence on the severity of atopic dermatitis in African American children. Pediatr Dermatol. 2020;37:142-146.
- Narla S, Silverberg JI. The role of environmental exposures in atopic dermatitis. Curr Allergy Asthma Rep. 2020;20:74.
- Bauer SJ, Spoer BR, Ehrman R, et al. A systematic review of historic neighborhood redlining and contemporary health outcomes. Public Health. 2025;238:181-187.
- Chung J, Simpson EL. The socioeconomics of atopic dermatitis. Ann Allergy Asthma Immunol. 2019;122:360-366.
- Martinez A, de la Rosa R, Mujahid M, et al. Structural racism and its pathways to asthma and atopic dermatitis. J Allergy Clin Immunol. 2021;148:1112-1120.
- Bell MA, Whang KA, Thomas J, et al. Racial and ethnic disparities in access to emerging and frontline therapies in common dermatological conditions: a cross-sectional study. J Natl Med Assoc. 2020;112:650-653.
Atopic dermatitis (AD) is a chronic skin condition generally characterized by pruritic and erythematous papules and plaques.1 While AD commonly manifests in childhood, 1 in 4 patients living with AD report adult onset of the disease.2 The clinical presentation and prevalence of AD vary across age groups, skin tones, and racial and ethnic groups. Globally, AD is estimated to have a prevalence of 2.6%; however, rates vary widely by region.1 Morphology and distribution of AD lesions also vary by population; therefore, defining one classic presentation of AD is not sufficient in diverse patient populations.3
Epidemiology
The prevalence of AD ranges from 0.2% to 24.6% worldwide, with higher rates in Africa and Oceania and lower rates in India and Northern and Eastern Europe.1 In the United States, AD affects all racial and ethnic groups; however, prevalence and severity are increased in Black children compared with White children.4 In one prospective cohort study, Hispanic children and non-Hispanic Black children aged 3 years and younger had greater odds of AD persisting into mid childhood (approximately age 7 years) compared with non-Hispanic White children.5,6
Key Clinical Features
Clinical features of AD are heterogeneous and may include differences in color, morphology, and distribution. Brown, hyperpigmented, gray, and/or violaceous plaques may predominate in patients with skin of color (SOC) compared with the erythematous plaques commonly described in lighter skin tones.1,3 Established scoring systems for AD rely on erythema as a key diagnostic feature, but because erythema can be difficult to detect in darker skin tones, disease severity may be underestimated and diagnosis may be delayed in this population.4
Atopic dermatitis in SOC may manifest as lichenoid plaques,7 prurigo nodules,7,8 lichenification,1 and follicular accentuation.9 Lichen planus–like AD is a distinct variant characterized by lichenoid plaques with a predilection for the extensor surfaces and face in patients with darker skin tones1,8 occurring in approximately 9% of patients in one study.10
Other key clinical features of AD in patients with SOC include pityriasis alba,10 increased risk for postinflammatory pigment alteration (including hyperpigmentation and/or hypopigmentation),1 and greater trunk and extensor involvement.1,11
Worth Noting
The scientific landscape for AD has grown rapidly, increasing our understanding of its pathophysiology, treatment, and social impact. Nonsteroidal treatments available for pediatric and adult patients with AD have increased in recent years, including crisaborole (approved for use in those ages ≥3 months), tacrolimus (≥2 years), and pimecrolimus (≥2 years). Injectable options include dupilumab (≥6 months), lebrikizumab (≥12 years), nemolizumab (≥12 years), and tralokinumab (≥12 years). Oral options include abrocitinib (≥12 years) and upadacitinib (≥12 years).12 Topical options include roflumilast 0.15% cream (≥6 years)12 and 0.05% cream (≥2-5 years),13 ruxolitinib 1.5% cream (≥2 years),14 and tapinarof 1% cream (≥2 years).12
For some patients, postinflammatory pigment alteration associated with AD has a higher impact on quality of life than the AD itself.7 In a study of 260 US adults with AD, the emotional impact of pigmentary changes was greatest in Black patients, with 53.3% reporting that pigment changes bothered them “a lot” or “very much.”15
Genome-wide association studies have not identified a single determinant that explains racial and ethnic differences in susceptibility to AD.4 Instead, social determinants of health are thought to play a role in the difference in AD prevalence and severity across groups in the United States.16
Health Disparity Highlight
In an analysis of 20 US metropolitan cities, urban and inner-city residence was associated with approximately 1.7-fold increased odds of AD.4 Among pediatric patients with moderate to severe AD, Black children were more likely to be exposed to tobacco smoke17 and traffic-related air pollution.18 Low socioeconomic status and low income also have been associated with moderate16 and severe19 AD. At the same education level, Black individuals in the United States receive less income than their White counterparts and have markedly less wealth at equivalent incomes.20
In utero exposure to maternal stress is associated with AD.4 Increased IgE levels have been recorded in children who develop AD, with Black children having the highest IgE levels overall compared to other children.18
An analysis of medical records from an urban medical center in Baltimore, Maryland, from 2013 through 2018 showed that Black patients with AD were less likely to receive topical corticosteroids, topical calcineurin inhibitors, a topical phosphodiesterase 4 inhibitor, and a biologic compared to White patients with AD.21
Since the disproportionate burden experienced by patients with AD is not physiologic, it is imperative to address these systemic complexities and address the barriers impacting treatment availability to improve health outcomes for all patients living with AD.
Atopic dermatitis (AD) is a chronic skin condition generally characterized by pruritic and erythematous papules and plaques.1 While AD commonly manifests in childhood, 1 in 4 patients living with AD report adult onset of the disease.2 The clinical presentation and prevalence of AD vary across age groups, skin tones, and racial and ethnic groups. Globally, AD is estimated to have a prevalence of 2.6%; however, rates vary widely by region.1 Morphology and distribution of AD lesions also vary by population; therefore, defining one classic presentation of AD is not sufficient in diverse patient populations.3
Epidemiology
The prevalence of AD ranges from 0.2% to 24.6% worldwide, with higher rates in Africa and Oceania and lower rates in India and Northern and Eastern Europe.1 In the United States, AD affects all racial and ethnic groups; however, prevalence and severity are increased in Black children compared with White children.4 In one prospective cohort study, Hispanic children and non-Hispanic Black children aged 3 years and younger had greater odds of AD persisting into mid childhood (approximately age 7 years) compared with non-Hispanic White children.5,6
Key Clinical Features
Clinical features of AD are heterogeneous and may include differences in color, morphology, and distribution. Brown, hyperpigmented, gray, and/or violaceous plaques may predominate in patients with skin of color (SOC) compared with the erythematous plaques commonly described in lighter skin tones.1,3 Established scoring systems for AD rely on erythema as a key diagnostic feature, but because erythema can be difficult to detect in darker skin tones, disease severity may be underestimated and diagnosis may be delayed in this population.4
Atopic dermatitis in SOC may manifest as lichenoid plaques,7 prurigo nodules,7,8 lichenification,1 and follicular accentuation.9 Lichen planus–like AD is a distinct variant characterized by lichenoid plaques with a predilection for the extensor surfaces and face in patients with darker skin tones1,8 occurring in approximately 9% of patients in one study.10
Other key clinical features of AD in patients with SOC include pityriasis alba,10 increased risk for postinflammatory pigment alteration (including hyperpigmentation and/or hypopigmentation),1 and greater trunk and extensor involvement.1,11
Worth Noting
The scientific landscape for AD has grown rapidly, increasing our understanding of its pathophysiology, treatment, and social impact. Nonsteroidal treatments available for pediatric and adult patients with AD have increased in recent years, including crisaborole (approved for use in those ages ≥3 months), tacrolimus (≥2 years), and pimecrolimus (≥2 years). Injectable options include dupilumab (≥6 months), lebrikizumab (≥12 years), nemolizumab (≥12 years), and tralokinumab (≥12 years). Oral options include abrocitinib (≥12 years) and upadacitinib (≥12 years).12 Topical options include roflumilast 0.15% cream (≥6 years)12 and 0.05% cream (≥2-5 years),13 ruxolitinib 1.5% cream (≥2 years),14 and tapinarof 1% cream (≥2 years).12
For some patients, postinflammatory pigment alteration associated with AD has a higher impact on quality of life than the AD itself.7 In a study of 260 US adults with AD, the emotional impact of pigmentary changes was greatest in Black patients, with 53.3% reporting that pigment changes bothered them “a lot” or “very much.”15
Genome-wide association studies have not identified a single determinant that explains racial and ethnic differences in susceptibility to AD.4 Instead, social determinants of health are thought to play a role in the difference in AD prevalence and severity across groups in the United States.16
Health Disparity Highlight
In an analysis of 20 US metropolitan cities, urban and inner-city residence was associated with approximately 1.7-fold increased odds of AD.4 Among pediatric patients with moderate to severe AD, Black children were more likely to be exposed to tobacco smoke17 and traffic-related air pollution.18 Low socioeconomic status and low income also have been associated with moderate16 and severe19 AD. At the same education level, Black individuals in the United States receive less income than their White counterparts and have markedly less wealth at equivalent incomes.20
In utero exposure to maternal stress is associated with AD.4 Increased IgE levels have been recorded in children who develop AD, with Black children having the highest IgE levels overall compared to other children.18
An analysis of medical records from an urban medical center in Baltimore, Maryland, from 2013 through 2018 showed that Black patients with AD were less likely to receive topical corticosteroids, topical calcineurin inhibitors, a topical phosphodiesterase 4 inhibitor, and a biologic compared to White patients with AD.21
Since the disproportionate burden experienced by patients with AD is not physiologic, it is imperative to address these systemic complexities and address the barriers impacting treatment availability to improve health outcomes for all patients living with AD.
- Kaufman BP, Guttman-Yassky E, Alexis AF. Atopic dermatitis in diverse racial and ethnic groups—variations in epidemiology, genetics, clinical presentation and treatment. Exp Dermatol. 2018;27:340-357.
- Lee HH, Patel KR, Singam V, et al. A systematic review and meta-analysis of the prevalence and phenotype of adult-onset atopic dermatitis. J Am Acad Dermatol. 2019;80:1526-1532.E7.
- Adawi W, Cornman H, Kambala A, et al. Diagnosing atopic dermatitis in skin of color. Dermatol Clin. 2023;41:417-429.
- Narla S, Silverberg JI. Current updates in the epidemiology and comorbidities of atopic dermatitis. Ann Allergy Asthma Immunol. 2025;135:511-520.
- Croce EA, Levy ML, Adamson AS, et al. Reframing racial and ethnic disparities in atopic dermatitis in Black and Latinx populations. J Allergy Clin Immunol. 2021;148:1104-1111.
- Kim Y, Blomberg M, Rifas-Shiman SL, et al. Racial/ethnic differences in incidence and persistence of childhood atopic dermatitis. J Invest Dermatol. 2019;139:827-834.
- Nomura T, Wu J, Kabashima K, et al. Endophenotypic variations of atopic dermatitis by age, race, and ethnicity. J Allergy Clin Immunol. 2020;8:1840-1852.
- McColl M, Boozalis E, Aguh C, et al. Pruritus in Black skin: unique molecular characteristics and clinical features. J Natl Med Assoc. 2021;114:30-38.
- Silverberg JI, Margolis DJ, Boguniewicz M, et al. Distribution of atopic dermatitis lesions in United States adults. J Eur Acad Dermatol Venereol. 2019;33:1341-1348.
- Summey BT, Bowen SE, Allen HB. Lichen planus-like atopic dermatitis: expanding the differential diagnosis of spongiotic dermatitis. J Cutan Pathol. 2008;35:311-314.
- Odhiambo JA, Williams HC, Clayton TO, et al; ISAAC Phase Three Study Group. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124:1251-1258.E23.
- Gallagher K, Halperin-Goldstein S, Paller AS. New treatments in atopic dermatitis update. Ann Allergy Asthma Immunol. 2025;135:498-510.E10.
- Shaw ML. FDA expands roflumilast use for atopic dermatitis to children aged 2 to 5 years. Am J Managed Care. October 6, 2025. Accessed April 30, 2026. https://www.ajmc.com/view/fda-expands -roflumilast-use-for-atopic-dermatitis-to-children-aged-2-to-5-years
- Eichenfield LF, Stein Gold LF, Simpson EL, et al. Efficacy and safety of ruxolitinib cream in children aged 2 to 11 years with atopic dermatitis: results from TRuE-AD3, a phase 3, randomized double-blind study. J Am Acad of Dermatol. 2025;93:689-698.
- Heath CR, Dosono B, Shi VY, et al. Variability in skin tone changes by race and ethnicity among US adults with atopic dermatitis. Presented at: Skin of Color Update 2024, September 13-15, 2024, New York, NY.
- Tackett KJ, Jenkins F, Morrell DS, et al. Structural racism and its influence on the severity of atopic dermatitis in African American children. Pediatr Dermatol. 2020;37:142-146.
- Narla S, Silverberg JI. The role of environmental exposures in atopic dermatitis. Curr Allergy Asthma Rep. 2020;20:74.
- Bauer SJ, Spoer BR, Ehrman R, et al. A systematic review of historic neighborhood redlining and contemporary health outcomes. Public Health. 2025;238:181-187.
- Chung J, Simpson EL. The socioeconomics of atopic dermatitis. Ann Allergy Asthma Immunol. 2019;122:360-366.
- Martinez A, de la Rosa R, Mujahid M, et al. Structural racism and its pathways to asthma and atopic dermatitis. J Allergy Clin Immunol. 2021;148:1112-1120.
- Bell MA, Whang KA, Thomas J, et al. Racial and ethnic disparities in access to emerging and frontline therapies in common dermatological conditions: a cross-sectional study. J Natl Med Assoc. 2020;112:650-653.
- Kaufman BP, Guttman-Yassky E, Alexis AF. Atopic dermatitis in diverse racial and ethnic groups—variations in epidemiology, genetics, clinical presentation and treatment. Exp Dermatol. 2018;27:340-357.
- Lee HH, Patel KR, Singam V, et al. A systematic review and meta-analysis of the prevalence and phenotype of adult-onset atopic dermatitis. J Am Acad Dermatol. 2019;80:1526-1532.E7.
- Adawi W, Cornman H, Kambala A, et al. Diagnosing atopic dermatitis in skin of color. Dermatol Clin. 2023;41:417-429.
- Narla S, Silverberg JI. Current updates in the epidemiology and comorbidities of atopic dermatitis. Ann Allergy Asthma Immunol. 2025;135:511-520.
- Croce EA, Levy ML, Adamson AS, et al. Reframing racial and ethnic disparities in atopic dermatitis in Black and Latinx populations. J Allergy Clin Immunol. 2021;148:1104-1111.
- Kim Y, Blomberg M, Rifas-Shiman SL, et al. Racial/ethnic differences in incidence and persistence of childhood atopic dermatitis. J Invest Dermatol. 2019;139:827-834.
- Nomura T, Wu J, Kabashima K, et al. Endophenotypic variations of atopic dermatitis by age, race, and ethnicity. J Allergy Clin Immunol. 2020;8:1840-1852.
