User login
Frequent cannabis use in depression tripled over past decade
Not only are individuals with depression at significantly higher risk for cannabis use, compared with those without depression, this trend has increased dramatically over the last decade, new research shows.
Investigators analyzed data from more than 16,000 U.S. adults between the ages of 20 and 59 years and found that those with depression had almost twice the odds of any past-month cannabis use compared with those without depression. Odds rose from 1.5 in the 2005-2006 period to 2.3 in the 2015-2016 period.
Moreover, the odds ratio for daily or near-daily use almost tripled for those with versus without depression between the two periods.

“Clinicians should screen their depressed patients for cannabis use, since this is becoming more common and could actually make their depressive symptoms worse rather than better,” senior author Deborah Hasin, PhD, professor of epidemiology, Columbia University Irving Medical Center, New York City, told Medscape Medical News.
The results were published online August 18 in JAMA Network Open.
Misleading advertising
“Cannabis use is increasing in the U.S. and the potency of cannabis products is increasing as well,” Dr. Hasin said.
“Misleading media information and advertising suggests that cannabis is a good treatment for depression, although studies show that cannabis use may actually worsen depression symptoms, [so] we were interested in whether U.S. adults were increasingly likely to be cannabis users if they were depressed,” she reported.
To investigate, the researchers assessed data from the National Health and Nutrition Examination Survey (NHANES), with a final study sample consisting of 16,216 U.S. adults. The mean age was 39.12 years, 48.9% were men, 66.4% were non-Hispanic White, 65.6% had at least some college education, and 62.4% had an annual family income of less than $75,000.
Of these participants, 7.5% had “probable depression,” based on the Patient Health Questionnaire–9, the investigators report.
Past-month cannabis use was defined as using cannabis at least once during the past 20 days. Daily or near-daily past-month use was defined as using cannabis at least 20 times in the past 30 days.
Covariates included age, gender, race, education, marital status, annual family income, and past-year use of other substances, such as alcohol, heroin, and methamphetamine.
The researchers note that because the NHANES data were divided into six survey years (2005-2006, 2007-2008, 2009-2010, 2011-2012, 2013-2014, and 2015-2016), their analysis was based on a “new sample weight” that combined the datasets.
Especially pronounced
Results showed that the prevalence of any past-month cannabis use in the overall sample group increased from 12.2% in the 2005-2006 period to 17.3% in the 2015-2016 period (P < .001).
The investigators characterized this change as “significant,” adding that the estimated odds of cannabis use increased by approximately 9% between every 2-year time period.
The change was even more dramatic when the increase was examined across survey time periods (OR, 1.12; P < .001). The estimated odds of daily or near-daily use increased by approximately 12% between every 2-year period.
Interestingly, however, there were no significant changes in odds for depression when consecutive survey years were compared.
When the researchers specifically focused on the association between any past-month cannabis use and depression versus no depression, they found an adjusted OR of 1.90 (95% CI, 1.62-2.12; P < .001).
Individuals with depression also had 2.29 (95% CI, 1.80-2.92) times the odds for daily or near-daily cannabis use, compared with those without depression.
A post-hoc analysis looked at time trends in a sample group that included those missing information on at least one covariate (n = 17,724 participants). It showed similar results to those in the final sample that included no missing data.
People with depression have increased risk of using “most substances that can be abused,” Dr. Hasin said. “However, with the overall rates of cannabis use increasing in the general population, this is becoming especially pronounced for cannabis.”
Clear implications
Commenting on the findings for Medscape Medical News, Deepak D’Souza, MD, professor of psychiatry, Yale University, New Haven, Conn., said there is “concern about the unsubstantiated claims of cannabis having a beneficial effect in psychiatric disorders, the most common being depression.”
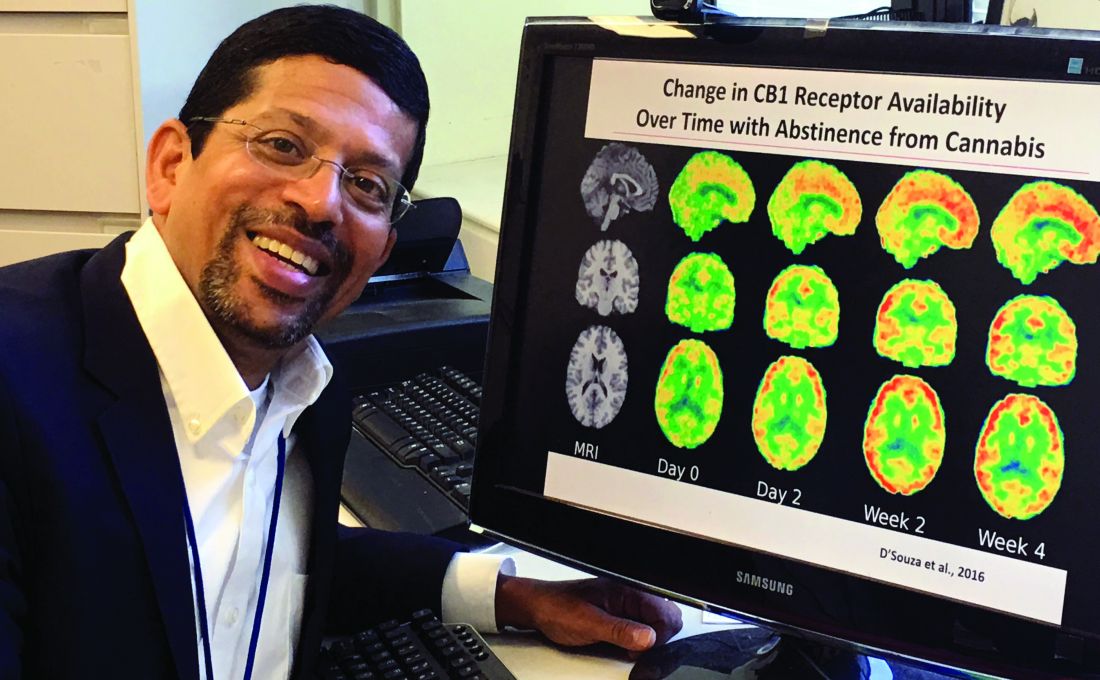
Dr. D’Souza, who was not involved with the study, called it “yet another piece of evidence suggesting that over the period of time during which cannabis laws have been liberalized, rates of past-month and daily cannabis use have increased, whereas rates of other substances, including alcohol, have remained stable.”
He suggested that a common limitation of epidemiological studies is that it is difficult to tell the direction of the association, “and it could be bidirectional.”
Nevertheless, there are clear implications for the practicing clinician, he added.
“If people have a history of depression, one should ask patients about the use of cannabis and also remind them about potential psychiatric negative effects of use,” Dr. D’Souza noted.
For the general public, “the point is that there is no good evidence to support cannabis use in depression treatment and, in fact, people with depression might be more likely to use it in problematic way,” he said.
Dr. Hasin agreed that it is “certainly possible that the relationship between cannabis use and depression is bidirectional, but the mechanism of this association requires more study.”
The study was supported by a grant from the National Institute on Drug Abuse to Dr. Hasin and by the New York State Psychiatric Institute. The study authors and Dr. D’Souza disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Not only are individuals with depression at significantly higher risk for cannabis use, compared with those without depression, this trend has increased dramatically over the last decade, new research shows.
Investigators analyzed data from more than 16,000 U.S. adults between the ages of 20 and 59 years and found that those with depression had almost twice the odds of any past-month cannabis use compared with those without depression. Odds rose from 1.5 in the 2005-2006 period to 2.3 in the 2015-2016 period.
Moreover, the odds ratio for daily or near-daily use almost tripled for those with versus without depression between the two periods.
“Clinicians should screen their depressed patients for cannabis use, since this is becoming more common and could actually make their depressive symptoms worse rather than better,” senior author Deborah Hasin, PhD, professor of epidemiology, Columbia University Irving Medical Center, New York City, told Medscape Medical News.
The results were published online August 18 in JAMA Network Open.
Misleading advertising
“Cannabis use is increasing in the U.S. and the potency of cannabis products is increasing as well,” Dr. Hasin said.
“Misleading media information and advertising suggests that cannabis is a good treatment for depression, although studies show that cannabis use may actually worsen depression symptoms, [so] we were interested in whether U.S. adults were increasingly likely to be cannabis users if they were depressed,” she reported.
To investigate, the researchers assessed data from the National Health and Nutrition Examination Survey (NHANES), with a final study sample consisting of 16,216 U.S. adults. The mean age was 39.12 years, 48.9% were men, 66.4% were non-Hispanic White, 65.6% had at least some college education, and 62.4% had an annual family income of less than $75,000.
Of these participants, 7.5% had “probable depression,” based on the Patient Health Questionnaire–9, the investigators report.
Past-month cannabis use was defined as using cannabis at least once during the past 20 days. Daily or near-daily past-month use was defined as using cannabis at least 20 times in the past 30 days.
Covariates included age, gender, race, education, marital status, annual family income, and past-year use of other substances, such as alcohol, heroin, and methamphetamine.
The researchers note that because the NHANES data were divided into six survey years (2005-2006, 2007-2008, 2009-2010, 2011-2012, 2013-2014, and 2015-2016), their analysis was based on a “new sample weight” that combined the datasets.
Especially pronounced
Results showed that the prevalence of any past-month cannabis use in the overall sample group increased from 12.2% in the 2005-2006 period to 17.3% in the 2015-2016 period (P < .001).
The investigators characterized this change as “significant,” adding that the estimated odds of cannabis use increased by approximately 9% between every 2-year time period.
The change was even more dramatic when the increase was examined across survey time periods (OR, 1.12; P < .001). The estimated odds of daily or near-daily use increased by approximately 12% between every 2-year period.
Interestingly, however, there were no significant changes in odds for depression when consecutive survey years were compared.
When the researchers specifically focused on the association between any past-month cannabis use and depression versus no depression, they found an adjusted OR of 1.90 (95% CI, 1.62-2.12; P < .001).
Individuals with depression also had 2.29 (95% CI, 1.80-2.92) times the odds for daily or near-daily cannabis use, compared with those without depression.
A post-hoc analysis looked at time trends in a sample group that included those missing information on at least one covariate (n = 17,724 participants). It showed similar results to those in the final sample that included no missing data.
People with depression have increased risk of using “most substances that can be abused,” Dr. Hasin said. “However, with the overall rates of cannabis use increasing in the general population, this is becoming especially pronounced for cannabis.”
Clear implications
Commenting on the findings for Medscape Medical News, Deepak D’Souza, MD, professor of psychiatry, Yale University, New Haven, Conn., said there is “concern about the unsubstantiated claims of cannabis having a beneficial effect in psychiatric disorders, the most common being depression.”
Dr. D’Souza, who was not involved with the study, called it “yet another piece of evidence suggesting that over the period of time during which cannabis laws have been liberalized, rates of past-month and daily cannabis use have increased, whereas rates of other substances, including alcohol, have remained stable.”
He suggested that a common limitation of epidemiological studies is that it is difficult to tell the direction of the association, “and it could be bidirectional.”
Nevertheless, there are clear implications for the practicing clinician, he added.
“If people have a history of depression, one should ask patients about the use of cannabis and also remind them about potential psychiatric negative effects of use,” Dr. D’Souza noted.
For the general public, “the point is that there is no good evidence to support cannabis use in depression treatment and, in fact, people with depression might be more likely to use it in problematic way,” he said.
Dr. Hasin agreed that it is “certainly possible that the relationship between cannabis use and depression is bidirectional, but the mechanism of this association requires more study.”
The study was supported by a grant from the National Institute on Drug Abuse to Dr. Hasin and by the New York State Psychiatric Institute. The study authors and Dr. D’Souza disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Not only are individuals with depression at significantly higher risk for cannabis use, compared with those without depression, this trend has increased dramatically over the last decade, new research shows.
Investigators analyzed data from more than 16,000 U.S. adults between the ages of 20 and 59 years and found that those with depression had almost twice the odds of any past-month cannabis use compared with those without depression. Odds rose from 1.5 in the 2005-2006 period to 2.3 in the 2015-2016 period.
Moreover, the odds ratio for daily or near-daily use almost tripled for those with versus without depression between the two periods.
“Clinicians should screen their depressed patients for cannabis use, since this is becoming more common and could actually make their depressive symptoms worse rather than better,” senior author Deborah Hasin, PhD, professor of epidemiology, Columbia University Irving Medical Center, New York City, told Medscape Medical News.
The results were published online August 18 in JAMA Network Open.
Misleading advertising
“Cannabis use is increasing in the U.S. and the potency of cannabis products is increasing as well,” Dr. Hasin said.
“Misleading media information and advertising suggests that cannabis is a good treatment for depression, although studies show that cannabis use may actually worsen depression symptoms, [so] we were interested in whether U.S. adults were increasingly likely to be cannabis users if they were depressed,” she reported.
To investigate, the researchers assessed data from the National Health and Nutrition Examination Survey (NHANES), with a final study sample consisting of 16,216 U.S. adults. The mean age was 39.12 years, 48.9% were men, 66.4% were non-Hispanic White, 65.6% had at least some college education, and 62.4% had an annual family income of less than $75,000.
Of these participants, 7.5% had “probable depression,” based on the Patient Health Questionnaire–9, the investigators report.
Past-month cannabis use was defined as using cannabis at least once during the past 20 days. Daily or near-daily past-month use was defined as using cannabis at least 20 times in the past 30 days.
Covariates included age, gender, race, education, marital status, annual family income, and past-year use of other substances, such as alcohol, heroin, and methamphetamine.
The researchers note that because the NHANES data were divided into six survey years (2005-2006, 2007-2008, 2009-2010, 2011-2012, 2013-2014, and 2015-2016), their analysis was based on a “new sample weight” that combined the datasets.
Especially pronounced
Results showed that the prevalence of any past-month cannabis use in the overall sample group increased from 12.2% in the 2005-2006 period to 17.3% in the 2015-2016 period (P < .001).
The investigators characterized this change as “significant,” adding that the estimated odds of cannabis use increased by approximately 9% between every 2-year time period.
The change was even more dramatic when the increase was examined across survey time periods (OR, 1.12; P < .001). The estimated odds of daily or near-daily use increased by approximately 12% between every 2-year period.
Interestingly, however, there were no significant changes in odds for depression when consecutive survey years were compared.
When the researchers specifically focused on the association between any past-month cannabis use and depression versus no depression, they found an adjusted OR of 1.90 (95% CI, 1.62-2.12; P < .001).
Individuals with depression also had 2.29 (95% CI, 1.80-2.92) times the odds for daily or near-daily cannabis use, compared with those without depression.
A post-hoc analysis looked at time trends in a sample group that included those missing information on at least one covariate (n = 17,724 participants). It showed similar results to those in the final sample that included no missing data.
People with depression have increased risk of using “most substances that can be abused,” Dr. Hasin said. “However, with the overall rates of cannabis use increasing in the general population, this is becoming especially pronounced for cannabis.”
Clear implications
Commenting on the findings for Medscape Medical News, Deepak D’Souza, MD, professor of psychiatry, Yale University, New Haven, Conn., said there is “concern about the unsubstantiated claims of cannabis having a beneficial effect in psychiatric disorders, the most common being depression.”
Dr. D’Souza, who was not involved with the study, called it “yet another piece of evidence suggesting that over the period of time during which cannabis laws have been liberalized, rates of past-month and daily cannabis use have increased, whereas rates of other substances, including alcohol, have remained stable.”
He suggested that a common limitation of epidemiological studies is that it is difficult to tell the direction of the association, “and it could be bidirectional.”
Nevertheless, there are clear implications for the practicing clinician, he added.
“If people have a history of depression, one should ask patients about the use of cannabis and also remind them about potential psychiatric negative effects of use,” Dr. D’Souza noted.
For the general public, “the point is that there is no good evidence to support cannabis use in depression treatment and, in fact, people with depression might be more likely to use it in problematic way,” he said.
Dr. Hasin agreed that it is “certainly possible that the relationship between cannabis use and depression is bidirectional, but the mechanism of this association requires more study.”
The study was supported by a grant from the National Institute on Drug Abuse to Dr. Hasin and by the New York State Psychiatric Institute. The study authors and Dr. D’Souza disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Humira topped drug-revenue list for 2019
Humira outsold all other drugs in 2019 in terms of revenue as cytokine inhibitor medications earned their way to three of the first four spots on the pharmaceutical best-seller list, according to a new analysis from the IQVIA Institute for Human Data Science.
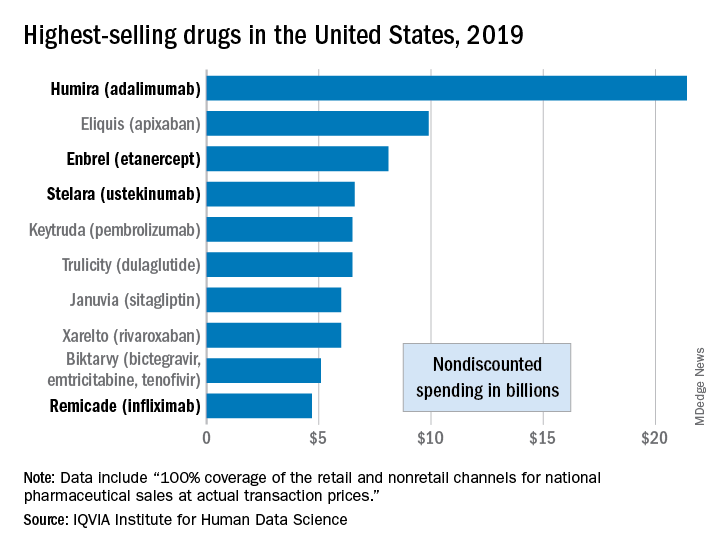
Sales of Humira (adalimumab) amounted to $21.4 billion before discounting, Murray Aitken, the institute’s executive director, and associates wrote in their analysis. That’s more than double the total of the anticoagulant Eliquis (apixaban), which brought in $9.9 billion in its last year before generic forms became available.
The next two spots were filled by the tumor necrosis factor inhibitor Enbrel (etanercept) with $8.1 billion in sales and the interleukin 12/23 inhibitor Stelara (ustekinumab) with sales totaling $6.6 billion, followed by the chemotherapy drug Keytruda (pembrolizumab) close behind after racking up $6.5 billion in sales, the researchers reported.
Total nondiscounted spending on all drugs in the U.S. market came to $511 billion in 2019, an increase of 5.7% over the $484 billion spent in 2018, based on data from the July 2020 IQVIA National Sales Perspectives.
These figures are “not adjusted for estimates of off-invoice discounts and rebates,” the authors noted, but they include “prescription and insulin products sold into chain and independent pharmacies, food store pharmacies, mail service pharmacies, long-term care facilities, hospitals, clinics, and other institutional settings.”
Those “discounts and rebates” do exist, however, and they can add up. Drug sales for 2019, “after deducting negotiated rebates, discounts, and other forms of price concessions, such as patient coupons or vouchers that offset out-of-pocket costs,” were $235 billion less than overall nondiscounted spending, the report noted.
Now that we’ve shown you the money, let’s take a quick look at volume. The leading drugs by number of dispensed prescriptions in 2019 were, not surprisingly, quite different. First, with 118 million prescriptions, was atorvastatin, followed by levothyroxine (113 million), lisinopril (96), amlodipine (89), and metoprolol (85), Mr. Aitken and associates reported.
Altogether, over 4.2 billion prescriptions were dispensed last year, with a couple of caveats: 90-day and 30-day fills were both counted as one prescription, and OTC drugs were not included, they pointed out.
Humira outsold all other drugs in 2019 in terms of revenue as cytokine inhibitor medications earned their way to three of the first four spots on the pharmaceutical best-seller list, according to a new analysis from the IQVIA Institute for Human Data Science.
Sales of Humira (adalimumab) amounted to $21.4 billion before discounting, Murray Aitken, the institute’s executive director, and associates wrote in their analysis. That’s more than double the total of the anticoagulant Eliquis (apixaban), which brought in $9.9 billion in its last year before generic forms became available.
The next two spots were filled by the tumor necrosis factor inhibitor Enbrel (etanercept) with $8.1 billion in sales and the interleukin 12/23 inhibitor Stelara (ustekinumab) with sales totaling $6.6 billion, followed by the chemotherapy drug Keytruda (pembrolizumab) close behind after racking up $6.5 billion in sales, the researchers reported.
Total nondiscounted spending on all drugs in the U.S. market came to $511 billion in 2019, an increase of 5.7% over the $484 billion spent in 2018, based on data from the July 2020 IQVIA National Sales Perspectives.
These figures are “not adjusted for estimates of off-invoice discounts and rebates,” the authors noted, but they include “prescription and insulin products sold into chain and independent pharmacies, food store pharmacies, mail service pharmacies, long-term care facilities, hospitals, clinics, and other institutional settings.”
Those “discounts and rebates” do exist, however, and they can add up. Drug sales for 2019, “after deducting negotiated rebates, discounts, and other forms of price concessions, such as patient coupons or vouchers that offset out-of-pocket costs,” were $235 billion less than overall nondiscounted spending, the report noted.
Now that we’ve shown you the money, let’s take a quick look at volume. The leading drugs by number of dispensed prescriptions in 2019 were, not surprisingly, quite different. First, with 118 million prescriptions, was atorvastatin, followed by levothyroxine (113 million), lisinopril (96), amlodipine (89), and metoprolol (85), Mr. Aitken and associates reported.
Altogether, over 4.2 billion prescriptions were dispensed last year, with a couple of caveats: 90-day and 30-day fills were both counted as one prescription, and OTC drugs were not included, they pointed out.
Humira outsold all other drugs in 2019 in terms of revenue as cytokine inhibitor medications earned their way to three of the first four spots on the pharmaceutical best-seller list, according to a new analysis from the IQVIA Institute for Human Data Science.
Sales of Humira (adalimumab) amounted to $21.4 billion before discounting, Murray Aitken, the institute’s executive director, and associates wrote in their analysis. That’s more than double the total of the anticoagulant Eliquis (apixaban), which brought in $9.9 billion in its last year before generic forms became available.
The next two spots were filled by the tumor necrosis factor inhibitor Enbrel (etanercept) with $8.1 billion in sales and the interleukin 12/23 inhibitor Stelara (ustekinumab) with sales totaling $6.6 billion, followed by the chemotherapy drug Keytruda (pembrolizumab) close behind after racking up $6.5 billion in sales, the researchers reported.
Total nondiscounted spending on all drugs in the U.S. market came to $511 billion in 2019, an increase of 5.7% over the $484 billion spent in 2018, based on data from the July 2020 IQVIA National Sales Perspectives.
These figures are “not adjusted for estimates of off-invoice discounts and rebates,” the authors noted, but they include “prescription and insulin products sold into chain and independent pharmacies, food store pharmacies, mail service pharmacies, long-term care facilities, hospitals, clinics, and other institutional settings.”
Those “discounts and rebates” do exist, however, and they can add up. Drug sales for 2019, “after deducting negotiated rebates, discounts, and other forms of price concessions, such as patient coupons or vouchers that offset out-of-pocket costs,” were $235 billion less than overall nondiscounted spending, the report noted.
Now that we’ve shown you the money, let’s take a quick look at volume. The leading drugs by number of dispensed prescriptions in 2019 were, not surprisingly, quite different. First, with 118 million prescriptions, was atorvastatin, followed by levothyroxine (113 million), lisinopril (96), amlodipine (89), and metoprolol (85), Mr. Aitken and associates reported.