- McColl M, Boozalis E, Aguh C, et al. Pruritus in Black skin: unique molecular characteristics and clinical features. J Natl Med Assoc. 2021;114:30-38.
- Silverberg JI, Margolis DJ, Boguniewicz M, et al. Distribution of atopic dermatitis lesions in United States adults. J Eur Acad Dermatol Venereol. 2019;33:1341-1348.
- Summey BT, Bowen SE, Allen HB. Lichen planus-like atopic dermatitis: expanding the differential diagnosis of spongiotic dermatitis. J Cutan Pathol. 2008;35:311-314.
- Odhiambo JA, Williams HC, Clayton TO, et al; ISAAC Phase Three Study Group. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124:1251-1258.E23.
- Gallagher K, Halperin-Goldstein S, Paller AS. New treatments in atopic dermatitis update. Ann Allergy Asthma Immunol. 2025;135:498-510.E10.
- Shaw ML. FDA expands roflumilast use for atopic dermatitis to children aged 2 to 5 years. Am J Managed Care. October 6, 2025. Accessed April 30, 2026. https://www.ajmc.com/view/fda-expands -roflumilast-use-for-atopic-dermatitis-to-children-aged-2-to-5-years
- Eichenfield LF, Stein Gold LF, Simpson EL, et al. Efficacy and safety of ruxolitinib cream in children aged 2 to 11 years with atopic dermatitis: results from TRuE-AD3, a phase 3, randomized double-blind study. J Am Acad of Dermatol. 2025;93:689-698.
- Heath CR, Dosono B, Shi VY, et al. Variability in skin tone changes by race and ethnicity among US adults with atopic dermatitis. Presented at: Skin of Color Update 2024, September 13-15, 2024, New York, NY.
- Tackett KJ, Jenkins F, Morrell DS, et al. Structural racism and its influence on the severity of atopic dermatitis in African American children. Pediatr Dermatol. 2020;37:142-146.
- Narla S, Silverberg JI. The role of environmental exposures in atopic dermatitis. Curr Allergy Asthma Rep. 2020;20:74.
- Bauer SJ, Spoer BR, Ehrman R, et al. A systematic review of historic neighborhood redlining and contemporary health outcomes. Public Health. 2025;238:181-187.
- Chung J, Simpson EL. The socioeconomics of atopic dermatitis. Ann Allergy Asthma Immunol. 2019;122:360-366.
- Martinez A, de la Rosa R, Mujahid M, et al. Structural racism and its pathways to asthma and atopic dermatitis. J Allergy Clin Immunol. 2021;148:1112-1120.
- Bell MA, Whang KA, Thomas J, et al. Racial and ethnic disparities in access to emerging and frontline therapies in common dermatological conditions: a cross-sectional study. J Natl Med Assoc. 2020;112:650-653.
Atopic Dermatitis: New Insights and Expanded Treatment Options
Atopic Dermatitis: New Insights and Expanded Treatment Options

Medical Decision-Making in Evaluation and Management Coding: Practical Applications and Key Clarifications
Medical Decision-Making in Evaluation and Management Coding: Practical Applications and Key Clarifications
The new coding guidelines for evaluation and management services have simplified coding by focusing on medical decision-making (MDM), but practicing clinicians often have questions about how to apply the rules. This article will focus on common mistakes and nuances clarified in communications from the American Medical Association. As before, the highest level of service in 2 of 3 categories—complexity, data, and risk—determine the level of service. Only medically necessary services should be reported, and all reported codes should accurately reflect the services provided.
Key Clarifications to MDM Criteria
Important clarifications that came after the initial distribution of the new coding rules include the following:
- An established problem not at treatment target requiring ongoing MDM counts as moderate complexity in column 1.
- Under the category of risk, prescription drug therapy includes discussion of risks, benefits, and alternatives with a decision to start, stop, or continue a prescription medication; this differs from a simple refill that does not require evaluation, discussion, and shared decision-making.
- Social determinants of health that are medically appropriate to address during the visit are considered moderate under the category of risk. These include issues that directly affect patient management (eg, transportation access, medication affordability, cultural norms, restrictions) and other factors influencing health and well-being (eg, income, education, occupation, environmental change, unemployment, working conditions, social support) when they impact the patient’s condition and inform treatment decisions.
- Independent interpretation of a laboratory test counts as moderate under the category of data; an example would be a biopsy reported as “consistent with lupus erythematosus” in a patient with a heliotrope rash and shawl sign, which may require clinicopathologic correlation and reinterpretation as “diagnostic of dermatomyositis.”
- The decision to perform a 0- or 10-day global procedure on the same date of service as the visit is already bundled in payment for the procedure and should not be reported as a separate service; however, if scheduled for a future date of service, it counts as low under the risk category if the patient has no unique risk factors or moderate if the patient does have unique risk factors that weigh into MDM. In contrast, the decision to perform a 90-day global procedure is reportable even on the same date of service (with modifier 57) and counts as moderate risk without unique risk factors and high with such factors.
Application of MDM Coding in Common Dermatology Encounters
Let’s look at some common scenarios and how they should be coded.
A patient presents with a new lesion of concern. On physical examination, it is a stuck-on keratotic papule with no inflammation, and you reassure them that it is merely a benign seborrheic keratosis. This encounter would be coded as straightforward MDM (level 2, new or established), reflecting the evaluation of a single minor problem.
A patient returns with localized eczema and is doing very well with triamcinolone cream applied as needed, and you simply refill the prescription. This encounter represents low-level MDM (level 3, established), reflecting a single stable problem managed with a simple prescription refill.
A patient presents with psoriasis that has had some response to topical therapy but is clearly not at target, and the patient now reports axial joint stiffness that is much worse in the morning and takes more than 30 minutes to resolve. You discuss risks, benefits, and alternatives; note that the patient already had recent screening for tuberculosis and other infectious diseases; and prescribe a T-helper 17 biologic because of the axial arthritis. This encounter represents moderate-level MDM (level 4, established), reflecting both a problem not at target and a new problem of uncertain prognosis under complexity, as well as shared decision-making to initiate prescription drug therapy under risk. Although review or ordering of 3 laboratory tests would also meet moderate criteria under data, only 2 of the 3 domains are required to establish the level of service.
A patient presents with a severe flare of eczema that requires treatment with cyclosporine. This encounter represents high-level MDM (level 5, established), reflecting a severe exacerbation of an existing condition requiring a high-risk medication with at least quarterly laboratory monitoring and uncertainty regarding long-term therapy needs.
Application of Moderate and High MDM in Dermatology
Many dermatology patients present with multiple problems, and the visit often falls into the moderate category for column 1 (complexity) of MDM (level 4, new or established). Under complexity, moderate could be 2 stable problems, one worsening problem, one new problem of uncertain prognosis, or one problem improved but not at target. Remember that moderate MDM also must be established in a second category, such as risk. Under the risk category, moderate could include prescription drug therapy, addressing a relevant social determinant of health, the decision to perform a 0- or 10-day global procedure not performed on the same date of service with unique patient risk factors, or the decision to perform a 90-day global procedure in a patient with no unique risk factors.
Don’t forget the data category, as it often is relevant to determining the correct level of service. Moderate under the data category includes review or ordering of 3 laboratory tests (a complete metabolic panel is a single laboratory test, and a complete blood count is a single laboratory test), independent interpretation of a laboratory test, or a phone call to another provider caring for the patient with a medically necessary discussion of management (eg, severe eczema in a patient on a calcium channel blocker—you call the primary care physician advising that calcium-channel blockers are the most common cause of eczematous drug eruption and advise a change in therapy). If 2 of the above categories were necessary (eg, order 3 laboratory tests and call the primary care physician), that would count as high MDM under the data category. Remember, 2 categories are required to establish the level of service.
Other examples of high MDM (level 5, new or established) include a new diagnosis with major risk to life or limb plus one of the following: high-risk medication requiring at least quarterly drug monitoring, the decision to perform a 90-day global surgery with documented additional patient risk factors, or the decision to admit to the hospital (eg, new invasive melanoma with excision and decision to perform adjacent tissue transfer in a patient who takes aspirin).
Final Thoughts
Physicians should perform medically necessary services that are in the best interest of their patients. Current coding rules focus on the complexity, risk, and data associated with MDM.
The new coding guidelines for evaluation and management services have simplified coding by focusing on medical decision-making (MDM), but practicing clinicians often have questions about how to apply the rules. This article will focus on common mistakes and nuances clarified in communications from the American Medical Association. As before, the highest level of service in 2 of 3 categories—complexity, data, and risk—determine the level of service. Only medically necessary services should be reported, and all reported codes should accurately reflect the services provided.
Key Clarifications to MDM Criteria
Important clarifications that came after the initial distribution of the new coding rules include the following:
- An established problem not at treatment target requiring ongoing MDM counts as moderate complexity in column 1.
- Under the category of risk, prescription drug therapy includes discussion of risks, benefits, and alternatives with a decision to start, stop, or continue a prescription medication; this differs from a simple refill that does not require evaluation, discussion, and shared decision-making.
- Social determinants of health that are medically appropriate to address during the visit are considered moderate under the category of risk. These include issues that directly affect patient management (eg, transportation access, medication affordability, cultural norms, restrictions) and other factors influencing health and well-being (eg, income, education, occupation, environmental change, unemployment, working conditions, social support) when they impact the patient’s condition and inform treatment decisions.
- Independent interpretation of a laboratory test counts as moderate under the category of data; an example would be a biopsy reported as “consistent with lupus erythematosus” in a patient with a heliotrope rash and shawl sign, which may require clinicopathologic correlation and reinterpretation as “diagnostic of dermatomyositis.”
- The decision to perform a 0- or 10-day global procedure on the same date of service as the visit is already bundled in payment for the procedure and should not be reported as a separate service; however, if scheduled for a future date of service, it counts as low under the risk category if the patient has no unique risk factors or moderate if the patient does have unique risk factors that weigh into MDM. In contrast, the decision to perform a 90-day global procedure is reportable even on the same date of service (with modifier 57) and counts as moderate risk without unique risk factors and high with such factors.
Application of MDM Coding in Common Dermatology Encounters
Let’s look at some common scenarios and how they should be coded.
A patient presents with a new lesion of concern. On physical examination, it is a stuck-on keratotic papule with no inflammation, and you reassure them that it is merely a benign seborrheic keratosis. This encounter would be coded as straightforward MDM (level 2, new or established), reflecting the evaluation of a single minor problem.
A patient returns with localized eczema and is doing very well with triamcinolone cream applied as needed, and you simply refill the prescription. This encounter represents low-level MDM (level 3, established), reflecting a single stable problem managed with a simple prescription refill.
A patient presents with psoriasis that has had some response to topical therapy but is clearly not at target, and the patient now reports axial joint stiffness that is much worse in the morning and takes more than 30 minutes to resolve. You discuss risks, benefits, and alternatives; note that the patient already had recent screening for tuberculosis and other infectious diseases; and prescribe a T-helper 17 biologic because of the axial arthritis. This encounter represents moderate-level MDM (level 4, established), reflecting both a problem not at target and a new problem of uncertain prognosis under complexity, as well as shared decision-making to initiate prescription drug therapy under risk. Although review or ordering of 3 laboratory tests would also meet moderate criteria under data, only 2 of the 3 domains are required to establish the level of service.
A patient presents with a severe flare of eczema that requires treatment with cyclosporine. This encounter represents high-level MDM (level 5, established), reflecting a severe exacerbation of an existing condition requiring a high-risk medication with at least quarterly laboratory monitoring and uncertainty regarding long-term therapy needs.
Application of Moderate and High MDM in Dermatology
Many dermatology patients present with multiple problems, and the visit often falls into the moderate category for column 1 (complexity) of MDM (level 4, new or established). Under complexity, moderate could be 2 stable problems, one worsening problem, one new problem of uncertain prognosis, or one problem improved but not at target. Remember that moderate MDM also must be established in a second category, such as risk. Under the risk category, moderate could include prescription drug therapy, addressing a relevant social determinant of health, the decision to perform a 0- or 10-day global procedure not performed on the same date of service with unique patient risk factors, or the decision to perform a 90-day global procedure in a patient with no unique risk factors.
Don’t forget the data category, as it often is relevant to determining the correct level of service. Moderate under the data category includes review or ordering of 3 laboratory tests (a complete metabolic panel is a single laboratory test, and a complete blood count is a single laboratory test), independent interpretation of a laboratory test, or a phone call to another provider caring for the patient with a medically necessary discussion of management (eg, severe eczema in a patient on a calcium channel blocker—you call the primary care physician advising that calcium-channel blockers are the most common cause of eczematous drug eruption and advise a change in therapy). If 2 of the above categories were necessary (eg, order 3 laboratory tests and call the primary care physician), that would count as high MDM under the data category. Remember, 2 categories are required to establish the level of service.
Other examples of high MDM (level 5, new or established) include a new diagnosis with major risk to life or limb plus one of the following: high-risk medication requiring at least quarterly drug monitoring, the decision to perform a 90-day global surgery with documented additional patient risk factors, or the decision to admit to the hospital (eg, new invasive melanoma with excision and decision to perform adjacent tissue transfer in a patient who takes aspirin).
Final Thoughts
Physicians should perform medically necessary services that are in the best interest of their patients. Current coding rules focus on the complexity, risk, and data associated with MDM.
The new coding guidelines for evaluation and management services have simplified coding by focusing on medical decision-making (MDM), but practicing clinicians often have questions about how to apply the rules. This article will focus on common mistakes and nuances clarified in communications from the American Medical Association. As before, the highest level of service in 2 of 3 categories—complexity, data, and risk—determine the level of service. Only medically necessary services should be reported, and all reported codes should accurately reflect the services provided.
Key Clarifications to MDM Criteria
Important clarifications that came after the initial distribution of the new coding rules include the following:
- An established problem not at treatment target requiring ongoing MDM counts as moderate complexity in column 1.
- Under the category of risk, prescription drug therapy includes discussion of risks, benefits, and alternatives with a decision to start, stop, or continue a prescription medication; this differs from a simple refill that does not require evaluation, discussion, and shared decision-making.
- Social determinants of health that are medically appropriate to address during the visit are considered moderate under the category of risk. These include issues that directly affect patient management (eg, transportation access, medication affordability, cultural norms, restrictions) and other factors influencing health and well-being (eg, income, education, occupation, environmental change, unemployment, working conditions, social support) when they impact the patient’s condition and inform treatment decisions.
- Independent interpretation of a laboratory test counts as moderate under the category of data; an example would be a biopsy reported as “consistent with lupus erythematosus” in a patient with a heliotrope rash and shawl sign, which may require clinicopathologic correlation and reinterpretation as “diagnostic of dermatomyositis.”
- The decision to perform a 0- or 10-day global procedure on the same date of service as the visit is already bundled in payment for the procedure and should not be reported as a separate service; however, if scheduled for a future date of service, it counts as low under the risk category if the patient has no unique risk factors or moderate if the patient does have unique risk factors that weigh into MDM. In contrast, the decision to perform a 90-day global procedure is reportable even on the same date of service (with modifier 57) and counts as moderate risk without unique risk factors and high with such factors.