Altogether, over 4.2 billion prescriptions were dispensed last year, with a couple of caveats: 90-day and 30-day fills were both counted as one prescription, and OTC drugs were not included, they pointed out.
The transitions of COVID-19
When I was preparing for the recent birth of my baby, I anticipated a period of transition for myself. As a reproductive psychiatrist, I have treated many women during the perinatal and postpartum periods, and have a unique appreciation for the life changes that accompany birth. What I did not expect, however, was the world transitioning with me.

“The new normal” is an economic phrase that describes the COVID-19 era. The pandemic has engendered economic instability, collapsed industries, challenged health care systems, and has led to many deaths worldwide. The COVID-19 pandemic also has been associated with overall increases in anxiety and depression.1 Emerging research suggests that frontline medical workers are especially at risk for developing psychological distress.2
COVID-19 has also created immense challenges for families. Because of concern for the spread of the virus, schools have been suspended, older grandparents isolated, and many parents continue to work remotely. For families in psychiatric care, this time has also been a time of change. Telepsychiatry might be more accessible, but the transition has been an adjustment for patients and clinicians.
As psychiatrists, how do we best treat families during this time? What are some ways to support our psychiatric colleagues? How do we ensure our own emotional well-being amid the tremendous changes occurring around us?
Background of interpersonal psychotherapy
Interpersonal psychotherapy (IPT) is a form of psychotherapy designed to treat depression following periods of transition. Its main goals include improving interpersonal connection and reducing psychological distress. Originally developed in the 1970s by Gerald Klerman, MD; Myrna Weissman, PhD; and Eugene Paykel, MD, IPT is a structured, time-limited form of psychotherapy.3
Conceptualizing depression as a treatable illness, Pim Cuijpers, PhD, and associates summarized the division of IPT into three phases.4 The initial phase involves history taking, forming an alliance, and choosing an interpersonal focus for treatment. The middle phase focuses on applying interpersonal problem-specific therapeutic techniques. The concluding phase of treatment involves consolidation of gains as well as formulating contingency plans for relapse of symptoms. Over the course of treatment, an IPT clinician focuses on life transitions and emphasizes that isolation and antagonistic relationships increase an individual’s vulnerability for a depressive episode.3
Randomized, controlled trials support IPT’s efficacy as a treatment for depression. Research also suggests it can possibly prevent the development of depression.4 Although IPT initially was designed as an individual form of psychotherapy, it has been adapted to both family and group contexts.5,6 IPT is also an empirically valid form of psychotherapy for postpartum depression.7
Interpersonal psychotherapy for families
Given IPT’s role for treating depression following times of transition, clinicians should consider adapting interpersonal psychotherapy to family treatment during this time. Addressing social isolation, managing complex family relationships, and monitoring the family’s overall emotional health should be prioritized. Families under quarantine or who are grieving the death of family members may especially benefit from improved interpersonal connection. Consistent with the IPT model, contingency plans for the family should also be explored to prepare for potential future waves of the pandemic.
In addition to supporting and strengthening families, psychiatrists can use IPT themes to identify positive changes for families tied to COVID-19. Despite its difficulties, the stay-at-home order provided some families a unique chance to slow down and adapt a more relaxed routine. Busy families were suddenly given the opportunity to spend more time with one another. Although many older grandparents were isolated, creative uses of technology provided a chance for grandparents to remain an integral part of family life. Psychiatrists can assist families in transitioning back to previous schedules, while also exploring ways to incorporate the positive changes gained during this time.
Interpersonal psychotherapy for psychiatrists
An interpersonal focus could also be helpful for clinicians to adapt to changes in psychiatric practice. Many clinicians have been thrust into telepsychiatry practice, some with little to no preparation. Because of the trauma associated with frontline work, some psychiatrists have expanded their patient panel to treat physician colleagues. For consult-liaison psychiatrists, the possible neuropsychiatric effects of COVID-19 are new symptoms to consider when evaluating patients in a medical hospital setting.8 Fundamentally, modern day psychiatrists have never encountered a pandemic nor attempted to treat its psychological implications. Prioritizing seeking support from colleagues and caring for one’s personal relationships are helpful tools for clinicians to maintain their own emotional health during this challenging period.
Personal reflection
When I reflect on my baby’s recent birth, I recognize the importance of interpersonal relationships. COVID-19 developed shortly after I gave birth, during the initial haze of the newborn period. Initially, I felt overwhelmed by the many transitions and emotions that were occurring simultaneously. However, as I began to prioritize socialization for myself and my family (albeit creatively at times while socially distancing), I witnessed its positive effects on my emotional well-being and recognized its value in managing times of transition.
Using IPT for families, colleagues, and ourselves
As general psychiatrists, there are several ways to utilize IPT-related themes during this time:
- Connect with families: Although families may recognize they are struggling emotionally, some may find it difficult to navigate the sea of mental health resources. This is particularly true when a family’s financial situation is also stressed. Reaching out to local religious services and community medical resources or inquiring about the mental health of other family members are ways for psychiatrists to engage more families in mental health treatment.
- Reach out to colleagues: Psychiatrists are not immune to developing psychiatric disorders,and it is important to support each other.9 This is also an unusual time when psychiatrists are treating symptoms in patients that they themselves may be also experiencing. Supporting help groups and hot lines, reaching out to colleagues who appear to be struggling and addressing interpersonal conflicts within one’s practice are crucial practices for psychiatrists during this time.
- Explore within ourselves: Evaluating our own interpersonal relationships as well as areas for improvement are critical skills to maintain our own emotional well-being. Setting aside time to connect with friends in a nonclinical setting and prioritizing our family connections are helpful tools. In addition, exploring our reactions to past life transitions could improve our own level of insight into our response to COVID-19.
Conclusion
Conceptualizing COVID-19 as a period of transition and using IPT themes are helpful tools to mitigate the potential adverse psychological effects of COVID-19 on families. Similarly, they can also be helpful in supporting our colleagues and helping ourselves cope during this difficult period.
References
1. Qiu J et al. Gen Psychiatr. 2020 Mar 6;33(2):e100213.
2. Gautam M et al. Psychosomatics. 2020 Apr 20. doi: 10.1016/j.psym.2020.04.009.
3. Markowitz JC, Weissman MM. Clin Psychol Psychother. 2012 Mar-Apr;19(2):99-105.
4. Cuijpers P et al. Am J Psychiatry. 2016 Jul;173(7):680-7.
5. Dietz LJ et al. J Am Acad Child Adolesc Psychiatry. 2015 Mar;54(3):191-9.
6. Verdeli H et al. Child Adolesc Psychiatr Clin N Am. 2008 Jul;17(3):605-24.
7. Stuart S. Clin Psychol Psychother. 2012 Mar-Apr;19(2):134-40.
8. Rogers JP et al. Lancet Psychiatry. 2020 Jul;7(7):611-27.
9. Korkeila JA et al. Scand J Public Health. 2003;31(2):85-91.
Dr. Reinstein is a psychiatry attending at Zucker Hillside Hospital, New York. Her clinical interests include reproductive psychiatry and family therapy, with a specific focus on maternal mental health. She is one of the recipients of the 4th Annual Resident Recognition Award for Excellence in Family Oriented Care. Dr. Reinstein has no conflicts of interest. Alison M. Heru, MD, the Families in Psychiatry columnist, invited Dr. Reinstein to address this topic.
When I was preparing for the recent birth of my baby, I anticipated a period of transition for myself. As a reproductive psychiatrist, I have treated many women during the perinatal and postpartum periods, and have a unique appreciation for the life changes that accompany birth. What I did not expect, however, was the world transitioning with me.
“The new normal” is an economic phrase that describes the COVID-19 era. The pandemic has engendered economic instability, collapsed industries, challenged health care systems, and has led to many deaths worldwide. The COVID-19 pandemic also has been associated with overall increases in anxiety and depression.1 Emerging research suggests that frontline medical workers are especially at risk for developing psychological distress.2
COVID-19 has also created immense challenges for families. Because of concern for the spread of the virus, schools have been suspended, older grandparents isolated, and many parents continue to work remotely. For families in psychiatric care, this time has also been a time of change. Telepsychiatry might be more accessible, but the transition has been an adjustment for patients and clinicians.
As psychiatrists, how do we best treat families during this time? What are some ways to support our psychiatric colleagues? How do we ensure our own emotional well-being amid the tremendous changes occurring around us?
Background of interpersonal psychotherapy
Interpersonal psychotherapy (IPT) is a form of psychotherapy designed to treat depression following periods of transition. Its main goals include improving interpersonal connection and reducing psychological distress. Originally developed in the 1970s by Gerald Klerman, MD; Myrna Weissman, PhD; and Eugene Paykel, MD, IPT is a structured, time-limited form of psychotherapy.3
Conceptualizing depression as a treatable illness, Pim Cuijpers, PhD, and associates summarized the division of IPT into three phases.4 The initial phase involves history taking, forming an alliance, and choosing an interpersonal focus for treatment. The middle phase focuses on applying interpersonal problem-specific therapeutic techniques. The concluding phase of treatment involves consolidation of gains as well as formulating contingency plans for relapse of symptoms. Over the course of treatment, an IPT clinician focuses on life transitions and emphasizes that isolation and antagonistic relationships increase an individual’s vulnerability for a depressive episode.3
Randomized, controlled trials support IPT’s efficacy as a treatment for depression. Research also suggests it can possibly prevent the development of depression.4 Although IPT initially was designed as an individual form of psychotherapy, it has been adapted to both family and group contexts.5,6 IPT is also an empirically valid form of psychotherapy for postpartum depression.7
Interpersonal psychotherapy for families
Given IPT’s role for treating depression following times of transition, clinicians should consider adapting interpersonal psychotherapy to family treatment during this time. Addressing social isolation, managing complex family relationships, and monitoring the family’s overall emotional health should be prioritized. Families under quarantine or who are grieving the death of family members may especially benefit from improved interpersonal connection. Consistent with the IPT model, contingency plans for the family should also be explored to prepare for potential future waves of the pandemic.
In addition to supporting and strengthening families, psychiatrists can use IPT themes to identify positive changes for families tied to COVID-19. Despite its difficulties, the stay-at-home order provided some families a unique chance to slow down and adapt a more relaxed routine. Busy families were suddenly given the opportunity to spend more time with one another. Although many older grandparents were isolated, creative uses of technology provided a chance for grandparents to remain an integral part of family life. Psychiatrists can assist families in transitioning back to previous schedules, while also exploring ways to incorporate the positive changes gained during this time.
Interpersonal psychotherapy for psychiatrists
An interpersonal focus could also be helpful for clinicians to adapt to changes in psychiatric practice. Many clinicians have been thrust into telepsychiatry practice, some with little to no preparation. Because of the trauma associated with frontline work, some psychiatrists have expanded their patient panel to treat physician colleagues. For consult-liaison psychiatrists, the possible neuropsychiatric effects of COVID-19 are new symptoms to consider when evaluating patients in a medical hospital setting.8 Fundamentally, modern day psychiatrists have never encountered a pandemic nor attempted to treat its psychological implications. Prioritizing seeking support from colleagues and caring for one’s personal relationships are helpful tools for clinicians to maintain their own emotional health during this challenging period.
Personal reflection
When I reflect on my baby’s recent birth, I recognize the importance of interpersonal relationships. COVID-19 developed shortly after I gave birth, during the initial haze of the newborn period. Initially, I felt overwhelmed by the many transitions and emotions that were occurring simultaneously. However, as I began to prioritize socialization for myself and my family (albeit creatively at times while socially distancing), I witnessed its positive effects on my emotional well-being and recognized its value in managing times of transition.
Using IPT for families, colleagues, and ourselves
As general psychiatrists, there are several ways to utilize IPT-related themes during this time:
- Connect with families: Although families may recognize they are struggling emotionally, some may find it difficult to navigate the sea of mental health resources. This is particularly true when a family’s financial situation is also stressed. Reaching out to local religious services and community medical resources or inquiring about the mental health of other family members are ways for psychiatrists to engage more families in mental health treatment.
- Reach out to colleagues: Psychiatrists are not immune to developing psychiatric disorders,and it is important to support each other.9 This is also an unusual time when psychiatrists are treating symptoms in patients that they themselves may be also experiencing. Supporting help groups and hot lines, reaching out to colleagues who appear to be struggling and addressing interpersonal conflicts within one’s practice are crucial practices for psychiatrists during this time.
- Explore within ourselves: Evaluating our own interpersonal relationships as well as areas for improvement are critical skills to maintain our own emotional well-being. Setting aside time to connect with friends in a nonclinical setting and prioritizing our family connections are helpful tools. In addition, exploring our reactions to past life transitions could improve our own level of insight into our response to COVID-19.
Conclusion
Conceptualizing COVID-19 as a period of transition and using IPT themes are helpful tools to mitigate the potential adverse psychological effects of COVID-19 on families. Similarly, they can also be helpful in supporting our colleagues and helping ourselves cope during this difficult period.
References
1. Qiu J et al. Gen Psychiatr. 2020 Mar 6;33(2):e100213.
2. Gautam M et al. Psychosomatics. 2020 Apr 20. doi: 10.1016/j.psym.2020.04.009.
3. Markowitz JC, Weissman MM. Clin Psychol Psychother. 2012 Mar-Apr;19(2):99-105.
4. Cuijpers P et al. Am J Psychiatry. 2016 Jul;173(7):680-7.
5. Dietz LJ et al. J Am Acad Child Adolesc Psychiatry. 2015 Mar;54(3):191-9.
6. Verdeli H et al. Child Adolesc Psychiatr Clin N Am. 2008 Jul;17(3):605-24.
7. Stuart S. Clin Psychol Psychother. 2012 Mar-Apr;19(2):134-40.
8. Rogers JP et al. Lancet Psychiatry. 2020 Jul;7(7):611-27.
9. Korkeila JA et al. Scand J Public Health. 2003;31(2):85-91.
Dr. Reinstein is a psychiatry attending at Zucker Hillside Hospital, New York. Her clinical interests include reproductive psychiatry and family therapy, with a specific focus on maternal mental health. She is one of the recipients of the 4th Annual Resident Recognition Award for Excellence in Family Oriented Care. Dr. Reinstein has no conflicts of interest. Alison M. Heru, MD, the Families in Psychiatry columnist, invited Dr. Reinstein to address this topic.
When I was preparing for the recent birth of my baby, I anticipated a period of transition for myself. As a reproductive psychiatrist, I have treated many women during the perinatal and postpartum periods, and have a unique appreciation for the life changes that accompany birth. What I did not expect, however, was the world transitioning with me.
“The new normal” is an economic phrase that describes the COVID-19 era. The pandemic has engendered economic instability, collapsed industries, challenged health care systems, and has led to many deaths worldwide. The COVID-19 pandemic also has been associated with overall increases in anxiety and depression.1 Emerging research suggests that frontline medical workers are especially at risk for developing psychological distress.2
COVID-19 has also created immense challenges for families. Because of concern for the spread of the virus, schools have been suspended, older grandparents isolated, and many parents continue to work remotely. For families in psychiatric care, this time has also been a time of change. Telepsychiatry might be more accessible, but the transition has been an adjustment for patients and clinicians.
As psychiatrists, how do we best treat families during this time? What are some ways to support our psychiatric colleagues? How do we ensure our own emotional well-being amid the tremendous changes occurring around us?
Background of interpersonal psychotherapy
Interpersonal psychotherapy (IPT) is a form of psychotherapy designed to treat depression following periods of transition. Its main goals include improving interpersonal connection and reducing psychological distress. Originally developed in the 1970s by Gerald Klerman, MD; Myrna Weissman, PhD; and Eugene Paykel, MD, IPT is a structured, time-limited form of psychotherapy.3
Conceptualizing depression as a treatable illness, Pim Cuijpers, PhD, and associates summarized the division of IPT into three phases.4 The initial phase involves history taking, forming an alliance, and choosing an interpersonal focus for treatment. The middle phase focuses on applying interpersonal problem-specific therapeutic techniques. The concluding phase of treatment involves consolidation of gains as well as formulating contingency plans for relapse of symptoms. Over the course of treatment, an IPT clinician focuses on life transitions and emphasizes that isolation and antagonistic relationships increase an individual’s vulnerability for a depressive episode.3
Randomized, controlled trials support IPT’s efficacy as a treatment for depression. Research also suggests it can possibly prevent the development of depression.4 Although IPT initially was designed as an individual form of psychotherapy, it has been adapted to both family and group contexts.5,6 IPT is also an empirically valid form of psychotherapy for postpartum depression.7
Interpersonal psychotherapy for families
Given IPT’s role for treating depression following times of transition, clinicians should consider adapting interpersonal psychotherapy to family treatment during this time. Addressing social isolation, managing complex family relationships, and monitoring the family’s overall emotional health should be prioritized. Families under quarantine or who are grieving the death of family members may especially benefit from improved interpersonal connection. Consistent with the IPT model, contingency plans for the family should also be explored to prepare for potential future waves of the pandemic.
In addition to supporting and strengthening families, psychiatrists can use IPT themes to identify positive changes for families tied to COVID-19. Despite its difficulties, the stay-at-home order provided some families a unique chance to slow down and adapt a more relaxed routine. Busy families were suddenly given the opportunity to spend more time with one another. Although many older grandparents were isolated, creative uses of technology provided a chance for grandparents to remain an integral part of family life. Psychiatrists can assist families in transitioning back to previous schedules, while also exploring ways to incorporate the positive changes gained during this time.
Interpersonal psychotherapy for psychiatrists
An interpersonal focus could also be helpful for clinicians to adapt to changes in psychiatric practice. Many clinicians have been thrust into telepsychiatry practice, some with little to no preparation. Because of the trauma associated with frontline work, some psychiatrists have expanded their patient panel to treat physician colleagues. For consult-liaison psychiatrists, the possible neuropsychiatric effects of COVID-19 are new symptoms to consider when evaluating patients in a medical hospital setting.8 Fundamentally, modern day psychiatrists have never encountered a pandemic nor attempted to treat its psychological implications. Prioritizing seeking support from colleagues and caring for one’s personal relationships are helpful tools for clinicians to maintain their own emotional health during this challenging period.
Personal reflection
When I reflect on my baby’s recent birth, I recognize the importance of interpersonal relationships. COVID-19 developed shortly after I gave birth, during the initial haze of the newborn period. Initially, I felt overwhelmed by the many transitions and emotions that were occurring simultaneously. However, as I began to prioritize socialization for myself and my family (albeit creatively at times while socially distancing), I witnessed its positive effects on my emotional well-being and recognized its value in managing times of transition.
Using IPT for families, colleagues, and ourselves
As general psychiatrists, there are several ways to utilize IPT-related themes during this time:
- Connect with families: Although families may recognize they are struggling emotionally, some may find it difficult to navigate the sea of mental health resources. This is particularly true when a family’s financial situation is also stressed. Reaching out to local religious services and community medical resources or inquiring about the mental health of other family members are ways for psychiatrists to engage more families in mental health treatment.
- Reach out to colleagues: Psychiatrists are not immune to developing psychiatric disorders,and it is important to support each other.9 This is also an unusual time when psychiatrists are treating symptoms in patients that they themselves may be also experiencing. Supporting help groups and hot lines, reaching out to colleagues who appear to be struggling and addressing interpersonal conflicts within one’s practice are crucial practices for psychiatrists during this time.
- Explore within ourselves: Evaluating our own interpersonal relationships as well as areas for improvement are critical skills to maintain our own emotional well-being. Setting aside time to connect with friends in a nonclinical setting and prioritizing our family connections are helpful tools. In addition, exploring our reactions to past life transitions could improve our own level of insight into our response to COVID-19.
Conclusion
Conceptualizing COVID-19 as a period of transition and using IPT themes are helpful tools to mitigate the potential adverse psychological effects of COVID-19 on families. Similarly, they can also be helpful in supporting our colleagues and helping ourselves cope during this difficult period.
References
1. Qiu J et al. Gen Psychiatr. 2020 Mar 6;33(2):e100213.
2. Gautam M et al. Psychosomatics. 2020 Apr 20. doi: 10.1016/j.psym.2020.04.009.
3. Markowitz JC, Weissman MM. Clin Psychol Psychother. 2012 Mar-Apr;19(2):99-105.
4. Cuijpers P et al. Am J Psychiatry. 2016 Jul;173(7):680-7.
5. Dietz LJ et al. J Am Acad Child Adolesc Psychiatry. 2015 Mar;54(3):191-9.
6. Verdeli H et al. Child Adolesc Psychiatr Clin N Am. 2008 Jul;17(3):605-24.
7. Stuart S. Clin Psychol Psychother. 2012 Mar-Apr;19(2):134-40.
8. Rogers JP et al. Lancet Psychiatry. 2020 Jul;7(7):611-27.
9. Korkeila JA et al. Scand J Public Health. 2003;31(2):85-91.
Dr. Reinstein is a psychiatry attending at Zucker Hillside Hospital, New York. Her clinical interests include reproductive psychiatry and family therapy, with a specific focus on maternal mental health. She is one of the recipients of the 4th Annual Resident Recognition Award for Excellence in Family Oriented Care. Dr. Reinstein has no conflicts of interest. Alison M. Heru, MD, the Families in Psychiatry columnist, invited Dr. Reinstein to address this topic.
Performance status, molecular testing key to metastatic cancer prognosis
according to Sam Brondfield, MD, MA, an inpatient medical oncologist at the University of California, San Francisco.

Oncologists have at their fingertips a voluminous and ever-growing body of clinical trials data to draw on for prognostication. Yet many hospitalists will be surprised to learn that this wealth of information is of little value in the inpatient settings where they work, he said at HM20 Virtual, hosted by the Society of Hospital Medicine.
“The applicability of clinical trials data to hospitalized patients is generally poor. That’s an important caveat to keep in mind,” Dr. Brondfield said.
Enrollment in clinical trials is usually restricted to patients with a score of 0 or 1 on the Eastern Clinical Oncology Group Performance Status, meaning their cancer is causing minimal or no disruption to their life (see graphic). Sometimes trials will include patients with a performance status of 2 on the ECOG scale, a tool developed nearly 40 years ago, but clinical trials virtually never enroll those with an ECOG status of 3 or 4. Yet most hospitalized patients with metastatic cancer have an ECOG performance status of 3 or worse. Thus, the clinical trials outcome data are of little relevance.
“In oncology the distinction between ECOG 2 and 3 is very important,” Dr. Brondfield emphasized.
When he talks about treatment options with hospitalized patients who have metastatic cancer and poor performance status – that is, ECOG 3 or 4 – he’ll often say: “Assuming you feel better and can go home, that’s when these clinical trial data may apply better to you.”
Dr. Brondfield cautioned against quoting the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) 5-year overall survival data when hospitalized patients with advanced cancer ask how long they have to live. For one thing, the national average 5-year overall survival figure is hardly an individualized assessment. Plus, oncology is a fast-moving field in which important treatment advances occur all the time, and the SEER data lag far behind. For example, when Dr. Brondfield recently looked up the current SEER 5-year survival for patients diagnosed with metastatic non–small cell lung cancer (NSCLC), the figure quoted was less than 6%, and it was drawn from data accrued in 2009-2015. That simply doesn’t reflect contemporary practice.
Indeed, it’s no longer true that the average survival of patients with metastatic NSCLC is less than a year. In the practice-changing KEYNOTE-189 randomized trial, which accrued participants in 2016-2017, the median overall survival of patients randomized to pembrolizumab (Keytruda) plus standard cytotoxic chemotherapy was 22 months, compared with 11 months with chemotherapy plus placebo (J Clin Oncol. 2020 May 10. doi: 10.1200/JCO.19.03136). As a result, immunotherapy with a programmed death–1 inhibitor such as pembrolizumab in combination with chemotherapy is now standard practice in patients with metastatic NSCLC without targetable mutations.