Application of MDM Coding in Common Dermatology Encounters
Let’s look at some common scenarios and how they should be coded.
A patient presents with a new lesion of concern. On physical examination, it is a stuck-on keratotic papule with no inflammation, and you reassure them that it is merely a benign seborrheic keratosis. This encounter would be coded as straightforward MDM (level 2, new or established), reflecting the evaluation of a single minor problem.
A patient returns with localized eczema and is doing very well with triamcinolone cream applied as needed, and you simply refill the prescription. This encounter represents low-level MDM (level 3, established), reflecting a single stable problem managed with a simple prescription refill.
A patient presents with psoriasis that has had some response to topical therapy but is clearly not at target, and the patient now reports axial joint stiffness that is much worse in the morning and takes more than 30 minutes to resolve. You discuss risks, benefits, and alternatives; note that the patient already had recent screening for tuberculosis and other infectious diseases; and prescribe a T-helper 17 biologic because of the axial arthritis. This encounter represents moderate-level MDM (level 4, established), reflecting both a problem not at target and a new problem of uncertain prognosis under complexity, as well as shared decision-making to initiate prescription drug therapy under risk. Although review or ordering of 3 laboratory tests would also meet moderate criteria under data, only 2 of the 3 domains are required to establish the level of service.
A patient presents with a severe flare of eczema that requires treatment with cyclosporine. This encounter represents high-level MDM (level 5, established), reflecting a severe exacerbation of an existing condition requiring a high-risk medication with at least quarterly laboratory monitoring and uncertainty regarding long-term therapy needs.
Application of Moderate and High MDM in Dermatology
Many dermatology patients present with multiple problems, and the visit often falls into the moderate category for column 1 (complexity) of MDM (level 4, new or established). Under complexity, moderate could be 2 stable problems, one worsening problem, one new problem of uncertain prognosis, or one problem improved but not at target. Remember that moderate MDM also must be established in a second category, such as risk. Under the risk category, moderate could include prescription drug therapy, addressing a relevant social determinant of health, the decision to perform a 0- or 10-day global procedure not performed on the same date of service with unique patient risk factors, or the decision to perform a 90-day global procedure in a patient with no unique risk factors.
Don’t forget the data category, as it often is relevant to determining the correct level of service. Moderate under the data category includes review or ordering of 3 laboratory tests (a complete metabolic panel is a single laboratory test, and a complete blood count is a single laboratory test), independent interpretation of a laboratory test, or a phone call to another provider caring for the patient with a medically necessary discussion of management (eg, severe eczema in a patient on a calcium channel blocker—you call the primary care physician advising that calcium-channel blockers are the most common cause of eczematous drug eruption and advise a change in therapy). If 2 of the above categories were necessary (eg, order 3 laboratory tests and call the primary care physician), that would count as high MDM under the data category. Remember, 2 categories are required to establish the level of service.
Other examples of high MDM (level 5, new or established) include a new diagnosis with major risk to life or limb plus one of the following: high-risk medication requiring at least quarterly drug monitoring, the decision to perform a 90-day global surgery with documented additional patient risk factors, or the decision to admit to the hospital (eg, new invasive melanoma with excision and decision to perform adjacent tissue transfer in a patient who takes aspirin).
Final Thoughts
Physicians should perform medically necessary services that are in the best interest of their patients. Current coding rules focus on the complexity, risk, and data associated with MDM.
Medical Decision-Making in Evaluation and Management Coding: Practical Applications and Key Clarifications
Medical Decision-Making in Evaluation and Management Coding: Practical Applications and Key Clarifications
PRACTICE POINTS
- Evaluation and management coding is now based on medical decision-making (MDM), requiring 2 of 3 categories (complexity, data, risk) and strict adherence to medical necessity.
- Moderate MDM includes problems not at target, shared decision-making for prescription therapy (not simple refills), relevant social determinants of health, and independent test interpretation.
- Common errors include misclassifying refills, overlooking the data category, and improperly reporting procedural decisions, especially same-day minor procedures.

Limitations of Fitzpatrick Skin Type as a Proxy for Skin Color and Race
Limitations of Fitzpatrick Skin Type as a Proxy for Skin Color and Race
Recognizing inflammation in darker skin tones has important implications for diagnosis and management of skin disease, particularly in patients with skin of color.1 In this context, classification systems commonly are used—both in research and clinical practice—to standardize descriptions of skin tone across diverse populations. Fitzpatrick skin type (FST) originally was developed to classify cutaneous response to UV radiation exposure and remains one of the most widely used frameworks in dermatology.2 However, FST often is used beyond its intended purpose as a proxy for differentiating skin color and race.3,4 This mismatch risks obscuring clinically meaningful differences and limiting the accuracy of dermatologic research. Herein, we review the intended use of FST, its limitations in representing skin color and race, and considerations for more accurate characterization of skin pigmentation in clinical practice and research.
Origins and Intended Use of the FST Scale
Fitzpatrick skin type was developed by Thomas B. Fitzpatrick in the 1970s to guide UVA dosing for psoralen plus UVA therapy in patients with psoriasis.5,6 The scale was intended to estimate an individual’s erythematous and pigmentary response to UV exposure.6,7 Early iterations of FST largely were based on lighter skin types, reflecting its initial use in predominantly White populations, which limited representation of the full spectrum of skin tone diversity.5
Clinical, Educational, and Research Limitations of FST
Fitzpatrick skin type now is widely, albeit inaccurately, used in both research and clinical practice as a proxy for skin color and race,7,8 which reflects its ease of use and the lack of standardized alternatives; however, FST does not adequately capture variability in baseline skin pigmentation, undertone, or inflammatory response. These limitations are especially pronounced in phototypes IV to VI, which encompass highly heterogeneous populations. As a result, grouping patients by FST alone to describe skin color and race may obscure important differences and limit meaningful interpretation of clinical and research findings.
Clinically, recognition of dermatologic conditions such as erythema may be more challenging in darker skin tones, in which classic visual cues are less apparent.1,7 Relying on FST to stratify skin color may further compound diagnostic uncertainty by oversimplifying the cutaneous presentation. In addition, treatment decisions, including laser settings and assessment of pigmentary risk, often are guided by FST despite within-group variability.7 Further, educational frameworks that rely heavily on FST may inadequately prepare clinicians to recognize disease across diverse skin tones, contributing to delayed diagnosis and disparities in care in populations with skin of color.
The implications also extend to dermatologic research. Fitzpatrick skin type frequently is used to assess study populations; however, its limited ability to reflect true variation in pigmentation and ethnicity introduces misclassification bias.3,7 The broad FST scale may group heterogeneous populations, obscuring differences in treatment response. As a result, studies relying on FST to represent skin color or race may have reduced generalizability across diverse populations. Importantly, these limitations are not merely conceptual but may contribute to measurable disparities in dermatologic diagnosis and outcomes.
Rethinking Skin Classification Frameworks
Despite these shortcomings, FST remains deeply embedded in dermatology. Its decades-long use has led to widespread familiarity and integration into clinical guidelines, education, and research. At the same time, the absence of a universally accepted alternative has reinforced the continued use of FST as a proxy for skin color and race.
Alternative strategies for characterizing skin pigmentation include objective measures such as spectrophotometry and melanin index assessment.9
Although these approaches may provide more precise quantification of pigmentation, their use may be limited by the need for specialized equipment and reduced feasibility in routine clinical settings. Other proposed approaches incorporate multidimensional factors such as pigmentation, photoreactivity, and genetic ancestry.4 While these techniques represent important advances, none has achieved widespread adoption yet, and each presents challenges related to feasibility and standardization.
In the absence of a single ideal system, a more nuanced approach is needed. Fitzpatrick skin type should be used in the context for which it was designed: estimating UV response. Incorporating additional descriptors, including self-identified race and ethnicity, alongside more detailed assessments of pigmentation may improve the accuracy and relevance of both clinical evaluation and research. Combining FST with more precise and inclusive frameworks represents a pragmatic step toward better reflecting patient diversity.
- Taylor SC. Recognizing erythema in skin of color. J Am Acad Dermatol.
- Fitzpatrick TB. The validity and practicality of sun-reactive skin types I through VI. Arch Dermatol. 1988;124:869-871. doi:10.1001 /archderm.124.6.869
- Eilers S, Bach DQ, Gaber R, et al. Accuracy of self-reported Fitzpatrick skin phototype classification in US Hispanic and Latino populations. JAMA Dermatol. 2013;149:797-803. doi:10.1001 /jamadermatol.2013.4091
- Del Bino S, Bernerd F. Variations in skin colour and the biological consequences of ultraviolet radiation exposure. Br J Dermatol. 2013;169(S3):33-40. doi:10.1111/bjd.12529
- Goon P, Banfield C, Bello O, et al. Skin cancers in skin types IV–VI: does the Fitzpatrick scale give a false sense of security? Clin Exp Dermatol. 2022;47:1112-1117. doi:10.1002/ski2.40
- Fitzpatrick TB. Soleil et peau. J Med Asthet. 1975;2:33-34.
- Ware OR, Dawson JE, Shinohara MM, et al. Racial limitations of Fitzpatrick skin type. Cutis. 2020;105:77-80.
- Lester JC, Taylor SC, Chren MM. Under-representation of skin of colour in dermatology images: not just an educational issue. Br J Dermatol. 2019;180:1521-1522. doi:10.1111/bjd.17608
- Fullerton A, Fischer T, Lahti A, et al. Guidelines for measurement of skin colour and erythema. a report from the Standardization Group of the European Society of Contact Dermatitis. Contact Dermatitis. 1996;35:1-10. doi:10.1111/j.1600-0536.1996.tb02242.x
Recognizing inflammation in darker skin tones has important implications for diagnosis and management of skin disease, particularly in patients with skin of color.1 In this context, classification systems commonly are used—both in research and clinical practice—to standardize descriptions of skin tone across diverse populations. Fitzpatrick skin type (FST) originally was developed to classify cutaneous response to UV radiation exposure and remains one of the most widely used frameworks in dermatology.2 However, FST often is used beyond its intended purpose as a proxy for differentiating skin color and race.3,4 This mismatch risks obscuring clinically meaningful differences and limiting the accuracy of dermatologic research. Herein, we review the intended use of FST, its limitations in representing skin color and race, and considerations for more accurate characterization of skin pigmentation in clinical practice and research.
Origins and Intended Use of the FST Scale
Fitzpatrick skin type was developed by Thomas B. Fitzpatrick in the 1970s to guide UVA dosing for psoralen plus UVA therapy in patients with psoriasis.5,6 The scale was intended to estimate an individual’s erythematous and pigmentary response to UV exposure.6,7 Early iterations of FST largely were based on lighter skin types, reflecting its initial use in predominantly White populations, which limited representation of the full spectrum of skin tone diversity.5
Clinical, Educational, and Research Limitations of FST
Fitzpatrick skin type now is widely, albeit inaccurately, used in both research and clinical practice as a proxy for skin color and race,7,8 which reflects its ease of use and the lack of standardized alternatives; however, FST does not adequately capture variability in baseline skin pigmentation, undertone, or inflammatory response. These limitations are especially pronounced in phototypes IV to VI, which encompass highly heterogeneous populations. As a result, grouping patients by FST alone to describe skin color and race may obscure important differences and limit meaningful interpretation of clinical and research findings.
Clinically, recognition of dermatologic conditions such as erythema may be more challenging in darker skin tones, in which classic visual cues are less apparent.1,7 Relying on FST to stratify skin color may further compound diagnostic uncertainty by oversimplifying the cutaneous presentation. In addition, treatment decisions, including laser settings and assessment of pigmentary risk, often are guided by FST despite within-group variability.7 Further, educational frameworks that rely heavily on FST may inadequately prepare clinicians to recognize disease across diverse skin tones, contributing to delayed diagnosis and disparities in care in populations with skin of color.
The implications also extend to dermatologic research. Fitzpatrick skin type frequently is used to assess study populations; however, its limited ability to reflect true variation in pigmentation and ethnicity introduces misclassification bias.3,7 The broad FST scale may group heterogeneous populations, obscuring differences in treatment response. As a result, studies relying on FST to represent skin color or race may have reduced generalizability across diverse populations. Importantly, these limitations are not merely conceptual but may contribute to measurable disparities in dermatologic diagnosis and outcomes.
Rethinking Skin Classification Frameworks
Despite these shortcomings, FST remains deeply embedded in dermatology. Its decades-long use has led to widespread familiarity and integration into clinical guidelines, education, and research. At the same time, the absence of a universally accepted alternative has reinforced the continued use of FST as a proxy for skin color and race.
Alternative strategies for characterizing skin pigmentation include objective measures such as spectrophotometry and melanin index assessment.9
Although these approaches may provide more precise quantification of pigmentation, their use may be limited by the need for specialized equipment and reduced feasibility in routine clinical settings. Other proposed approaches incorporate multidimensional factors such as pigmentation, photoreactivity, and genetic ancestry.4 While these techniques represent important advances, none has achieved widespread adoption yet, and each presents challenges related to feasibility and standardization.
In the absence of a single ideal system, a more nuanced approach is needed. Fitzpatrick skin type should be used in the context for which it was designed: estimating UV response. Incorporating additional descriptors, including self-identified race and ethnicity, alongside more detailed assessments of pigmentation may improve the accuracy and relevance of both clinical evaluation and research. Combining FST with more precise and inclusive frameworks represents a pragmatic step toward better reflecting patient diversity.
Recognizing inflammation in darker skin tones has important implications for diagnosis and management of skin disease, particularly in patients with skin of color.1 In this context, classification systems commonly are used—both in research and clinical practice—to standardize descriptions of skin tone across diverse populations. Fitzpatrick skin type (FST) originally was developed to classify cutaneous response to UV radiation exposure and remains one of the most widely used frameworks in dermatology.2 However, FST often is used beyond its intended purpose as a proxy for differentiating skin color and race.3,4 This mismatch risks obscuring clinically meaningful differences and limiting the accuracy of dermatologic research. Herein, we review the intended use of FST, its limitations in representing skin color and race, and considerations for more accurate characterization of skin pigmentation in clinical practice and research.
Origins and Intended Use of the FST Scale
Fitzpatrick skin type was developed by Thomas B. Fitzpatrick in the 1970s to guide UVA dosing for psoralen plus UVA therapy in patients with psoriasis.5,6 The scale was intended to estimate an individual’s erythematous and pigmentary response to UV exposure.6,7 Early iterations of FST largely were based on lighter skin types, reflecting its initial use in predominantly White populations, which limited representation of the full spectrum of skin tone diversity.5
Clinical, Educational, and Research Limitations of FST
Fitzpatrick skin type now is widely, albeit inaccurately, used in both research and clinical practice as a proxy for skin color and race,7,8 which reflects its ease of use and the lack of standardized alternatives; however, FST does not adequately capture variability in baseline skin pigmentation, undertone, or inflammatory response. These limitations are especially pronounced in phototypes IV to VI, which encompass highly heterogeneous populations. As a result, grouping patients by FST alone to describe skin color and race may obscure important differences and limit meaningful interpretation of clinical and research findings.