Performance status guides treatment decision-making
Hospitalists can help oncologists in decision-making regarding whether to offer palliative systemic therapy to patients with advanced metastatic cancer and poor performance status by determining whether that status is caused by the cancer itself or some other cause that’s not easily reversible, such as liver failure.
Take, for example, the inpatient with advanced SCLC. This is an aggressive and chemosensitive cancer. Dr. Brondfield said he is among many medical oncologists who are convinced that, if poor performance status in a patient with advanced SCLC is caused by the cancer itself, prompt initiation of inpatient chemotherapy should be recommended to elicit a response that improves quality of life and performance status in the short term. If, on the other hand, the poor performance status is caused by organ failure or some other issue that can’t easily be improved, hospice may be more appropriate.
“The contour of SCLC over time is that despite its treatment responsiveness it inevitably recurs. But with chemotherapy you can give people in this situation months of quality time, so we generally try to treat these sorts of patients,” Dr. Brondfield explained.
The National Comprehensive Cancer Network guidelines upon which oncologists rely leave lots of room for interpretation regarding the appropriateness of inpatient chemotherapy in patients with advanced cancer and poor patient performance status. Citing “knowledge that’s been passed down across oncology generations,” Dr. Brondfield said he and many of his colleagues believe early palliative supportive care rather than systemic cytotoxic cancer-directed therapy is appropriate for patients with poor performance status who have one of several specific relatively nonchemoresponsive types of metastatic cancer. These include esophageal, gastric, and head and neck cancers.
On the other hand, advanced SCLC isn’t the only type of metastatic cancer that’s so chemosensitive that he and many other oncologists believe aggressive chemotherapy should be offered even in the face of poor patient performance status attributable to the cancer itself.
Take, for example, colorectal cancer with no more than five metastases to the lung or liver, provided those metastases are treatable with resection or radiation. “Those patients are actually curable at a high rate. They have about a 30%-40% cure rate. So those patients, even if they have poor performance status, if we can get them up for surgery or radiation, we usually do try to treat them aggressively,” Dr. Brondfield said.
There are other often chemoresponsive metastatic cancers for which oncologists frequently recommend aggressive treatment to improve quality of life in patients with poor performance status. These cancers include aggressive lymphomas, which are actually often curable; multiple myeloma; testicular and germ cell cancers; NSCLC with a targetable mutation, which is often responsive to oral medications; and prostate and well-differentiated thyroid cancers, which can usually be treated with hormone- or iodine-based therapies rather than more toxic intravenous cytotoxic chemotherapy.
The impact of inpatient palliative chemotherapy in patients with poor performance status and advanced solid cancers not on the short list of highly chemosensitive cancers has not been well studied. A recent retrospective study of 228 such patients who received inpatient palliative chemotherapy at a large Brazilian academic medical center provided little reason for enthusiasm regarding the practice. Survival was short, with 30- and 60-day survival rates of 56% and 39%, respectively. Plus, 30% of patients were admitted to the ICU, where they received aggressive and costly end-of-life care. The investigators found these results suggestive of overprescribing of inpatient palliative chemotherapy (BMC Palliat Care. 2019 May 20;18[1]:42. doi: 10.1186/s12904-019-0427-4).
Of note, the investigators found in a multivariate analysis that an elevated bilirubin was associated with a 217% increased risk of 30-day mortality, and hypercalcemia was associated with a 119% increased risk.
“That’s something to take into account when these decisions are being made,” Dr. Brondfield advised.
In response to an audience comment that oncologists often seem overly optimistic about prognosis, Dr. Brondfield observed, “I think it’s very common for there to be a disagreement between the oncologist wanting to be aggressive for a sick inpatient and the hospitalist or generalist provider thinking: ‘This person looks way too sick for chemotherapy.’ ”
For this reason he is a firm believer in having multidisciplinary conversations regarding prognosis in challenging situations involving hospitalized patients with advanced cancer. An oncologist can bring to such discussions a detailed understanding of clinical trial and molecular data as well as information about the patient’s response to the first round of therapy. But lots of other factors are relevant to prognosis, including nutritional status, comorbidities, and the intuitive eyeball test of how a patient might do. The patient’s family, primary care provider, oncologist, the hospitalist, and the palliative care team will have perspectives of their own.
Molecular testing is now the norm in metastatic cancers
These days oncologists order molecular testing for most patients with metastatic carcinomas to determine eligibility for targeted therapy, suitability for participation in clinical trials, prognostication, and/or assistance in determining the site of origin if that’s unclear.
A single-pass fine needle aspiration biopsy doesn’t provide enough tissue for molecular testing. It’s therefore important to order initially a multipass fine needle aspiration to avoid the need for a repeat biopsy, which is uncomfortable for the patient and can delay diagnosis and treatment.
Dr. Brondfield advised waiting for molecular testing results to come in before trying to prognosticate in patients with a metastatic cancer for which targetable mutations might be present. Survival rates can vary substantially depending upon those test results. Take, for example, metastatic NSCLC: Just within the past year, clinical trials have been published reporting overall survival rates of 39 months in patients with treatable mutations in epidermal growth factor receptor, 42 months with anaplastic lymphoma kinase mutations, and 51 months in patients whose tumor signature features mutations in c-ros oncogene 1, as compared with 22 months with no targetable mutations in the KEYNOTE-189 trial.
“There’s a lot of heterogeneity around how metastatic tumors behave and respond to therapy. Not all metastatic cancers are the same,” the oncologist emphasized.
according to Sam Brondfield, MD, MA, an inpatient medical oncologist at the University of California, San Francisco.
Oncologists have at their fingertips a voluminous and ever-growing body of clinical trials data to draw on for prognostication. Yet many hospitalists will be surprised to learn that this wealth of information is of little value in the inpatient settings where they work, he said at HM20 Virtual, hosted by the Society of Hospital Medicine.
“The applicability of clinical trials data to hospitalized patients is generally poor. That’s an important caveat to keep in mind,” Dr. Brondfield said.
Enrollment in clinical trials is usually restricted to patients with a score of 0 or 1 on the Eastern Clinical Oncology Group Performance Status, meaning their cancer is causing minimal or no disruption to their life (see graphic). Sometimes trials will include patients with a performance status of 2 on the ECOG scale, a tool developed nearly 40 years ago, but clinical trials virtually never enroll those with an ECOG status of 3 or 4. Yet most hospitalized patients with metastatic cancer have an ECOG performance status of 3 or worse. Thus, the clinical trials outcome data are of little relevance.
“In oncology the distinction between ECOG 2 and 3 is very important,” Dr. Brondfield emphasized.
When he talks about treatment options with hospitalized patients who have metastatic cancer and poor performance status – that is, ECOG 3 or 4 – he’ll often say: “Assuming you feel better and can go home, that’s when these clinical trial data may apply better to you.”
Dr. Brondfield cautioned against quoting the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) 5-year overall survival data when hospitalized patients with advanced cancer ask how long they have to live. For one thing, the national average 5-year overall survival figure is hardly an individualized assessment. Plus, oncology is a fast-moving field in which important treatment advances occur all the time, and the SEER data lag far behind. For example, when Dr. Brondfield recently looked up the current SEER 5-year survival for patients diagnosed with metastatic non–small cell lung cancer (NSCLC), the figure quoted was less than 6%, and it was drawn from data accrued in 2009-2015. That simply doesn’t reflect contemporary practice.
Indeed, it’s no longer true that the average survival of patients with metastatic NSCLC is less than a year. In the practice-changing KEYNOTE-189 randomized trial, which accrued participants in 2016-2017, the median overall survival of patients randomized to pembrolizumab (Keytruda) plus standard cytotoxic chemotherapy was 22 months, compared with 11 months with chemotherapy plus placebo (J Clin Oncol. 2020 May 10. doi: 10.1200/JCO.19.03136). As a result, immunotherapy with a programmed death–1 inhibitor such as pembrolizumab in combination with chemotherapy is now standard practice in patients with metastatic NSCLC without targetable mutations.
Performance status guides treatment decision-making
Hospitalists can help oncologists in decision-making regarding whether to offer palliative systemic therapy to patients with advanced metastatic cancer and poor performance status by determining whether that status is caused by the cancer itself or some other cause that’s not easily reversible, such as liver failure.
Take, for example, the inpatient with advanced SCLC. This is an aggressive and chemosensitive cancer. Dr. Brondfield said he is among many medical oncologists who are convinced that, if poor performance status in a patient with advanced SCLC is caused by the cancer itself, prompt initiation of inpatient chemotherapy should be recommended to elicit a response that improves quality of life and performance status in the short term. If, on the other hand, the poor performance status is caused by organ failure or some other issue that can’t easily be improved, hospice may be more appropriate.
“The contour of SCLC over time is that despite its treatment responsiveness it inevitably recurs. But with chemotherapy you can give people in this situation months of quality time, so we generally try to treat these sorts of patients,” Dr. Brondfield explained.
The National Comprehensive Cancer Network guidelines upon which oncologists rely leave lots of room for interpretation regarding the appropriateness of inpatient chemotherapy in patients with advanced cancer and poor patient performance status. Citing “knowledge that’s been passed down across oncology generations,” Dr. Brondfield said he and many of his colleagues believe early palliative supportive care rather than systemic cytotoxic cancer-directed therapy is appropriate for patients with poor performance status who have one of several specific relatively nonchemoresponsive types of metastatic cancer. These include esophageal, gastric, and head and neck cancers.
On the other hand, advanced SCLC isn’t the only type of metastatic cancer that’s so chemosensitive that he and many other oncologists believe aggressive chemotherapy should be offered even in the face of poor patient performance status attributable to the cancer itself.
Take, for example, colorectal cancer with no more than five metastases to the lung or liver, provided those metastases are treatable with resection or radiation. “Those patients are actually curable at a high rate. They have about a 30%-40% cure rate. So those patients, even if they have poor performance status, if we can get them up for surgery or radiation, we usually do try to treat them aggressively,” Dr. Brondfield said.
There are other often chemoresponsive metastatic cancers for which oncologists frequently recommend aggressive treatment to improve quality of life in patients with poor performance status. These cancers include aggressive lymphomas, which are actually often curable; multiple myeloma; testicular and germ cell cancers; NSCLC with a targetable mutation, which is often responsive to oral medications; and prostate and well-differentiated thyroid cancers, which can usually be treated with hormone- or iodine-based therapies rather than more toxic intravenous cytotoxic chemotherapy.
The impact of inpatient palliative chemotherapy in patients with poor performance status and advanced solid cancers not on the short list of highly chemosensitive cancers has not been well studied. A recent retrospective study of 228 such patients who received inpatient palliative chemotherapy at a large Brazilian academic medical center provided little reason for enthusiasm regarding the practice. Survival was short, with 30- and 60-day survival rates of 56% and 39%, respectively. Plus, 30% of patients were admitted to the ICU, where they received aggressive and costly end-of-life care. The investigators found these results suggestive of overprescribing of inpatient palliative chemotherapy (BMC Palliat Care. 2019 May 20;18[1]:42. doi: 10.1186/s12904-019-0427-4).
Of note, the investigators found in a multivariate analysis that an elevated bilirubin was associated with a 217% increased risk of 30-day mortality, and hypercalcemia was associated with a 119% increased risk.
“That’s something to take into account when these decisions are being made,” Dr. Brondfield advised.
In response to an audience comment that oncologists often seem overly optimistic about prognosis, Dr. Brondfield observed, “I think it’s very common for there to be a disagreement between the oncologist wanting to be aggressive for a sick inpatient and the hospitalist or generalist provider thinking: ‘This person looks way too sick for chemotherapy.’ ”
For this reason he is a firm believer in having multidisciplinary conversations regarding prognosis in challenging situations involving hospitalized patients with advanced cancer. An oncologist can bring to such discussions a detailed understanding of clinical trial and molecular data as well as information about the patient’s response to the first round of therapy. But lots of other factors are relevant to prognosis, including nutritional status, comorbidities, and the intuitive eyeball test of how a patient might do. The patient’s family, primary care provider, oncologist, the hospitalist, and the palliative care team will have perspectives of their own.
Molecular testing is now the norm in metastatic cancers
These days oncologists order molecular testing for most patients with metastatic carcinomas to determine eligibility for targeted therapy, suitability for participation in clinical trials, prognostication, and/or assistance in determining the site of origin if that’s unclear.
A single-pass fine needle aspiration biopsy doesn’t provide enough tissue for molecular testing. It’s therefore important to order initially a multipass fine needle aspiration to avoid the need for a repeat biopsy, which is uncomfortable for the patient and can delay diagnosis and treatment.
Dr. Brondfield advised waiting for molecular testing results to come in before trying to prognosticate in patients with a metastatic cancer for which targetable mutations might be present. Survival rates can vary substantially depending upon those test results. Take, for example, metastatic NSCLC: Just within the past year, clinical trials have been published reporting overall survival rates of 39 months in patients with treatable mutations in epidermal growth factor receptor, 42 months with anaplastic lymphoma kinase mutations, and 51 months in patients whose tumor signature features mutations in c-ros oncogene 1, as compared with 22 months with no targetable mutations in the KEYNOTE-189 trial.
“There’s a lot of heterogeneity around how metastatic tumors behave and respond to therapy. Not all metastatic cancers are the same,” the oncologist emphasized.
according to Sam Brondfield, MD, MA, an inpatient medical oncologist at the University of California, San Francisco.
Oncologists have at their fingertips a voluminous and ever-growing body of clinical trials data to draw on for prognostication. Yet many hospitalists will be surprised to learn that this wealth of information is of little value in the inpatient settings where they work, he said at HM20 Virtual, hosted by the Society of Hospital Medicine.
“The applicability of clinical trials data to hospitalized patients is generally poor. That’s an important caveat to keep in mind,” Dr. Brondfield said.
Enrollment in clinical trials is usually restricted to patients with a score of 0 or 1 on the Eastern Clinical Oncology Group Performance Status, meaning their cancer is causing minimal or no disruption to their life (see graphic). Sometimes trials will include patients with a performance status of 2 on the ECOG scale, a tool developed nearly 40 years ago, but clinical trials virtually never enroll those with an ECOG status of 3 or 4. Yet most hospitalized patients with metastatic cancer have an ECOG performance status of 3 or worse. Thus, the clinical trials outcome data are of little relevance.
“In oncology the distinction between ECOG 2 and 3 is very important,” Dr. Brondfield emphasized.
When he talks about treatment options with hospitalized patients who have metastatic cancer and poor performance status – that is, ECOG 3 or 4 – he’ll often say: “Assuming you feel better and can go home, that’s when these clinical trial data may apply better to you.”
Dr. Brondfield cautioned against quoting the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) 5-year overall survival data when hospitalized patients with advanced cancer ask how long they have to live. For one thing, the national average 5-year overall survival figure is hardly an individualized assessment. Plus, oncology is a fast-moving field in which important treatment advances occur all the time, and the SEER data lag far behind. For example, when Dr. Brondfield recently looked up the current SEER 5-year survival for patients diagnosed with metastatic non–small cell lung cancer (NSCLC), the figure quoted was less than 6%, and it was drawn from data accrued in 2009-2015. That simply doesn’t reflect contemporary practice.
Indeed, it’s no longer true that the average survival of patients with metastatic NSCLC is less than a year. In the practice-changing KEYNOTE-189 randomized trial, which accrued participants in 2016-2017, the median overall survival of patients randomized to pembrolizumab (Keytruda) plus standard cytotoxic chemotherapy was 22 months, compared with 11 months with chemotherapy plus placebo (J Clin Oncol. 2020 May 10. doi: 10.1200/JCO.19.03136). As a result, immunotherapy with a programmed death–1 inhibitor such as pembrolizumab in combination with chemotherapy is now standard practice in patients with metastatic NSCLC without targetable mutations.
Performance status guides treatment decision-making
Hospitalists can help oncologists in decision-making regarding whether to offer palliative systemic therapy to patients with advanced metastatic cancer and poor performance status by determining whether that status is caused by the cancer itself or some other cause that’s not easily reversible, such as liver failure.
Take, for example, the inpatient with advanced SCLC. This is an aggressive and chemosensitive cancer. Dr. Brondfield said he is among many medical oncologists who are convinced that, if poor performance status in a patient with advanced SCLC is caused by the cancer itself, prompt initiation of inpatient chemotherapy should be recommended to elicit a response that improves quality of life and performance status in the short term. If, on the other hand, the poor performance status is caused by organ failure or some other issue that can’t easily be improved, hospice may be more appropriate.
“The contour of SCLC over time is that despite its treatment responsiveness it inevitably recurs. But with chemotherapy you can give people in this situation months of quality time, so we generally try to treat these sorts of patients,” Dr. Brondfield explained.
The National Comprehensive Cancer Network guidelines upon which oncologists rely leave lots of room for interpretation regarding the appropriateness of inpatient chemotherapy in patients with advanced cancer and poor patient performance status. Citing “knowledge that’s been passed down across oncology generations,” Dr. Brondfield said he and many of his colleagues believe early palliative supportive care rather than systemic cytotoxic cancer-directed therapy is appropriate for patients with poor performance status who have one of several specific relatively nonchemoresponsive types of metastatic cancer. These include esophageal, gastric, and head and neck cancers.
On the other hand, advanced SCLC isn’t the only type of metastatic cancer that’s so chemosensitive that he and many other oncologists believe aggressive chemotherapy should be offered even in the face of poor patient performance status attributable to the cancer itself.
Take, for example, colorectal cancer with no more than five metastases to the lung or liver, provided those metastases are treatable with resection or radiation. “Those patients are actually curable at a high rate. They have about a 30%-40% cure rate. So those patients, even if they have poor performance status, if we can get them up for surgery or radiation, we usually do try to treat them aggressively,” Dr. Brondfield said.
There are other often chemoresponsive metastatic cancers for which oncologists frequently recommend aggressive treatment to improve quality of life in patients with poor performance status. These cancers include aggressive lymphomas, which are actually often curable; multiple myeloma; testicular and germ cell cancers; NSCLC with a targetable mutation, which is often responsive to oral medications; and prostate and well-differentiated thyroid cancers, which can usually be treated with hormone- or iodine-based therapies rather than more toxic intravenous cytotoxic chemotherapy.
The impact of inpatient palliative chemotherapy in patients with poor performance status and advanced solid cancers not on the short list of highly chemosensitive cancers has not been well studied. A recent retrospective study of 228 such patients who received inpatient palliative chemotherapy at a large Brazilian academic medical center provided little reason for enthusiasm regarding the practice. Survival was short, with 30- and 60-day survival rates of 56% and 39%, respectively. Plus, 30% of patients were admitted to the ICU, where they received aggressive and costly end-of-life care. The investigators found these results suggestive of overprescribing of inpatient palliative chemotherapy (BMC Palliat Care. 2019 May 20;18[1]:42. doi: 10.1186/s12904-019-0427-4).
Of note, the investigators found in a multivariate analysis that an elevated bilirubin was associated with a 217% increased risk of 30-day mortality, and hypercalcemia was associated with a 119% increased risk.
“That’s something to take into account when these decisions are being made,” Dr. Brondfield advised.
In response to an audience comment that oncologists often seem overly optimistic about prognosis, Dr. Brondfield observed, “I think it’s very common for there to be a disagreement between the oncologist wanting to be aggressive for a sick inpatient and the hospitalist or generalist provider thinking: ‘This person looks way too sick for chemotherapy.’ ”
For this reason he is a firm believer in having multidisciplinary conversations regarding prognosis in challenging situations involving hospitalized patients with advanced cancer. An oncologist can bring to such discussions a detailed understanding of clinical trial and molecular data as well as information about the patient’s response to the first round of therapy. But lots of other factors are relevant to prognosis, including nutritional status, comorbidities, and the intuitive eyeball test of how a patient might do. The patient’s family, primary care provider, oncologist, the hospitalist, and the palliative care team will have perspectives of their own.
Molecular testing is now the norm in metastatic cancers
These days oncologists order molecular testing for most patients with metastatic carcinomas to determine eligibility for targeted therapy, suitability for participation in clinical trials, prognostication, and/or assistance in determining the site of origin if that’s unclear.
A single-pass fine needle aspiration biopsy doesn’t provide enough tissue for molecular testing. It’s therefore important to order initially a multipass fine needle aspiration to avoid the need for a repeat biopsy, which is uncomfortable for the patient and can delay diagnosis and treatment.
Dr. Brondfield advised waiting for molecular testing results to come in before trying to prognosticate in patients with a metastatic cancer for which targetable mutations might be present. Survival rates can vary substantially depending upon those test results. Take, for example, metastatic NSCLC: Just within the past year, clinical trials have been published reporting overall survival rates of 39 months in patients with treatable mutations in epidermal growth factor receptor, 42 months with anaplastic lymphoma kinase mutations, and 51 months in patients whose tumor signature features mutations in c-ros oncogene 1, as compared with 22 months with no targetable mutations in the KEYNOTE-189 trial.
“There’s a lot of heterogeneity around how metastatic tumors behave and respond to therapy. Not all metastatic cancers are the same,” the oncologist emphasized.
FROM HM20 VIRTUAL
Oleander extract for COVID-19? That’s a hard ‘no’ experts say

“Though renowned for its beauty and use in landscaping, this Mediterranean shrub is responsible for cases of accidental poisoning across the globe. All parts of the plant are poisonous,” Cassandra Quave, PhD, ethnobotanist and herbarium curator at Emory University, Atlanta, cautioned in an article in The Conversation, an independent, not-for-profit publication.
Oleandrin has properties similar to digoxin; the onset of toxicity occurs several hours after consumption.
The first symptoms of oleandrin poisoning may be gastrointestinal, such as nausea, vomiting, abdominal pain, diarrhea (which may contain blood), and loss of appetite.
After these first symptoms, the heart may be affected by tachyarrhythmia, bradyarrhythmia, premature ventricular contractions, or atrioventricular blockage. Xanthopsia (yellow vision), a burning sensation in the eyes, paralysis of the gastrointestinal tract, and respiratory symptoms may also occur.
Oleandrin poisoning may affect the central nervous system, as evidenced by drowsiness, tremors, seizures, collapse, and coma leading to death. When applied to the skin, oleander sap can cause skin irritations and allergic reactions characterized by dermatitis.
Diagnosis of oleandrin poisoning is mainly made on the basis of a description of the plant, how much of it was ingested, how much time has elapsed since ingestion, and symptoms. Confirmation of oleandrin in blood involves fluorescence polarization immunoassay, digoxin immunoassay, or liquid chromatography-electrospray tandem mass spectrometry.
Neither oleander nor oleandrin is approved by regulatory agencies as a prescription drug or dietary supplement.
In vitro study
Oleandrin for COVID-19 made headlines after President Trump met in the Oval Office with Andrew Whitney, vice chairman and director of Phoenix Biotechnology, along with Housing and Urban Development Secretary Ben Carson, MD, and MyPillow founder/CEO Mike Lindell, a strong supporter of Trump and an investor in the biotech company, to learn about oleandrin, which Whitney called a “cure” for COVID-19, Axios reported.
In an in vitro study, researchers from Phoenix Biotechnology and the University of Texas Medical Branch, Galveston, tested oleandrin against SARS-CoV-2 in cultured Vero cells.