Clinically, recognition of dermatologic conditions such as erythema may be more challenging in darker skin tones, in which classic visual cues are less apparent.1,7 Relying on FST to stratify skin color may further compound diagnostic uncertainty by oversimplifying the cutaneous presentation. In addition, treatment decisions, including laser settings and assessment of pigmentary risk, often are guided by FST despite within-group variability.7 Further, educational frameworks that rely heavily on FST may inadequately prepare clinicians to recognize disease across diverse skin tones, contributing to delayed diagnosis and disparities in care in populations with skin of color.
The implications also extend to dermatologic research. Fitzpatrick skin type frequently is used to assess study populations; however, its limited ability to reflect true variation in pigmentation and ethnicity introduces misclassification bias.3,7 The broad FST scale may group heterogeneous populations, obscuring differences in treatment response. As a result, studies relying on FST to represent skin color or race may have reduced generalizability across diverse populations. Importantly, these limitations are not merely conceptual but may contribute to measurable disparities in dermatologic diagnosis and outcomes.
Rethinking Skin Classification Frameworks
Despite these shortcomings, FST remains deeply embedded in dermatology. Its decades-long use has led to widespread familiarity and integration into clinical guidelines, education, and research. At the same time, the absence of a universally accepted alternative has reinforced the continued use of FST as a proxy for skin color and race.
Alternative strategies for characterizing skin pigmentation include objective measures such as spectrophotometry and melanin index assessment.9
Although these approaches may provide more precise quantification of pigmentation, their use may be limited by the need for specialized equipment and reduced feasibility in routine clinical settings. Other proposed approaches incorporate multidimensional factors such as pigmentation, photoreactivity, and genetic ancestry.4 While these techniques represent important advances, none has achieved widespread adoption yet, and each presents challenges related to feasibility and standardization.
In the absence of a single ideal system, a more nuanced approach is needed. Fitzpatrick skin type should be used in the context for which it was designed: estimating UV response. Incorporating additional descriptors, including self-identified race and ethnicity, alongside more detailed assessments of pigmentation may improve the accuracy and relevance of both clinical evaluation and research. Combining FST with more precise and inclusive frameworks represents a pragmatic step toward better reflecting patient diversity.
- Taylor SC. Recognizing erythema in skin of color. J Am Acad Dermatol.
- Fitzpatrick TB. The validity and practicality of sun-reactive skin types I through VI. Arch Dermatol. 1988;124:869-871. doi:10.1001 /archderm.124.6.869
- Eilers S, Bach DQ, Gaber R, et al. Accuracy of self-reported Fitzpatrick skin phototype classification in US Hispanic and Latino populations. JAMA Dermatol. 2013;149:797-803. doi:10.1001 /jamadermatol.2013.4091
- Del Bino S, Bernerd F. Variations in skin colour and the biological consequences of ultraviolet radiation exposure. Br J Dermatol. 2013;169(S3):33-40. doi:10.1111/bjd.12529
- Goon P, Banfield C, Bello O, et al. Skin cancers in skin types IV–VI: does the Fitzpatrick scale give a false sense of security? Clin Exp Dermatol. 2022;47:1112-1117. doi:10.1002/ski2.40
- Fitzpatrick TB. Soleil et peau. J Med Asthet. 1975;2:33-34.
- Ware OR, Dawson JE, Shinohara MM, et al. Racial limitations of Fitzpatrick skin type. Cutis. 2020;105:77-80.
- Lester JC, Taylor SC, Chren MM. Under-representation of skin of colour in dermatology images: not just an educational issue. Br J Dermatol. 2019;180:1521-1522. doi:10.1111/bjd.17608
- Fullerton A, Fischer T, Lahti A, et al. Guidelines for measurement of skin colour and erythema. a report from the Standardization Group of the European Society of Contact Dermatitis. Contact Dermatitis. 1996;35:1-10. doi:10.1111/j.1600-0536.1996.tb02242.x
- Taylor SC. Recognizing erythema in skin of color. J Am Acad Dermatol.
- Fitzpatrick TB. The validity and practicality of sun-reactive skin types I through VI. Arch Dermatol. 1988;124:869-871. doi:10.1001 /archderm.124.6.869
- Eilers S, Bach DQ, Gaber R, et al. Accuracy of self-reported Fitzpatrick skin phototype classification in US Hispanic and Latino populations. JAMA Dermatol. 2013;149:797-803. doi:10.1001 /jamadermatol.2013.4091
- Del Bino S, Bernerd F. Variations in skin colour and the biological consequences of ultraviolet radiation exposure. Br J Dermatol. 2013;169(S3):33-40. doi:10.1111/bjd.12529
- Goon P, Banfield C, Bello O, et al. Skin cancers in skin types IV–VI: does the Fitzpatrick scale give a false sense of security? Clin Exp Dermatol. 2022;47:1112-1117. doi:10.1002/ski2.40
- Fitzpatrick TB. Soleil et peau. J Med Asthet. 1975;2:33-34.
- Ware OR, Dawson JE, Shinohara MM, et al. Racial limitations of Fitzpatrick skin type. Cutis. 2020;105:77-80.
- Lester JC, Taylor SC, Chren MM. Under-representation of skin of colour in dermatology images: not just an educational issue. Br J Dermatol. 2019;180:1521-1522. doi:10.1111/bjd.17608
- Fullerton A, Fischer T, Lahti A, et al. Guidelines for measurement of skin colour and erythema. a report from the Standardization Group of the European Society of Contact Dermatitis. Contact Dermatitis. 1996;35:1-10. doi:10.1111/j.1600-0536.1996.tb02242.x
Limitations of Fitzpatrick Skin Type as a Proxy for Skin Color and Race
Limitations of Fitzpatrick Skin Type as a Proxy for Skin Color and Race

Exophytic Papule on the Hand
Exophytic Papule on the Hand
THE DIAGNOSIS: Kaposi Sarcoma
Histopathology revealed a serum crust on the surface of the specimen, and the dermis contained compact collections of spindled cells with interspersed erythrocytes (Figure 1). Human herpesvirus 8–stained sections highlighted many lesional cell nuclei (Figure 2). A diagnosis of Kaposi sarcoma (KS) was made based on these findings. The patient expressed interest in surgical excision; however, he was lost to follow-up.
Kaposi sarcoma is an indolent, multifocal, angioproliferative tumor that predominantly affects mucocutaneous sites with less frequent involvement of visceral organs. Kaposi sarcoma is categorized into 4 subtypes: epidemic, iatrogenic, endemic, and classic. Human herpesvirus 8, primarily transmitted through saliva or sexual contact, plays a central role in the pathogenesis of KS, as it drives disease development across all subtypes. The virus causes proliferation of endothelial cells and the formation of angioproliferative lesions characteristic of KS.1
Prevalence is highest in the epidemic subtype, in which patients with advanced HIV and low CD4 T-cell counts may develop KS lesions. Although KS is associated most commonly with HIV, it also has been observed in men who have sex with men regardless of their HIV status.2 Patients undergoing immunosuppressive therapy also may not maintain immune tolerance to previously or newly acquired human herpesvirus 8, leading to the development of iatrogenic KS. This subtype particularly manifests in patients receiving therapy for autoimmune conditions or organ transplants and often only regresses if immunosuppressive therapy is withdrawn.3,4
The endemic and classic subtypes of KS may occur in patients without any known immunocompromise. Endemic KS demonstrates a predilection for pediatric populations in Africa and exhibits less pronounced sex disparity.5 In Uganda and Zimbabwe, endemic KS is the leading cancer in men and the second most frequently occurring cancer in women.6 In contrast, classic KS generally affects older men of Eastern European and Mediterranean descent or Ashkenazi Jewish ancestry. Patients with classic KS generally exhibit a less aggressive disease trajectory relative to other subtypes; however, these patients have a substantial risk for a secondary hematologic malignancy, which may already coexist at the time of diagnosis or emerge subsequently.1,7
Our patient, a native of Eastern Europe, was negative for HIV and was in a monogamous relationship with his wife; therefore, he was likely to have had the classic subtype of KS. As KS is a multifocal disease, lesions may independently emerge at different times and locations on the body. Our patient presented with a new lesion on the hand several years after excision of a similar lesion on the face. Lesions suspicious for KS include slow-growing, painless, red or violaceous patches, nodules, plaques, or patches on the extremities, most commonly manifesting on the feet and ankles. Our differential diagnosis included pyogenic granuloma, amelanotic melanoma, squamous cell carcinoma, and angiosarcoma.
The prognosis in patients with classic KS is favorable, as it often is limited to cutaneous sites and less commonly manifests on visceral organs. Nonetheless, pulmonary and gastrointestinal involvement manifesting as hemoptysis and rectal bleeding, respectively, can occur. This underscores the potential for more serious complications in instances with visceral involvement. Treatment focuses on managing symptoms and preventing growth and progression of individual lesions. Additionally, treatment strategies aim to improve cosmetic outcomes and address any underlying immunosuppression that may exacerbate the condition.8
For most patients, local therapies such as surgical excision, cryotherapy, laser therapy, or intralesional chemotherapy will remove or reduce individual lesions. Patients with widespread cutaneous or extracutaneous disease may require immunomodulatory agents such as interferon α or chemotherapeutic agents such as anthracyclines or paclitaxel.8
Our case highlights the importance of considering risk factors beyond HIV status when including KS as part of the differential diagnosis in patients with atypical vascular lesions. Early recognition enables timely evaluation of potential associated conditions and informs subsequent management decisions.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Lanternier F, Lebbé C, Schartz N, et al. Kaposi’s sarcoma in HIV-negative men having sex with men. AIDS. 2008;22:1163-1168. doi:10.1097/QAD.0b013e3283031a8a
- Penn I. Kaposi’s sarcoma in transplant recipients. Transplantation. 1997;64:669-673. doi:10.1097/00007890-199709150-00001
- Gallo Marin B, Maymone MBC, El Rayess F, et al. Kaposi sarcoma associated with tofacitinib use in a patient with rheumatoid arthritis. R I Med J (2013). 2023;106:18-20.
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. Updated June 5, 2023. Accessed May 15, 2026. https://www.ncbi.nlm .nih.gov/books/NBK534839/
- Dedicoat M, Newton R. Review of the distribution of Kaposi’s sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi’s sarcoma. Br J Cancer. 2003;88:1-3. doi:10.1038 /sj.bjc.6600745
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi sarcoma patients with comparison to HIV-related Kaposi sarcoma. Mod Pathol. 2008;21:572-582. doi:10.1038/modpathol.2008.15
- Ceccarelli M, Facciolà A, Taibi R, et al. The treatment of Kaposi’s sarcoma: present and future options, a review of the literature. Eur Rev Med Pharmacol Sci. 2019;23:7488-7497. doi:10.26355 /eurrev_201909_18860
THE DIAGNOSIS: Kaposi Sarcoma
Histopathology revealed a serum crust on the surface of the specimen, and the dermis contained compact collections of spindled cells with interspersed erythrocytes (Figure 1). Human herpesvirus 8–stained sections highlighted many lesional cell nuclei (Figure 2). A diagnosis of Kaposi sarcoma (KS) was made based on these findings. The patient expressed interest in surgical excision; however, he was lost to follow-up.
Kaposi sarcoma is an indolent, multifocal, angioproliferative tumor that predominantly affects mucocutaneous sites with less frequent involvement of visceral organs. Kaposi sarcoma is categorized into 4 subtypes: epidemic, iatrogenic, endemic, and classic. Human herpesvirus 8, primarily transmitted through saliva or sexual contact, plays a central role in the pathogenesis of KS, as it drives disease development across all subtypes. The virus causes proliferation of endothelial cells and the formation of angioproliferative lesions characteristic of KS.1
Prevalence is highest in the epidemic subtype, in which patients with advanced HIV and low CD4 T-cell counts may develop KS lesions. Although KS is associated most commonly with HIV, it also has been observed in men who have sex with men regardless of their HIV status.2 Patients undergoing immunosuppressive therapy also may not maintain immune tolerance to previously or newly acquired human herpesvirus 8, leading to the development of iatrogenic KS. This subtype particularly manifests in patients receiving therapy for autoimmune conditions or organ transplants and often only regresses if immunosuppressive therapy is withdrawn.3,4
The endemic and classic subtypes of KS may occur in patients without any known immunocompromise. Endemic KS demonstrates a predilection for pediatric populations in Africa and exhibits less pronounced sex disparity.5 In Uganda and Zimbabwe, endemic KS is the leading cancer in men and the second most frequently occurring cancer in women.6 In contrast, classic KS generally affects older men of Eastern European and Mediterranean descent or Ashkenazi Jewish ancestry. Patients with classic KS generally exhibit a less aggressive disease trajectory relative to other subtypes; however, these patients have a substantial risk for a secondary hematologic malignancy, which may already coexist at the time of diagnosis or emerge subsequently.1,7
Our patient, a native of Eastern Europe, was negative for HIV and was in a monogamous relationship with his wife; therefore, he was likely to have had the classic subtype of KS. As KS is a multifocal disease, lesions may independently emerge at different times and locations on the body. Our patient presented with a new lesion on the hand several years after excision of a similar lesion on the face. Lesions suspicious for KS include slow-growing, painless, red or violaceous patches, nodules, plaques, or patches on the extremities, most commonly manifesting on the feet and ankles. Our differential diagnosis included pyogenic granuloma, amelanotic melanoma, squamous cell carcinoma, and angiosarcoma.
The prognosis in patients with classic KS is favorable, as it often is limited to cutaneous sites and less commonly manifests on visceral organs. Nonetheless, pulmonary and gastrointestinal involvement manifesting as hemoptysis and rectal bleeding, respectively, can occur. This underscores the potential for more serious complications in instances with visceral involvement. Treatment focuses on managing symptoms and preventing growth and progression of individual lesions. Additionally, treatment strategies aim to improve cosmetic outcomes and address any underlying immunosuppression that may exacerbate the condition.8
For most patients, local therapies such as surgical excision, cryotherapy, laser therapy, or intralesional chemotherapy will remove or reduce individual lesions. Patients with widespread cutaneous or extracutaneous disease may require immunomodulatory agents such as interferon α or chemotherapeutic agents such as anthracyclines or paclitaxel.8
Our case highlights the importance of considering risk factors beyond HIV status when including KS as part of the differential diagnosis in patients with atypical vascular lesions. Early recognition enables timely evaluation of potential associated conditions and informs subsequent management decisions.
THE DIAGNOSIS: Kaposi Sarcoma
Histopathology revealed a serum crust on the surface of the specimen, and the dermis contained compact collections of spindled cells with interspersed erythrocytes (Figure 1). Human herpesvirus 8–stained sections highlighted many lesional cell nuclei (Figure 2). A diagnosis of Kaposi sarcoma (KS) was made based on these findings. The patient expressed interest in surgical excision; however, he was lost to follow-up.