“When administered both before and after virus infection, nanogram doses of oleandrin significantly inhibited replication by 45 to 3000-fold,” the researchers said in an article posted on bioRxiv, a free online archive and distribution service for unpublished preprints in the life sciences. The study has not been peer reviewed.
On the basis of these in vitro findings, the researchers said the plant extract has “potential to prevent disease and virus spread in persons recently exposed to SARS-CoV-2, as well as to prevent severe disease in persons at high risk.”
But it’s a far cry from test tube to human, one expert cautioned.
“This is an understatement: Care must be taken when inferring potential therapeutic benefits from in vitro antiviral effects,” Harlan Krumholz, MD, cardiologist and director, Yale New Haven Hospital Center for Outcomes Research and Evaluation, New Haven, Connecticut, told Medscape Medical News.
“There is a chasm between a single in vitro study and any use in humans outside of a protocol. People should be cautioned about that distance and the need [to] avoid such remedies unless part of a credible research project,” said Krumholz.
Yet Lindell told Axios that, in the Oval Office meeting, Trump expressed enthusiasm for the Food and Drug Administration to allow oleandrin to be marketed as a dietary supplement or approved for COVID-19.
“This is really just nonsense and a distraction,” Jonathan Reiner, MD, of George Washington University Medical Center, Washington, DC, said on CNN.
This article first appeared on Medscape.com.
“Though renowned for its beauty and use in landscaping, this Mediterranean shrub is responsible for cases of accidental poisoning across the globe. All parts of the plant are poisonous,” Cassandra Quave, PhD, ethnobotanist and herbarium curator at Emory University, Atlanta, cautioned in an article in The Conversation, an independent, not-for-profit publication.
Oleandrin has properties similar to digoxin; the onset of toxicity occurs several hours after consumption.
The first symptoms of oleandrin poisoning may be gastrointestinal, such as nausea, vomiting, abdominal pain, diarrhea (which may contain blood), and loss of appetite.
After these first symptoms, the heart may be affected by tachyarrhythmia, bradyarrhythmia, premature ventricular contractions, or atrioventricular blockage. Xanthopsia (yellow vision), a burning sensation in the eyes, paralysis of the gastrointestinal tract, and respiratory symptoms may also occur.
Oleandrin poisoning may affect the central nervous system, as evidenced by drowsiness, tremors, seizures, collapse, and coma leading to death. When applied to the skin, oleander sap can cause skin irritations and allergic reactions characterized by dermatitis.
Diagnosis of oleandrin poisoning is mainly made on the basis of a description of the plant, how much of it was ingested, how much time has elapsed since ingestion, and symptoms. Confirmation of oleandrin in blood involves fluorescence polarization immunoassay, digoxin immunoassay, or liquid chromatography-electrospray tandem mass spectrometry.
Neither oleander nor oleandrin is approved by regulatory agencies as a prescription drug or dietary supplement.
In vitro study
Oleandrin for COVID-19 made headlines after President Trump met in the Oval Office with Andrew Whitney, vice chairman and director of Phoenix Biotechnology, along with Housing and Urban Development Secretary Ben Carson, MD, and MyPillow founder/CEO Mike Lindell, a strong supporter of Trump and an investor in the biotech company, to learn about oleandrin, which Whitney called a “cure” for COVID-19, Axios reported.
In an in vitro study, researchers from Phoenix Biotechnology and the University of Texas Medical Branch, Galveston, tested oleandrin against SARS-CoV-2 in cultured Vero cells.
“When administered both before and after virus infection, nanogram doses of oleandrin significantly inhibited replication by 45 to 3000-fold,” the researchers said in an article posted on bioRxiv, a free online archive and distribution service for unpublished preprints in the life sciences. The study has not been peer reviewed.
On the basis of these in vitro findings, the researchers said the plant extract has “potential to prevent disease and virus spread in persons recently exposed to SARS-CoV-2, as well as to prevent severe disease in persons at high risk.”
But it’s a far cry from test tube to human, one expert cautioned.
“This is an understatement: Care must be taken when inferring potential therapeutic benefits from in vitro antiviral effects,” Harlan Krumholz, MD, cardiologist and director, Yale New Haven Hospital Center for Outcomes Research and Evaluation, New Haven, Connecticut, told Medscape Medical News.
“There is a chasm between a single in vitro study and any use in humans outside of a protocol. People should be cautioned about that distance and the need [to] avoid such remedies unless part of a credible research project,” said Krumholz.
Yet Lindell told Axios that, in the Oval Office meeting, Trump expressed enthusiasm for the Food and Drug Administration to allow oleandrin to be marketed as a dietary supplement or approved for COVID-19.
“This is really just nonsense and a distraction,” Jonathan Reiner, MD, of George Washington University Medical Center, Washington, DC, said on CNN.
This article first appeared on Medscape.com.
“Though renowned for its beauty and use in landscaping, this Mediterranean shrub is responsible for cases of accidental poisoning across the globe. All parts of the plant are poisonous,” Cassandra Quave, PhD, ethnobotanist and herbarium curator at Emory University, Atlanta, cautioned in an article in The Conversation, an independent, not-for-profit publication.
Oleandrin has properties similar to digoxin; the onset of toxicity occurs several hours after consumption.
The first symptoms of oleandrin poisoning may be gastrointestinal, such as nausea, vomiting, abdominal pain, diarrhea (which may contain blood), and loss of appetite.
After these first symptoms, the heart may be affected by tachyarrhythmia, bradyarrhythmia, premature ventricular contractions, or atrioventricular blockage. Xanthopsia (yellow vision), a burning sensation in the eyes, paralysis of the gastrointestinal tract, and respiratory symptoms may also occur.
Oleandrin poisoning may affect the central nervous system, as evidenced by drowsiness, tremors, seizures, collapse, and coma leading to death. When applied to the skin, oleander sap can cause skin irritations and allergic reactions characterized by dermatitis.
Diagnosis of oleandrin poisoning is mainly made on the basis of a description of the plant, how much of it was ingested, how much time has elapsed since ingestion, and symptoms. Confirmation of oleandrin in blood involves fluorescence polarization immunoassay, digoxin immunoassay, or liquid chromatography-electrospray tandem mass spectrometry.
Neither oleander nor oleandrin is approved by regulatory agencies as a prescription drug or dietary supplement.
In vitro study
Oleandrin for COVID-19 made headlines after President Trump met in the Oval Office with Andrew Whitney, vice chairman and director of Phoenix Biotechnology, along with Housing and Urban Development Secretary Ben Carson, MD, and MyPillow founder/CEO Mike Lindell, a strong supporter of Trump and an investor in the biotech company, to learn about oleandrin, which Whitney called a “cure” for COVID-19, Axios reported.
In an in vitro study, researchers from Phoenix Biotechnology and the University of Texas Medical Branch, Galveston, tested oleandrin against SARS-CoV-2 in cultured Vero cells.
“When administered both before and after virus infection, nanogram doses of oleandrin significantly inhibited replication by 45 to 3000-fold,” the researchers said in an article posted on bioRxiv, a free online archive and distribution service for unpublished preprints in the life sciences. The study has not been peer reviewed.
On the basis of these in vitro findings, the researchers said the plant extract has “potential to prevent disease and virus spread in persons recently exposed to SARS-CoV-2, as well as to prevent severe disease in persons at high risk.”
But it’s a far cry from test tube to human, one expert cautioned.
“This is an understatement: Care must be taken when inferring potential therapeutic benefits from in vitro antiviral effects,” Harlan Krumholz, MD, cardiologist and director, Yale New Haven Hospital Center for Outcomes Research and Evaluation, New Haven, Connecticut, told Medscape Medical News.
“There is a chasm between a single in vitro study and any use in humans outside of a protocol. People should be cautioned about that distance and the need [to] avoid such remedies unless part of a credible research project,” said Krumholz.
Yet Lindell told Axios that, in the Oval Office meeting, Trump expressed enthusiasm for the Food and Drug Administration to allow oleandrin to be marketed as a dietary supplement or approved for COVID-19.
“This is really just nonsense and a distraction,” Jonathan Reiner, MD, of George Washington University Medical Center, Washington, DC, said on CNN.
This article first appeared on Medscape.com.
Pulmonary artery denervation eases PAH after endarterectomy
Pulmonary artery denervation (PADN) provides persistent and clinically significant hemodynamic improvements in patients with persistent chronic thromboembolic hypertension (CTEPH) after pulmonary endarterectomy (PEA), according to a randomized, sham-controlled trial.
“PADN in patients with CTEPH after PEA was safe and effective,” according to an investigating team led by Alexander Romanov, MD, PhD.
The mean reduction in pulmonary vascular resistance (PVR) was 258 dyn/sec per cm–5 for those randomized to PADN versus 149 dyn/sec per cm–5 (P = .001) for those randomized to the sham procedure, according to the newly published findings.
For the 6-minute walk test (6MWT), the mean distance was 470 m for the experimental group versus 399 m (P = .03) for the controls.
Several secondary endpoints measuring hemodynamics also favored PADN relative to the sham procedure at 12 months. This included the relative increase in tricuspid annular systolic excursion (P = .03) and the increase in the right ventricular fraction area (P < .001).
A total of 50 patients with residual CTEPH for at least 6 months after PEA despite medical therapy were enrolled and randomized. Entry criteria included a mean pulmonary artery pressure (PAP) of 25 mm Hg or greater or PVR greater than 400 dyn/sec per cm–5 on right heart catheterization. Patients with comorbidities associated with a life expectancy of less than 1 year were excluded.
Those randomized to the sham group were treated with riociguat over the course of follow-up. This therapy was not offered to patients in the PADN group, but all patients were blinded to the procedure and told that riociguat might or might not be administered.
Following the procedure, participating clinicians, who were also blinded to the procedure, were instructed to provide standard therapies for heart failure, such beta-blockers, diuretics, or digoxin, as needed. All patients were placed on an oral anticoagulant.
At 12 months the mean PAP (26 vs. 35 mm Hg; P < .001) and the mean systolic PAP (46 vs. 54 mm Hg; P = .01) were significantly lower in the PADN group versus those who underwent a sham procedure.
About 52% of the PADN group versus 12% of the sham group were classified as responders by the definition of a PVR reduction of at least 150 dyn/sec per cm–5 and 6MWT improvement of at least 20%, compared with baseline, reported Dr. Romanov, of the E. Meshalkin National Medical Research Center, ministry of health, Novosibirsk, Russia, and coinvestigators.
Of the three deaths caused by heart failure over the course of follow-up, two occurred in the sham group. Of the eight hospitalizations for heart failure, seven (29% of the sham group) occurred among controls versus one in those treated with PADN (4% of this group; P = .049).
There was one groin hematoma at the puncture site in each group. Both resolved without any consequences prior to hospital discharge. There were no other significant procedure-related complications in either group.
Larger multicenter trials are needed to confirm these findings, according to both the trial investigators and Marius M. Hoeper, MD, who is charge of the pulmonary hypertension program at the Hannover (Germany) Medical School.
In an editorial that accompanied publication of these findings, Dr. Hoeper identified the small sample size of this study as one of its limitations, but he said the results are consistent with several other small studies associating pulmonary artery denervation with benefit in pulmonary hypertension.
“It appears as if we are currently witnessing the emergence of a new treatment option for various forms of pulmonary hypertension,” Dr. Hoeper wrote. In his critique of the study, he suggested that it would have been “more informative” if both groups were on background riociguat, but the data from this and other studies so far indicates that ablation to achieve denervation “is safe and feasible.”
The PADN technique used in this study might be relevant to the results. Dr. Hoeper noted that the investigators employed catheter tip–based electroanatomic mapping with a novel remote navigation system with three-dimensional imaging of the right ventricle and central pulmonary arteries.
“Apparently, this approach minimizes radiation exposure and provides precise location of ablation sites,” Dr. Hoeper observed. However, he called for direct comparisons of this tool to the guidance systems used in other studies.