Kaposi sarcoma is an indolent, multifocal, angioproliferative tumor that predominantly affects mucocutaneous sites with less frequent involvement of visceral organs. Kaposi sarcoma is categorized into 4 subtypes: epidemic, iatrogenic, endemic, and classic. Human herpesvirus 8, primarily transmitted through saliva or sexual contact, plays a central role in the pathogenesis of KS, as it drives disease development across all subtypes. The virus causes proliferation of endothelial cells and the formation of angioproliferative lesions characteristic of KS.1
Prevalence is highest in the epidemic subtype, in which patients with advanced HIV and low CD4 T-cell counts may develop KS lesions. Although KS is associated most commonly with HIV, it also has been observed in men who have sex with men regardless of their HIV status.2 Patients undergoing immunosuppressive therapy also may not maintain immune tolerance to previously or newly acquired human herpesvirus 8, leading to the development of iatrogenic KS. This subtype particularly manifests in patients receiving therapy for autoimmune conditions or organ transplants and often only regresses if immunosuppressive therapy is withdrawn.3,4
The endemic and classic subtypes of KS may occur in patients without any known immunocompromise. Endemic KS demonstrates a predilection for pediatric populations in Africa and exhibits less pronounced sex disparity.5 In Uganda and Zimbabwe, endemic KS is the leading cancer in men and the second most frequently occurring cancer in women.6 In contrast, classic KS generally affects older men of Eastern European and Mediterranean descent or Ashkenazi Jewish ancestry. Patients with classic KS generally exhibit a less aggressive disease trajectory relative to other subtypes; however, these patients have a substantial risk for a secondary hematologic malignancy, which may already coexist at the time of diagnosis or emerge subsequently.1,7
Our patient, a native of Eastern Europe, was negative for HIV and was in a monogamous relationship with his wife; therefore, he was likely to have had the classic subtype of KS. As KS is a multifocal disease, lesions may independently emerge at different times and locations on the body. Our patient presented with a new lesion on the hand several years after excision of a similar lesion on the face. Lesions suspicious for KS include slow-growing, painless, red or violaceous patches, nodules, plaques, or patches on the extremities, most commonly manifesting on the feet and ankles. Our differential diagnosis included pyogenic granuloma, amelanotic melanoma, squamous cell carcinoma, and angiosarcoma.
The prognosis in patients with classic KS is favorable, as it often is limited to cutaneous sites and less commonly manifests on visceral organs. Nonetheless, pulmonary and gastrointestinal involvement manifesting as hemoptysis and rectal bleeding, respectively, can occur. This underscores the potential for more serious complications in instances with visceral involvement. Treatment focuses on managing symptoms and preventing growth and progression of individual lesions. Additionally, treatment strategies aim to improve cosmetic outcomes and address any underlying immunosuppression that may exacerbate the condition.8
For most patients, local therapies such as surgical excision, cryotherapy, laser therapy, or intralesional chemotherapy will remove or reduce individual lesions. Patients with widespread cutaneous or extracutaneous disease may require immunomodulatory agents such as interferon α or chemotherapeutic agents such as anthracyclines or paclitaxel.8
Our case highlights the importance of considering risk factors beyond HIV status when including KS as part of the differential diagnosis in patients with atypical vascular lesions. Early recognition enables timely evaluation of potential associated conditions and informs subsequent management decisions.
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Lanternier F, Lebbé C, Schartz N, et al. Kaposi’s sarcoma in HIV-negative men having sex with men. AIDS. 2008;22:1163-1168. doi:10.1097/QAD.0b013e3283031a8a
- Penn I. Kaposi’s sarcoma in transplant recipients. Transplantation. 1997;64:669-673. doi:10.1097/00007890-199709150-00001
- Gallo Marin B, Maymone MBC, El Rayess F, et al. Kaposi sarcoma associated with tofacitinib use in a patient with rheumatoid arthritis. R I Med J (2013). 2023;106:18-20.
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. Updated June 5, 2023. Accessed May 15, 2026. https://www.ncbi.nlm .nih.gov/books/NBK534839/
- Dedicoat M, Newton R. Review of the distribution of Kaposi’s sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi’s sarcoma. Br J Cancer. 2003;88:1-3. doi:10.1038 /sj.bjc.6600745
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi sarcoma patients with comparison to HIV-related Kaposi sarcoma. Mod Pathol. 2008;21:572-582. doi:10.1038/modpathol.2008.15
- Ceccarelli M, Facciolà A, Taibi R, et al. The treatment of Kaposi’s sarcoma: present and future options, a review of the literature. Eur Rev Med Pharmacol Sci. 2019;23:7488-7497. doi:10.26355 /eurrev_201909_18860
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Lanternier F, Lebbé C, Schartz N, et al. Kaposi’s sarcoma in HIV-negative men having sex with men. AIDS. 2008;22:1163-1168. doi:10.1097/QAD.0b013e3283031a8a
- Penn I. Kaposi’s sarcoma in transplant recipients. Transplantation. 1997;64:669-673. doi:10.1097/00007890-199709150-00001
- Gallo Marin B, Maymone MBC, El Rayess F, et al. Kaposi sarcoma associated with tofacitinib use in a patient with rheumatoid arthritis. R I Med J (2013). 2023;106:18-20.
- Bishop BN, Lynch DT. Kaposi sarcoma. StatPearls [Internet]. Updated June 5, 2023. Accessed May 15, 2026. https://www.ncbi.nlm .nih.gov/books/NBK534839/
- Dedicoat M, Newton R. Review of the distribution of Kaposi’s sarcoma-associated herpesvirus (KSHV) in Africa in relation to the incidence of Kaposi’s sarcoma. Br J Cancer. 2003;88:1-3. doi:10.1038 /sj.bjc.6600745
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV-related Kaposi sarcoma patients with comparison to HIV-related Kaposi sarcoma. Mod Pathol. 2008;21:572-582. doi:10.1038/modpathol.2008.15
- Ceccarelli M, Facciolà A, Taibi R, et al. The treatment of Kaposi’s sarcoma: present and future options, a review of the literature. Eur Rev Med Pharmacol Sci. 2019;23:7488-7497. doi:10.26355 /eurrev_201909_18860
Exophytic Papule on the Hand
Exophytic Papule on the Hand
A man in his 70s with a history of hypertension was admitted to the hospital for symptomatic bradycardia. On the day of admission, he reported a growth on the left second digit of 1 month’s duration, for which dermatology was consulted. The patient said the growth was asymptomatic but occasionally would get caught on objects. He denied any recent fevers, weight loss, or fatigue. He also denied any trauma to the area or other inciting factors. The patient reported there were no lesions anywhere else on the body, but he did mention a similar mass had been excised from his face several years prior. He noted that he had immigrated to the United States from Eastern Europe within the past several years.
Results of laboratory testing at the current presentation, including a basic metabolic panel, complete blood count with differential, hepatic function panel, thyroid-stimulating hormone level, and HIV antigen/antibody testing, were unremarkable. Physical examination revealed a single, well-circumscribed, 6×6–mm, round, red, exophytic papule with a collarette of scale on the volar surface of the left second digit. The skin on both arms was otherwise unremarkable. There was no evidence of lymphadenopathy or mucosal involvement. A shave biopsy of the lesion was performed.
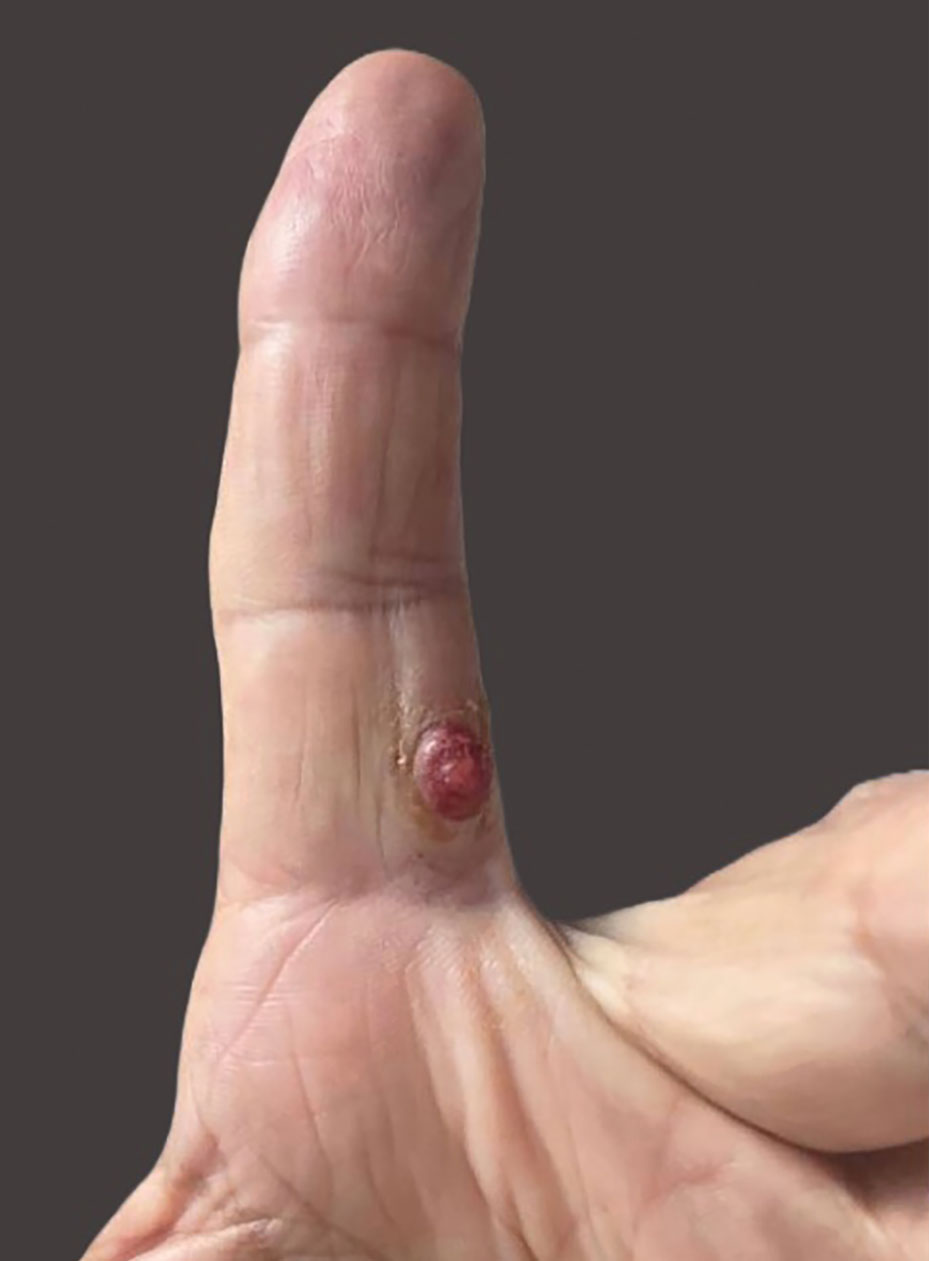

The Effect of GLP-1 Receptor Agonists on Hidradenitis Suppurativa: A Comprehensive Systematic Review
The Effect of GLP-1 Receptor Agonists on Hidradenitis Suppurativa: A Comprehensive Systematic Review
Hidradenitis suppurativa (HS) is a chronic relapsing inflammatory skin disorder affecting apocrine gland–bearing areas such as the axillae, inguinal regions, and anogenital area.1 It manifests with painful nodules, abscesses, sinus tract formation, and scarring.2 The disease strongly impacts patients’ quality of life due to pain, malodor, and psychosocial burden.3
The exact etiology of HS is multifactorial, involving genetic predisposition, mechanical stress, hormonal influences, dysbiosis, and immune dysregulation.4 Obesity and metabolic syndrome are highly prevalent among patients with HS and are considered exacerbating factors.5 Adipose tissue contributes to systemic inflammation through the secretion of proinflammatory cytokines such as tumor necrosis factor (TNF) α and interleukins (ILs).6
Management of HS includes lifestyle modifications, medical therapy, and surgical interventions. Medical treatments encompass antibiotics, retinoids, hormonal therapy, immunosuppressants, and immunomodulators such as anti-TNF and anti–IL-17 agents.7 Despite available therapies, many patients have suboptimal responses or experience adverse effects and dramatic reductions in their quality of life.3
Glucagonlike peptide 1 receptor agonists (GLP-1 RAs) are incretin-based therapies used in type 2 diabetes and obesity management.8 They enhance insulin secretion, suppress glucagon release, delay gastric emptying, and promote satiety.9 Beyond glycemic control, GLP-1 RAs exhibit anti-inflammatory properties and cardiovascular benefits.10
Given the high prevalence of obesity and metabolic syndrome in patients with HS as well as the anti-inflammatory effects of GLP-1 RAs, these agents may offer therapeutic benefits in HS.11 We conducted a systematic review to evaluate the existing evidence on the efficacy and safety of GLP-1 RAs in the treatment of HS.
Methods
A systematic review was conducted via a PubMed search of articles indexed for MEDLINE in October 2024, following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines12 using the terms hidradenitis suppurativa OR acne inversa AND GLP-1 receptor agonist OR glucagon-like peptide-1 receptor agonist OR liraglutide OR semaglutide OR exenatide OR dulaglutide. No filters were applied to limit the search by language or publication date.
Inclusion criteria were clinical trials, observational studies (cohort, case control, cross-sectional), and case reports/series involving patients diagnosed with HS treated with GLP-1 RAs. Outcomes of interest included clinical improvement in HS severity (eg, lesion count, pain assessment, HS-specific scores), safety, and adverse events. Exclusion criteria included animal studies or in vitro experiments, reviews, editorials, and opinion pieces without original patient data; studies not in English; and studies not reporting clinical outcomes related to HS.
Two independent reviewers (N.R.K. and S.K.C.) screened the titles and abstracts for relevance. Full-text articles of potentially eligible studies were retrieved for detailed evaluation. Data extracted included study design, patient demographics, intervention details, outcomes, and adverse events. Discrepancies were resolved through discussion.
Results
The initial search yielded 11 articles (Figure). After screening titles and abstracts, 9 articles were selected for full-text review. Of these, 3 articles met the inclusion criteria. These studies included 3 case reports. Interventions involved liraglutide (2 reports)13,14 and semaglutide15 (1 report)(Table). The patient population consisted of adult patients with HS with comorbid diabetes, obesity, and/or metabolic syndrome.
Jennings et al13 reported a 31-year-old obese woman with a history of smoking and Hurley stage 2 HS, a Hidradenitis Suppurativa Physician’s Global Assessment score of 4, a Dermatology Life Quality Index score of 24, and a body mass index of 45.3. She was treated with liraglutide monotherapy, starting with 0.6 mg subcutaneously once daily then titrating weekly to 1.8 mg subcutaneously. After 4 weeks, outcomes showed a reduction in Hidradenitis Suppurativa Physician’s Global Assessment (score=1) and Dermatology Life Quality Index (score=14) scores, and the patient lost 4.5 kg from baseline. The patient’s Hurley stage decreased from 2 to 1. After another 4 weeks, the patient’s weight decreased by a further 2 kg and HS remained controlled. No adverse events were recorded.
Khandalavala14 reported a single case of a 19-year-old woman with severe HS, obesity, and metabolic syndrome of 8 years’ duration treated with liraglutide. The patient had a weight of 215 lb with a body mass index of 37. With a combination of metformin 2000 mg/d, liraglutide 0.6 mg/d subcutaneously increased to 1.8 mg/d over 2 months, levonorgestrel-ethinyl estradiol (no dosage listed), dapsone 100 mg/d, and finasteride 5 mg/d, there was a marked reduction in nodules and abscesses after 6 months, with a weight loss of 40 lb (19% body weight). No adverse events were reported.
Mainville et al15 described a 59-year-old woman with refractory HS who showed improvement with a combination of intravenous ertapenem 1 g/d for 6 weeks, minocycline 100 mg/d for 3 months, metformin 500 mg three times daily for 2 months, doxycycline 100 mg/d to bridge to adalimumab (160 mg subcutaneously starting dose then 80 mg subcutaneously), and semaglutide (no dosage listed). After semaglutide was introduced, the patient lost 10 kg. The only adverse event was diarrhea.
Comment
The limited but growing body of evidence suggests that GLP-1 RAs may be beneficial in managing HS, particularly in patients with comorbid obesity. Treatment with liraglutide or semaglutide was associated with marked improvements in clinical severity scores, lesion counts, pain reduction, and quality of life.
As adjunct therapy, GLP-1 RAs could serve alongside standard HS treatments such as antibiotics and biologics. Addressing obesity, a known risk factor and disease modifier in HS, may lead to better disease control. The therapeutic benefits of GLP-1 RAs in HS are attributed to weight loss, which reduces adipose tissue and systemic inflammation.16 The anti-inflammatory effects of GLP-1 RAs involve the reduction of proinflammatory cytokines such as IL-6 and TNF-α.17 Metabolic improvements, including enhanced insulin sensitivity and lipid profile, also may contribute to disease modulation.17
Limitations—Because our analysis was limited to 3 case reports, the strength of the evidence is limited. These case reports also lack the standardized use of the Hidradenitis Suppurativa Clinical Response scoring system that generally is found in randomized controlled trials (RCTs). The lack of RCTs precludes definitive conclusions about efficacy. Future directions include the need for well-designed RCTs with large sample sizes to confirm findings, assessment of long-term safety and tolerability in patients with HS, and further research into the molecular mechanisms by which GLP-1 RAs affect HS pathophysiology. Of note, it is imperative to be aware of the medication shortage for all GLP-1 RAs when prescribing these medications for patients with HS.
Conclusion
Glucagonlike peptide 1 RAs show promise as a therapeutic option for HS, especially in patients with obesity and metabolic disturbances. The observed benefits likely result from weight loss and anti-inflammatory effects. Other drugs targeting glucose-dependent insulinotropic polypeptide and glucagon also are being studied thoroughly as options for managing HS. Although preliminary results are encouraging, robust clinical trials are needed to establish efficacy, optimal dosing, and safety in this patient population.
- Vinkel C, Thomsen SF. Hidradenitis suppurativa: causes, features, and current treatments. J Clin Aesthet Dermatol. 2018;11:17-23.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi:10.2147/CCID.S111019
- Chernyshov PV, Finlay AY, Tomas-Aragones L, et al. Quality of life in hidradenitis suppurativa: an update. Int J Environ Res Public Health. 2021;18:6131. doi:10.3390/ijerph18116131
- Seyed Jafari SM, Hunger RE, Schlapbach C. Hidradenitis suppurativa: current understanding of pathogenic mechanisms and suggestion for treatment algorithm. Front Med (Lausanne). 2020;7:68. doi:10.3389/fmed.2020.00068
- Alotaibi HM. Incidence, risk factors, and prognosis of hidradenitis suppurativa across the globe: insights from the literature. Clin Cosmet Investig Dermatol. 2023;16:545-552. doi:10.2147/CCID.S402453
- Vossen ARJV, van der Zee HH, Prens EP. Hidradenitis suppurativa: a systematic review integrating inflammatory pathways into a cohesive pathogenic model. Front Immunol. 2018;9:2965. doi:10.3389/fimmu.2018.02965
- Orenstein LAV, Nguyen TV, Damiani G, et al. Medical and surgical management of hidradenitis suppurativa: a review of international treatment guidelines and implementation in general dermatology practice. Dermatology. 2020;236:393-412. doi:10.1159/000507323
- Brown E, Cuthbertson DJ, Wilding JP. Newer GLP-1 receptor agonists and obesity-diabetes. Peptides. 2018;100:61-67. doi:10.1016/j.peptides.2017.12.009
- Cornell S. A review of GLP‐1 receptor agonists in type 2 diabetes: a focus on the mechanism of action of once‐weekly agents. J Clin Pharm Ther. 2020;45(suppl 1):17-27. doi:10.1111/jcpt.13230
- Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators Inflamm. 2016;2016:3094642. doi:10.1155/2016/3094642
- Mintoff D, Benhadou F, Pace NP, et al. Metabolic syndrome and hidradenitis suppurativa: epidemiological, molecular, and therapeutic aspects. Int J Dermatol. 2022;61:1175-1186. doi:10.1111/ijd.15910
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71
- Jennings L, Nestor L, Molloy O, et al. The treatment of hidradenitis suppurativa with the glucagon-like peptide-1 agonist liraglutide. Br J Dermatol. 2017;177:858-859. doi:10.1111/bjd.15233
- Khandalavala BN. A disease-modifying approach for advanced hidradenitis suppurativa (regimen with metformin, liraglutide, dapsone, and finasteride): a case report. Case Rep Dermatol. 2017;9:70-78. doi:10.1159/000473873
- Mainville L, MacHaalany J, Veillette H. Hidradenitis suppurativa patient requiring cardiac procedure with inguinal access: case management with ertapenem. SAGE Open Med Case Rep. 2024;12:2050313X241274819. doi:10.1177/2050313X241274819
- Hamed K, Alosaimi MN, Ali BA, et al. Glucagon-like peptide-1 (GLP-1) receptor agonists: exploring their impact on diabetes, obesity, and cardiovascular health through a comprehensive literature review. Cureus. 2024;16:E68390. doi:10.7759/cureus.68390
- Alharbi SH. Anti-inflammatory role of glucagon-like peptide 1 receptor agonists and its clinical implications. Ther Adv Endocrinol Metab. 2024;15:20420188231222367. doi:10.1177/20420188231222367
Hidradenitis suppurativa (HS) is a chronic relapsing inflammatory skin disorder affecting apocrine gland–bearing areas such as the axillae, inguinal regions, and anogenital area.1 It manifests with painful nodules, abscesses, sinus tract formation, and scarring.2 The disease strongly impacts patients’ quality of life due to pain, malodor, and psychosocial burden.3
The exact etiology of HS is multifactorial, involving genetic predisposition, mechanical stress, hormonal influences, dysbiosis, and immune dysregulation.4 Obesity and metabolic syndrome are highly prevalent among patients with HS and are considered exacerbating factors.5 Adipose tissue contributes to systemic inflammation through the secretion of proinflammatory cytokines such as tumor necrosis factor (TNF) α and interleukins (ILs).6
Management of HS includes lifestyle modifications, medical therapy, and surgical interventions. Medical treatments encompass antibiotics, retinoids, hormonal therapy, immunosuppressants, and immunomodulators such as anti-TNF and anti–IL-17 agents.7 Despite available therapies, many patients have suboptimal responses or experience adverse effects and dramatic reductions in their quality of life.3
Glucagonlike peptide 1 receptor agonists (GLP-1 RAs) are incretin-based therapies used in type 2 diabetes and obesity management.8 They enhance insulin secretion, suppress glucagon release, delay gastric emptying, and promote satiety.9 Beyond glycemic control, GLP-1 RAs exhibit anti-inflammatory properties and cardiovascular benefits.10
Given the high prevalence of obesity and metabolic syndrome in patients with HS as well as the anti-inflammatory effects of GLP-1 RAs, these agents may offer therapeutic benefits in HS.11 We conducted a systematic review to evaluate the existing evidence on the efficacy and safety of GLP-1 RAs in the treatment of HS.
Methods
A systematic review was conducted via a PubMed search of articles indexed for MEDLINE in October 2024, following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines12 using the terms hidradenitis suppurativa OR acne inversa AND GLP-1 receptor agonist OR glucagon-like peptide-1 receptor agonist OR liraglutide OR semaglutide OR exenatide OR dulaglutide. No filters were applied to limit the search by language or publication date.
Inclusion criteria were clinical trials, observational studies (cohort, case control, cross-sectional), and case reports/series involving patients diagnosed with HS treated with GLP-1 RAs. Outcomes of interest included clinical improvement in HS severity (eg, lesion count, pain assessment, HS-specific scores), safety, and adverse events. Exclusion criteria included animal studies or in vitro experiments, reviews, editorials, and opinion pieces without original patient data; studies not in English; and studies not reporting clinical outcomes related to HS.
Two independent reviewers (N.R.K. and S.K.C.) screened the titles and abstracts for relevance. Full-text articles of potentially eligible studies were retrieved for detailed evaluation. Data extracted included study design, patient demographics, intervention details, outcomes, and adverse events. Discrepancies were resolved through discussion.
Results
The initial search yielded 11 articles (Figure). After screening titles and abstracts, 9 articles were selected for full-text review. Of these, 3 articles met the inclusion criteria. These studies included 3 case reports. Interventions involved liraglutide (2 reports)13,14 and semaglutide15 (1 report)(Table). The patient population consisted of adult patients with HS with comorbid diabetes, obesity, and/or metabolic syndrome.
Jennings et al13 reported a 31-year-old obese woman with a history of smoking and Hurley stage 2 HS, a Hidradenitis Suppurativa Physician’s Global Assessment score of 4, a Dermatology Life Quality Index score of 24, and a body mass index of 45.3. She was treated with liraglutide monotherapy, starting with 0.6 mg subcutaneously once daily then titrating weekly to 1.8 mg subcutaneously. After 4 weeks, outcomes showed a reduction in Hidradenitis Suppurativa Physician’s Global Assessment (score=1) and Dermatology Life Quality Index (score=14) scores, and the patient lost 4.5 kg from baseline. The patient’s Hurley stage decreased from 2 to 1. After another 4 weeks, the patient’s weight decreased by a further 2 kg and HS remained controlled. No adverse events were recorded.
Khandalavala14 reported a single case of a 19-year-old woman with severe HS, obesity, and metabolic syndrome of 8 years’ duration treated with liraglutide. The patient had a weight of 215 lb with a body mass index of 37. With a combination of metformin 2000 mg/d, liraglutide 0.6 mg/d subcutaneously increased to 1.8 mg/d over 2 months, levonorgestrel-ethinyl estradiol (no dosage listed), dapsone 100 mg/d, and finasteride 5 mg/d, there was a marked reduction in nodules and abscesses after 6 months, with a weight loss of 40 lb (19% body weight). No adverse events were reported.
Mainville et al15 described a 59-year-old woman with refractory HS who showed improvement with a combination of intravenous ertapenem 1 g/d for 6 weeks, minocycline 100 mg/d for 3 months, metformin 500 mg three times daily for 2 months, doxycycline 100 mg/d to bridge to adalimumab (160 mg subcutaneously starting dose then 80 mg subcutaneously), and semaglutide (no dosage listed). After semaglutide was introduced, the patient lost 10 kg. The only adverse event was diarrhea.
Comment
The limited but growing body of evidence suggests that GLP-1 RAs may be beneficial in managing HS, particularly in patients with comorbid obesity. Treatment with liraglutide or semaglutide was associated with marked improvements in clinical severity scores, lesion counts, pain reduction, and quality of life.
As adjunct therapy, GLP-1 RAs could serve alongside standard HS treatments such as antibiotics and biologics. Addressing obesity, a known risk factor and disease modifier in HS, may lead to better disease control. The therapeutic benefits of GLP-1 RAs in HS are attributed to weight loss, which reduces adipose tissue and systemic inflammation.16 The anti-inflammatory effects of GLP-1 RAs involve the reduction of proinflammatory cytokines such as IL-6 and TNF-α.17 Metabolic improvements, including enhanced insulin sensitivity and lipid profile, also may contribute to disease modulation.17
Limitations—Because our analysis was limited to 3 case reports, the strength of the evidence is limited. These case reports also lack the standardized use of the Hidradenitis Suppurativa Clinical Response scoring system that generally is found in randomized controlled trials (RCTs). The lack of RCTs precludes definitive conclusions about efficacy. Future directions include the need for well-designed RCTs with large sample sizes to confirm findings, assessment of long-term safety and tolerability in patients with HS, and further research into the molecular mechanisms by which GLP-1 RAs affect HS pathophysiology. Of note, it is imperative to be aware of the medication shortage for all GLP-1 RAs when prescribing these medications for patients with HS.
Conclusion
Glucagonlike peptide 1 RAs show promise as a therapeutic option for HS, especially in patients with obesity and metabolic disturbances. The observed benefits likely result from weight loss and anti-inflammatory effects. Other drugs targeting glucose-dependent insulinotropic polypeptide and glucagon also are being studied thoroughly as options for managing HS. Although preliminary results are encouraging, robust clinical trials are needed to establish efficacy, optimal dosing, and safety in this patient population.
Hidradenitis suppurativa (HS) is a chronic relapsing inflammatory skin disorder affecting apocrine gland–bearing areas such as the axillae, inguinal regions, and anogenital area.1 It manifests with painful nodules, abscesses, sinus tract formation, and scarring.2 The disease strongly impacts patients’ quality of life due to pain, malodor, and psychosocial burden.3
The exact etiology of HS is multifactorial, involving genetic predisposition, mechanical stress, hormonal influences, dysbiosis, and immune dysregulation.4 Obesity and metabolic syndrome are highly prevalent among patients with HS and are considered exacerbating factors.5 Adipose tissue contributes to systemic inflammation through the secretion of proinflammatory cytokines such as tumor necrosis factor (TNF) α and interleukins (ILs).6
Management of HS includes lifestyle modifications, medical therapy, and surgical interventions. Medical treatments encompass antibiotics, retinoids, hormonal therapy, immunosuppressants, and immunomodulators such as anti-TNF and anti–IL-17 agents.7 Despite available therapies, many patients have suboptimal responses or experience adverse effects and dramatic reductions in their quality of life.3
Glucagonlike peptide 1 receptor agonists (GLP-1 RAs) are incretin-based therapies used in type 2 diabetes and obesity management.8 They enhance insulin secretion, suppress glucagon release, delay gastric emptying, and promote satiety.9 Beyond glycemic control, GLP-1 RAs exhibit anti-inflammatory properties and cardiovascular benefits.10
Given the high prevalence of obesity and metabolic syndrome in patients with HS as well as the anti-inflammatory effects of GLP-1 RAs, these agents may offer therapeutic benefits in HS.11 We conducted a systematic review to evaluate the existing evidence on the efficacy and safety of GLP-1 RAs in the treatment of HS.
Methods
A systematic review was conducted via a PubMed search of articles indexed for MEDLINE in October 2024, following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines12 using the terms hidradenitis suppurativa OR acne inversa AND GLP-1 receptor agonist OR glucagon-like peptide-1 receptor agonist OR liraglutide OR semaglutide OR exenatide OR dulaglutide. No filters were applied to limit the search by language or publication date.
Inclusion criteria were clinical trials, observational studies (cohort, case control, cross-sectional), and case reports/series involving patients diagnosed with HS treated with GLP-1 RAs. Outcomes of interest included clinical improvement in HS severity (eg, lesion count, pain assessment, HS-specific scores), safety, and adverse events. Exclusion criteria included animal studies or in vitro experiments, reviews, editorials, and opinion pieces without original patient data; studies not in English; and studies not reporting clinical outcomes related to HS.
Two independent reviewers (N.R.K. and S.K.C.) screened the titles and abstracts for relevance. Full-text articles of potentially eligible studies were retrieved for detailed evaluation. Data extracted included study design, patient demographics, intervention details, outcomes, and adverse events. Discrepancies were resolved through discussion.
Results
The initial search yielded 11 articles (Figure). After screening titles and abstracts, 9 articles were selected for full-text review. Of these, 3 articles met the inclusion criteria. These studies included 3 case reports. Interventions involved liraglutide (2 reports)13,14 and semaglutide15 (1 report)(Table). The patient population consisted of adult patients with HS with comorbid diabetes, obesity, and/or metabolic syndrome.
Jennings et al13 reported a 31-year-old obese woman with a history of smoking and Hurley stage 2 HS, a Hidradenitis Suppurativa Physician’s Global Assessment score of 4, a Dermatology Life Quality Index score of 24, and a body mass index of 45.3. She was treated with liraglutide monotherapy, starting with 0.6 mg subcutaneously once daily then titrating weekly to 1.8 mg subcutaneously. After 4 weeks, outcomes showed a reduction in Hidradenitis Suppurativa Physician’s Global Assessment (score=1) and Dermatology Life Quality Index (score=14) scores, and the patient lost 4.5 kg from baseline. The patient’s Hurley stage decreased from 2 to 1. After another 4 weeks, the patient’s weight decreased by a further 2 kg and HS remained controlled. No adverse events were recorded.
Khandalavala14 reported a single case of a 19-year-old woman with severe HS, obesity, and metabolic syndrome of 8 years’ duration treated with liraglutide. The patient had a weight of 215 lb with a body mass index of 37. With a combination of metformin 2000 mg/d, liraglutide 0.6 mg/d subcutaneously increased to 1.8 mg/d over 2 months, levonorgestrel-ethinyl estradiol (no dosage listed), dapsone 100 mg/d, and finasteride 5 mg/d, there was a marked reduction in nodules and abscesses after 6 months, with a weight loss of 40 lb (19% body weight). No adverse events were reported.
Mainville et al15 described a 59-year-old woman with refractory HS who showed improvement with a combination of intravenous ertapenem 1 g/d for 6 weeks, minocycline 100 mg/d for 3 months, metformin 500 mg three times daily for 2 months, doxycycline 100 mg/d to bridge to adalimumab (160 mg subcutaneously starting dose then 80 mg subcutaneously), and semaglutide (no dosage listed). After semaglutide was introduced, the patient lost 10 kg. The only adverse event was diarrhea.
Comment
The limited but growing body of evidence suggests that GLP-1 RAs may be beneficial in managing HS, particularly in patients with comorbid obesity. Treatment with liraglutide or semaglutide was associated with marked improvements in clinical severity scores, lesion counts, pain reduction, and quality of life.
As adjunct therapy, GLP-1 RAs could serve alongside standard HS treatments such as antibiotics and biologics. Addressing obesity, a known risk factor and disease modifier in HS, may lead to better disease control. The therapeutic benefits of GLP-1 RAs in HS are attributed to weight loss, which reduces adipose tissue and systemic inflammation.16 The anti-inflammatory effects of GLP-1 RAs involve the reduction of proinflammatory cytokines such as IL-6 and TNF-α.17 Metabolic improvements, including enhanced insulin sensitivity and lipid profile, also may contribute to disease modulation.17
Limitations—Because our analysis was limited to 3 case reports, the strength of the evidence is limited. These case reports also lack the standardized use of the Hidradenitis Suppurativa Clinical Response scoring system that generally is found in randomized controlled trials (RCTs). The lack of RCTs precludes definitive conclusions about efficacy. Future directions include the need for well-designed RCTs with large sample sizes to confirm findings, assessment of long-term safety and tolerability in patients with HS, and further research into the molecular mechanisms by which GLP-1 RAs affect HS pathophysiology. Of note, it is imperative to be aware of the medication shortage for all GLP-1 RAs when prescribing these medications for patients with HS.
Conclusion
Glucagonlike peptide 1 RAs show promise as a therapeutic option for HS, especially in patients with obesity and metabolic disturbances. The observed benefits likely result from weight loss and anti-inflammatory effects. Other drugs targeting glucose-dependent insulinotropic polypeptide and glucagon also are being studied thoroughly as options for managing HS. Although preliminary results are encouraging, robust clinical trials are needed to establish efficacy, optimal dosing, and safety in this patient population.
- Vinkel C, Thomsen SF. Hidradenitis suppurativa: causes, features, and current treatments. J Clin Aesthet Dermatol. 2018;11:17-23.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi:10.2147/CCID.S111019
- Chernyshov PV, Finlay AY, Tomas-Aragones L, et al. Quality of life in hidradenitis suppurativa: an update. Int J Environ Res Public Health. 2021;18:6131. doi:10.3390/ijerph18116131
- Seyed Jafari SM, Hunger RE, Schlapbach C. Hidradenitis suppurativa: current understanding of pathogenic mechanisms and suggestion for treatment algorithm. Front Med (Lausanne). 2020;7:68. doi:10.3389/fmed.2020.00068
- Alotaibi HM. Incidence, risk factors, and prognosis of hidradenitis suppurativa across the globe: insights from the literature. Clin Cosmet Investig Dermatol. 2023;16:545-552. doi:10.2147/CCID.S402453
- Vossen ARJV, van der Zee HH, Prens EP. Hidradenitis suppurativa: a systematic review integrating inflammatory pathways into a cohesive pathogenic model. Front Immunol. 2018;9:2965. doi:10.3389/fimmu.2018.02965
- Orenstein LAV, Nguyen TV, Damiani G, et al. Medical and surgical management of hidradenitis suppurativa: a review of international treatment guidelines and implementation in general dermatology practice. Dermatology. 2020;236:393-412. doi:10.1159/000507323
- Brown E, Cuthbertson DJ, Wilding JP. Newer GLP-1 receptor agonists and obesity-diabetes. Peptides. 2018;100:61-67. doi:10.1016/j.peptides.2017.12.009
- Cornell S. A review of GLP‐1 receptor agonists in type 2 diabetes: a focus on the mechanism of action of once‐weekly agents. J Clin Pharm Ther. 2020;45(suppl 1):17-27. doi:10.1111/jcpt.13230
- Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators Inflamm. 2016;2016:3094642. doi:10.1155/2016/3094642
- Mintoff D, Benhadou F, Pace NP, et al. Metabolic syndrome and hidradenitis suppurativa: epidemiological, molecular, and therapeutic aspects. Int J Dermatol. 2022;61:1175-1186. doi:10.1111/ijd.15910
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71
- Jennings L, Nestor L, Molloy O, et al. The treatment of hidradenitis suppurativa with the glucagon-like peptide-1 agonist liraglutide. Br J Dermatol. 2017;177:858-859. doi:10.1111/bjd.15233
- Khandalavala BN. A disease-modifying approach for advanced hidradenitis suppurativa (regimen with metformin, liraglutide, dapsone, and finasteride): a case report. Case Rep Dermatol. 2017;9:70-78. doi:10.1159/000473873
- Mainville L, MacHaalany J, Veillette H. Hidradenitis suppurativa patient requiring cardiac procedure with inguinal access: case management with ertapenem. SAGE Open Med Case Rep. 2024;12:2050313X241274819. doi:10.1177/2050313X241274819
- Hamed K, Alosaimi MN, Ali BA, et al. Glucagon-like peptide-1 (GLP-1) receptor agonists: exploring their impact on diabetes, obesity, and cardiovascular health through a comprehensive literature review. Cureus. 2024;16:E68390. doi:10.7759/cureus.68390
- Alharbi SH. Anti-inflammatory role of glucagon-like peptide 1 receptor agonists and its clinical implications. Ther Adv Endocrinol Metab. 2024;15:20420188231222367. doi:10.1177/20420188231222367
- Vinkel C, Thomsen SF. Hidradenitis suppurativa: causes, features, and current treatments. J Clin Aesthet Dermatol. 2018;11:17-23.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi:10.2147/CCID.S111019
- Chernyshov PV, Finlay AY, Tomas-Aragones L, et al. Quality of life in hidradenitis suppurativa: an update. Int J Environ Res Public Health. 2021;18:6131. doi:10.3390/ijerph18116131
- Seyed Jafari SM, Hunger RE, Schlapbach C. Hidradenitis suppurativa: current understanding of pathogenic mechanisms and suggestion for treatment algorithm. Front Med (Lausanne). 2020;7:68. doi:10.3389/fmed.2020.00068
- Alotaibi HM. Incidence, risk factors, and prognosis of hidradenitis suppurativa across the globe: insights from the literature. Clin Cosmet Investig Dermatol. 2023;16:545-552. doi:10.2147/CCID.S402453
- Vossen ARJV, van der Zee HH, Prens EP. Hidradenitis suppurativa: a systematic review integrating inflammatory pathways into a cohesive pathogenic model. Front Immunol. 2018;9:2965. doi:10.3389/fimmu.2018.02965
- Orenstein LAV, Nguyen TV, Damiani G, et al. Medical and surgical management of hidradenitis suppurativa: a review of international treatment guidelines and implementation in general dermatology practice. Dermatology. 2020;236:393-412. doi:10.1159/000507323
- Brown E, Cuthbertson DJ, Wilding JP. Newer GLP-1 receptor agonists and obesity-diabetes. Peptides. 2018;100:61-67. doi:10.1016/j.peptides.2017.12.009
- Cornell S. A review of GLP‐1 receptor agonists in type 2 diabetes: a focus on the mechanism of action of once‐weekly agents. J Clin Pharm Ther. 2020;45(suppl 1):17-27. doi:10.1111/jcpt.13230
- Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators Inflamm. 2016;2016:3094642. doi:10.1155/2016/3094642
- Mintoff D, Benhadou F, Pace NP, et al. Metabolic syndrome and hidradenitis suppurativa: epidemiological, molecular, and therapeutic aspects. Int J Dermatol. 2022;61:1175-1186. doi:10.1111/ijd.15910
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71
- Jennings L, Nestor L, Molloy O, et al. The treatment of hidradenitis suppurativa with the glucagon-like peptide-1 agonist liraglutide. Br J Dermatol. 2017;177:858-859. doi:10.1111/bjd.15233
- Khandalavala BN. A disease-modifying approach for advanced hidradenitis suppurativa (regimen with metformin, liraglutide, dapsone, and finasteride): a case report. Case Rep Dermatol. 2017;9:70-78. doi:10.1159/000473873
- Mainville L, MacHaalany J, Veillette H. Hidradenitis suppurativa patient requiring cardiac procedure with inguinal access: case management with ertapenem. SAGE Open Med Case Rep. 2024;12:2050313X241274819. doi:10.1177/2050313X241274819
- Hamed K, Alosaimi MN, Ali BA, et al. Glucagon-like peptide-1 (GLP-1) receptor agonists: exploring their impact on diabetes, obesity, and cardiovascular health through a comprehensive literature review. Cureus. 2024;16:E68390. doi:10.7759/cureus.68390
- Alharbi SH. Anti-inflammatory role of glucagon-like peptide 1 receptor agonists and its clinical implications. Ther Adv Endocrinol Metab. 2024;15:20420188231222367. doi:10.1177/20420188231222367
The Effect of GLP-1 Receptor Agonists on Hidradenitis Suppurativa: A Comprehensive Systematic Review
The Effect of GLP-1 Receptor Agonists on Hidradenitis Suppurativa: A Comprehensive Systematic Review
Practice Points
- Glucagonlike peptide 1 receptor agonists (GLP-1 RAs) can be used adjunctively to manage hidradenitis suppurativa (HS) symptoms.
- The anti-inflammatory properties of GLP-1 RAs as well as their tendency to cause weight loss and manage metabolic syndrome improve the outcome of HS.
- Although current evidence is limited to case reports, these agents can be successfully integrated with existing protocols (biologics, antibiotics, or metformin); however, clinicians should monitor for gastrointestinal adverse effects.
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
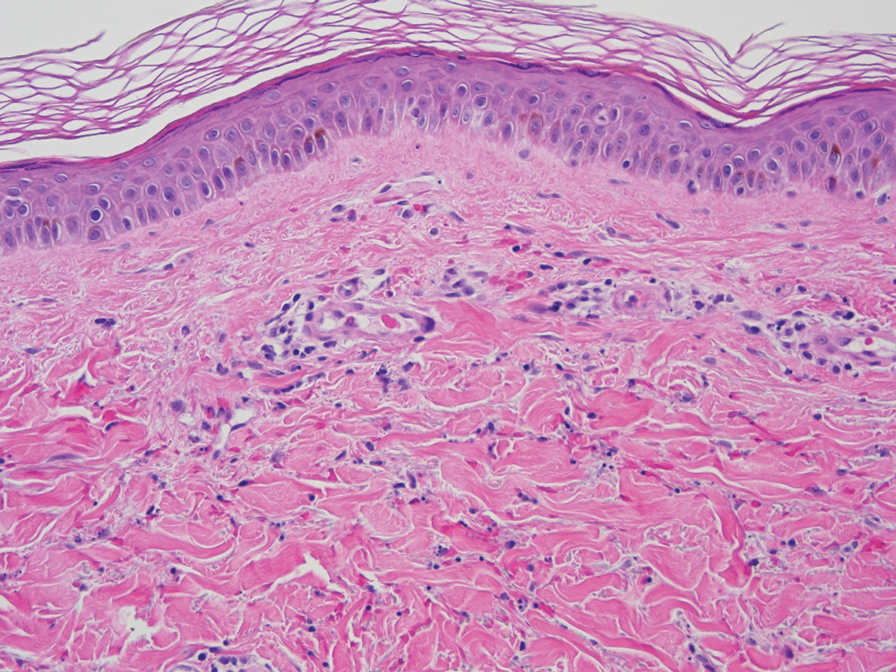
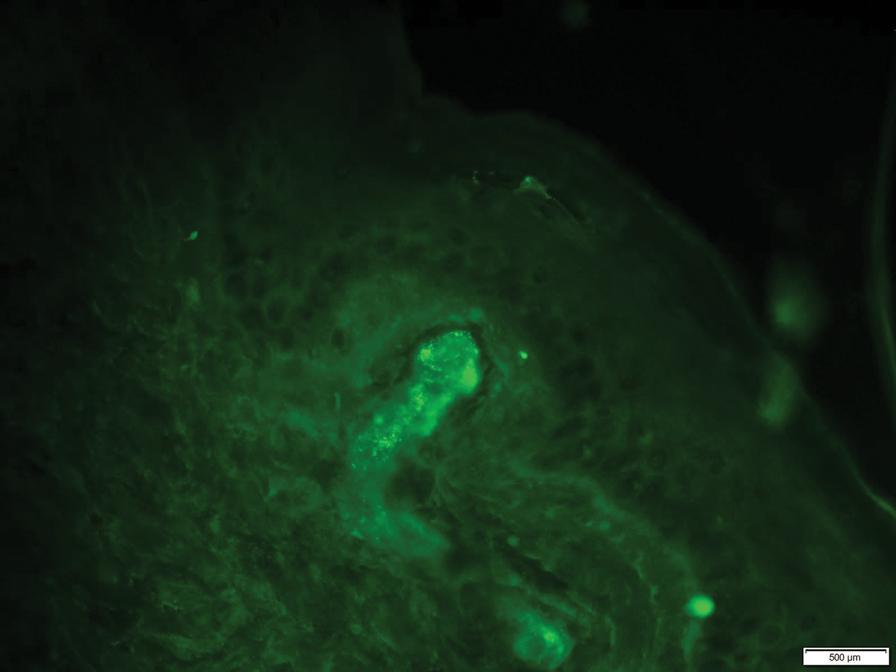
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
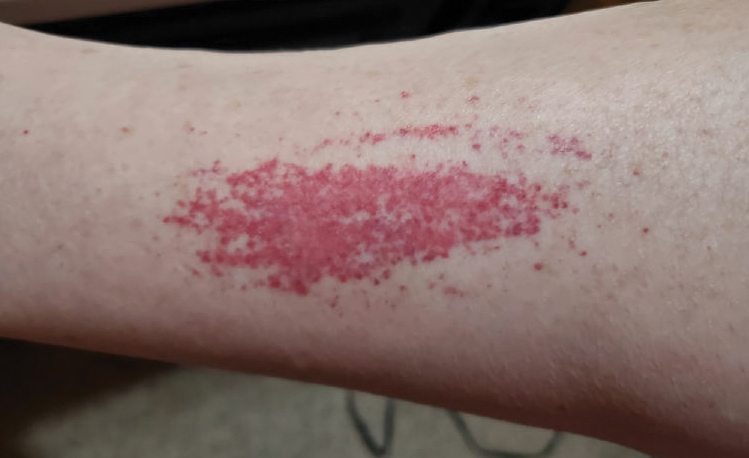
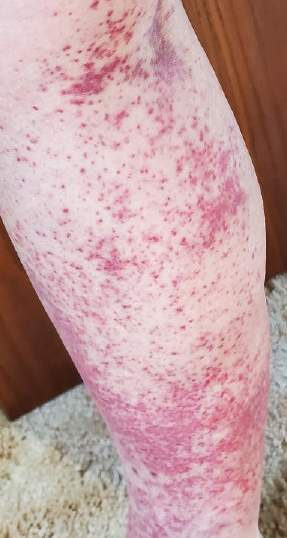
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Practice Points
- Elevation of the erythrocyte sedimentation rate (ESR) is nonspecific for inflammation and may be observed in the setting of increased immunoglobulin levels.
- Patients with elevated ESR and clinical evidence of recurrent petechiae and purpura should be screened for monoclonal and polyclonal gammopathies.

A Solitary Axillary Subcutaneous Mass
A Solitary Axillary Subcutaneous Mass
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
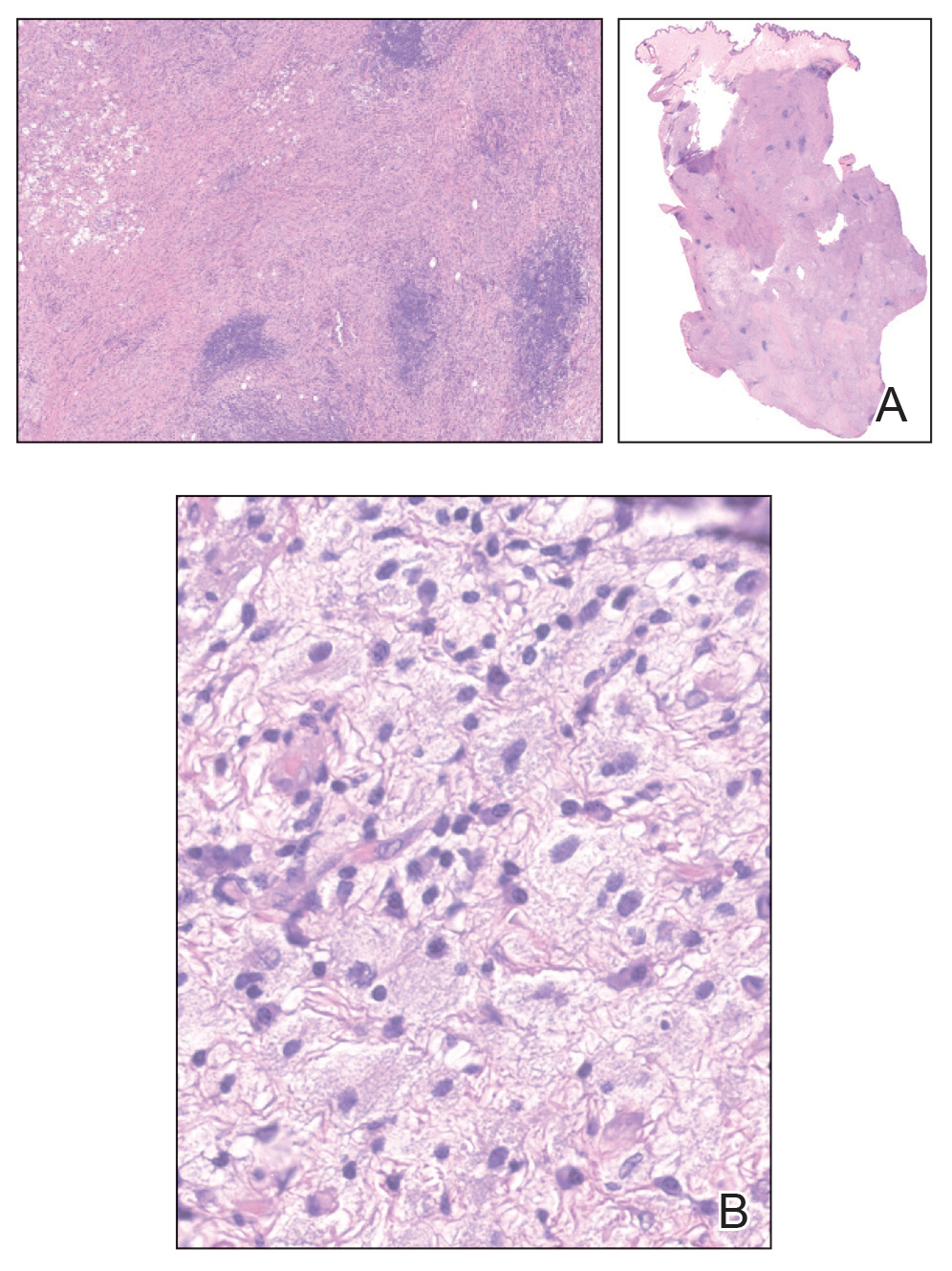
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
A Solitary Axillary Subcutaneous Mass
A Solitary Axillary Subcutaneous Mass
A 34-year-old man presented to our dermatology clinic for evaluation of a lesion in the right axilla of 1 year’s duration that had recently increased in size. The lesion was nontender and intermittently pruritic and was associated with focal hypohidrosis. The patient denied any fevers, chills, or recent weight change. His medical history was otherwise unremarkable. His only medications were daily ashwagandha and vitamin B and C supplements. On physical examination, a firm, 6-cm, subcutaneous nodule was noted in the right axilla with central alopecia and without a clear punctum. He had no palpable cervical, postauricular, or inguinal lymphadenopathy. The left axilla was clear, and there were no other relevant skin findings. Laboratory testing including a complete blood count, comprehensive metabolic panel, and sexually transmitted infections panel was unremarkable. Ultrasonography and subsequent magnetic resonance imaging of the right axilla showed a 4.9-cm nodule located in the subcutaneous fat with minimal deep infiltration and relatively smooth margins. An incisional biopsy of the lesion was performed.
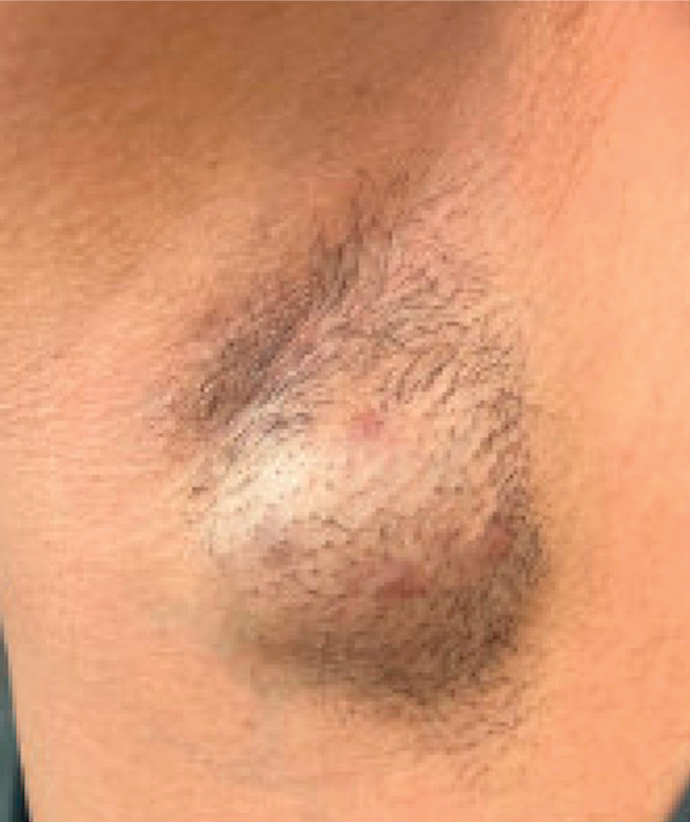

Xylazine-Induced Skin Necrosis
Xylazine-Induced Skin Necrosis
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
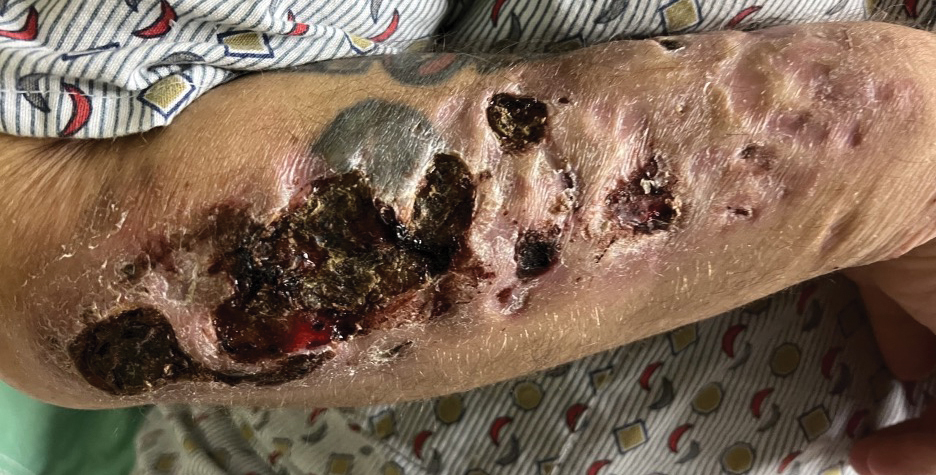
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
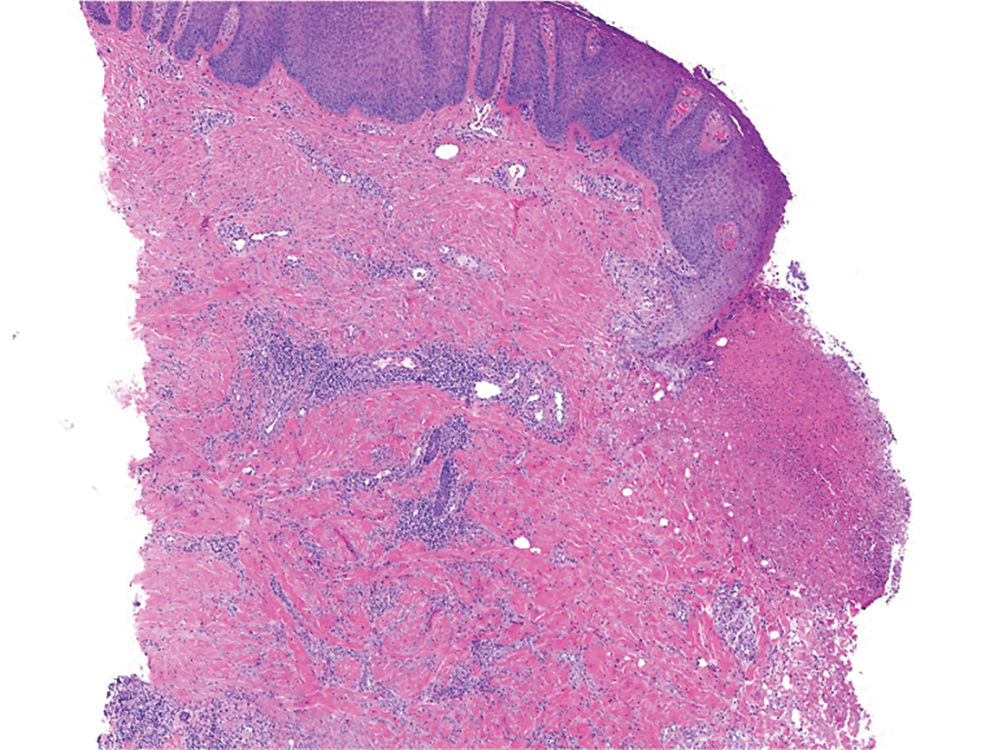
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
Xylazine-Induced Skin Necrosis
Xylazine-Induced Skin Necrosis
Practice Points
- Dermatologists should be aware of the potential for xylazine to cause ulcers in patients with a history of intravenous drug use.
- Early recognition of xylazine skin ulcers is imperative, as delayed diagnosis increases morbidity such as soft-tissue and bone infection, sepsis, and death.