In an interview, Dr. Hoeper acknowledged that it is not yet clear that a large-scale trial of pulmonary artery denervation for the indication evaluated in this study is coming. He noted several strategies in CTEPH are widely used without trials confirming a reduction in clinical events.
“Balloon pulmonary angioplasty for CTEPH has become an established treatment around the world without any randomized, controlled trial and without demonstration of improved outcomes. A couple of well-conducted observational trials might be sufficient to convince physicians to introduce PADN as well,” he said. If such studies associated PADN with “improvements in hemodynamics, exercise capacity, and patient-reported outcomes, it might be sufficient.”
Currently, Dr. Hoeper is most concerned about obtaining further evidence of safety, which he characterized as a “major issue.”
If a multicenter trial is conducted “the primary endpoint should be focused on clinical events,” according to Dr. Romanov, who was asked to comment on the next steps in validating PADN for the treatment of CTEPH-associated pulmonary hypertension persisting after endarterectomy.
“The mortality rate during 1-year long-term follow-up is not so high, but heart failure progression is a problem. So in my view, the primary endpoint should be a composite of death and heart failure hospitalization,” he said. He called for follow-up duration of 2-3 years.
Jonathan Steinberg, MD, director of cardiac clinical trials and education, Summit Medical Group, Montclair, N.J., also called a trial with hard endpoints, such as death, the ideal.
In the meantime, hemodynamic and functional measures “are still quite valuable and move the ball forward for this intervention,” he said in an interview. Senior author of this trial and principle investigator of the recent ERADICATE-AF trial, which evaluated renal denervation in preventing recurrence of atrial fibrillation (JAMA. 2020;323:248-55), Dr. Steinberg predicted, “I do indeed suspect we will see trials that are more accomplishable [than a large-scale, randomized, controlled trial] in the not too distant future.”
Dr. Romanov received funding from Biosense Webster. Dr. Hoeper has received fees for lectures and/or consultations from Acceleron, Actelion, Bayer, Janssen, Merck Sharp & Dohme, and Pfizer.
SOURCE: Romanov A et al. J Am Coll Cardiol. 2020 Aug 17;76:916-26.
Pulmonary artery denervation (PADN) provides persistent and clinically significant hemodynamic improvements in patients with persistent chronic thromboembolic hypertension (CTEPH) after pulmonary endarterectomy (PEA), according to a randomized, sham-controlled trial.
“PADN in patients with CTEPH after PEA was safe and effective,” according to an investigating team led by Alexander Romanov, MD, PhD.
The mean reduction in pulmonary vascular resistance (PVR) was 258 dyn/sec per cm–5 for those randomized to PADN versus 149 dyn/sec per cm–5 (P = .001) for those randomized to the sham procedure, according to the newly published findings.
For the 6-minute walk test (6MWT), the mean distance was 470 m for the experimental group versus 399 m (P = .03) for the controls.
Several secondary endpoints measuring hemodynamics also favored PADN relative to the sham procedure at 12 months. This included the relative increase in tricuspid annular systolic excursion (P = .03) and the increase in the right ventricular fraction area (P < .001).
A total of 50 patients with residual CTEPH for at least 6 months after PEA despite medical therapy were enrolled and randomized. Entry criteria included a mean pulmonary artery pressure (PAP) of 25 mm Hg or greater or PVR greater than 400 dyn/sec per cm–5 on right heart catheterization. Patients with comorbidities associated with a life expectancy of less than 1 year were excluded.
Those randomized to the sham group were treated with riociguat over the course of follow-up. This therapy was not offered to patients in the PADN group, but all patients were blinded to the procedure and told that riociguat might or might not be administered.
Following the procedure, participating clinicians, who were also blinded to the procedure, were instructed to provide standard therapies for heart failure, such beta-blockers, diuretics, or digoxin, as needed. All patients were placed on an oral anticoagulant.
At 12 months the mean PAP (26 vs. 35 mm Hg; P < .001) and the mean systolic PAP (46 vs. 54 mm Hg; P = .01) were significantly lower in the PADN group versus those who underwent a sham procedure.
About 52% of the PADN group versus 12% of the sham group were classified as responders by the definition of a PVR reduction of at least 150 dyn/sec per cm–5 and 6MWT improvement of at least 20%, compared with baseline, reported Dr. Romanov, of the E. Meshalkin National Medical Research Center, ministry of health, Novosibirsk, Russia, and coinvestigators.
Of the three deaths caused by heart failure over the course of follow-up, two occurred in the sham group. Of the eight hospitalizations for heart failure, seven (29% of the sham group) occurred among controls versus one in those treated with PADN (4% of this group; P = .049).
There was one groin hematoma at the puncture site in each group. Both resolved without any consequences prior to hospital discharge. There were no other significant procedure-related complications in either group.
Larger multicenter trials are needed to confirm these findings, according to both the trial investigators and Marius M. Hoeper, MD, who is charge of the pulmonary hypertension program at the Hannover (Germany) Medical School.
In an editorial that accompanied publication of these findings, Dr. Hoeper identified the small sample size of this study as one of its limitations, but he said the results are consistent with several other small studies associating pulmonary artery denervation with benefit in pulmonary hypertension.
“It appears as if we are currently witnessing the emergence of a new treatment option for various forms of pulmonary hypertension,” Dr. Hoeper wrote. In his critique of the study, he suggested that it would have been “more informative” if both groups were on background riociguat, but the data from this and other studies so far indicates that ablation to achieve denervation “is safe and feasible.”
The PADN technique used in this study might be relevant to the results. Dr. Hoeper noted that the investigators employed catheter tip–based electroanatomic mapping with a novel remote navigation system with three-dimensional imaging of the right ventricle and central pulmonary arteries.
“Apparently, this approach minimizes radiation exposure and provides precise location of ablation sites,” Dr. Hoeper observed. However, he called for direct comparisons of this tool to the guidance systems used in other studies.
In an interview, Dr. Hoeper acknowledged that it is not yet clear that a large-scale trial of pulmonary artery denervation for the indication evaluated in this study is coming. He noted several strategies in CTEPH are widely used without trials confirming a reduction in clinical events.
“Balloon pulmonary angioplasty for CTEPH has become an established treatment around the world without any randomized, controlled trial and without demonstration of improved outcomes. A couple of well-conducted observational trials might be sufficient to convince physicians to introduce PADN as well,” he said. If such studies associated PADN with “improvements in hemodynamics, exercise capacity, and patient-reported outcomes, it might be sufficient.”
Currently, Dr. Hoeper is most concerned about obtaining further evidence of safety, which he characterized as a “major issue.”
If a multicenter trial is conducted “the primary endpoint should be focused on clinical events,” according to Dr. Romanov, who was asked to comment on the next steps in validating PADN for the treatment of CTEPH-associated pulmonary hypertension persisting after endarterectomy.
“The mortality rate during 1-year long-term follow-up is not so high, but heart failure progression is a problem. So in my view, the primary endpoint should be a composite of death and heart failure hospitalization,” he said. He called for follow-up duration of 2-3 years.
Jonathan Steinberg, MD, director of cardiac clinical trials and education, Summit Medical Group, Montclair, N.J., also called a trial with hard endpoints, such as death, the ideal.
In the meantime, hemodynamic and functional measures “are still quite valuable and move the ball forward for this intervention,” he said in an interview. Senior author of this trial and principle investigator of the recent ERADICATE-AF trial, which evaluated renal denervation in preventing recurrence of atrial fibrillation (JAMA. 2020;323:248-55), Dr. Steinberg predicted, “I do indeed suspect we will see trials that are more accomplishable [than a large-scale, randomized, controlled trial] in the not too distant future.”
Dr. Romanov received funding from Biosense Webster. Dr. Hoeper has received fees for lectures and/or consultations from Acceleron, Actelion, Bayer, Janssen, Merck Sharp & Dohme, and Pfizer.
SOURCE: Romanov A et al. J Am Coll Cardiol. 2020 Aug 17;76:916-26.
Pulmonary artery denervation (PADN) provides persistent and clinically significant hemodynamic improvements in patients with persistent chronic thromboembolic hypertension (CTEPH) after pulmonary endarterectomy (PEA), according to a randomized, sham-controlled trial.
“PADN in patients with CTEPH after PEA was safe and effective,” according to an investigating team led by Alexander Romanov, MD, PhD.
The mean reduction in pulmonary vascular resistance (PVR) was 258 dyn/sec per cm–5 for those randomized to PADN versus 149 dyn/sec per cm–5 (P = .001) for those randomized to the sham procedure, according to the newly published findings.
For the 6-minute walk test (6MWT), the mean distance was 470 m for the experimental group versus 399 m (P = .03) for the controls.
Several secondary endpoints measuring hemodynamics also favored PADN relative to the sham procedure at 12 months. This included the relative increase in tricuspid annular systolic excursion (P = .03) and the increase in the right ventricular fraction area (P < .001).
A total of 50 patients with residual CTEPH for at least 6 months after PEA despite medical therapy were enrolled and randomized. Entry criteria included a mean pulmonary artery pressure (PAP) of 25 mm Hg or greater or PVR greater than 400 dyn/sec per cm–5 on right heart catheterization. Patients with comorbidities associated with a life expectancy of less than 1 year were excluded.
Those randomized to the sham group were treated with riociguat over the course of follow-up. This therapy was not offered to patients in the PADN group, but all patients were blinded to the procedure and told that riociguat might or might not be administered.
Following the procedure, participating clinicians, who were also blinded to the procedure, were instructed to provide standard therapies for heart failure, such beta-blockers, diuretics, or digoxin, as needed. All patients were placed on an oral anticoagulant.
At 12 months the mean PAP (26 vs. 35 mm Hg; P < .001) and the mean systolic PAP (46 vs. 54 mm Hg; P = .01) were significantly lower in the PADN group versus those who underwent a sham procedure.
About 52% of the PADN group versus 12% of the sham group were classified as responders by the definition of a PVR reduction of at least 150 dyn/sec per cm–5 and 6MWT improvement of at least 20%, compared with baseline, reported Dr. Romanov, of the E. Meshalkin National Medical Research Center, ministry of health, Novosibirsk, Russia, and coinvestigators.
Of the three deaths caused by heart failure over the course of follow-up, two occurred in the sham group. Of the eight hospitalizations for heart failure, seven (29% of the sham group) occurred among controls versus one in those treated with PADN (4% of this group; P = .049).
There was one groin hematoma at the puncture site in each group. Both resolved without any consequences prior to hospital discharge. There were no other significant procedure-related complications in either group.
Larger multicenter trials are needed to confirm these findings, according to both the trial investigators and Marius M. Hoeper, MD, who is charge of the pulmonary hypertension program at the Hannover (Germany) Medical School.
In an editorial that accompanied publication of these findings, Dr. Hoeper identified the small sample size of this study as one of its limitations, but he said the results are consistent with several other small studies associating pulmonary artery denervation with benefit in pulmonary hypertension.
“It appears as if we are currently witnessing the emergence of a new treatment option for various forms of pulmonary hypertension,” Dr. Hoeper wrote. In his critique of the study, he suggested that it would have been “more informative” if both groups were on background riociguat, but the data from this and other studies so far indicates that ablation to achieve denervation “is safe and feasible.”
The PADN technique used in this study might be relevant to the results. Dr. Hoeper noted that the investigators employed catheter tip–based electroanatomic mapping with a novel remote navigation system with three-dimensional imaging of the right ventricle and central pulmonary arteries.
“Apparently, this approach minimizes radiation exposure and provides precise location of ablation sites,” Dr. Hoeper observed. However, he called for direct comparisons of this tool to the guidance systems used in other studies.
In an interview, Dr. Hoeper acknowledged that it is not yet clear that a large-scale trial of pulmonary artery denervation for the indication evaluated in this study is coming. He noted several strategies in CTEPH are widely used without trials confirming a reduction in clinical events.
“Balloon pulmonary angioplasty for CTEPH has become an established treatment around the world without any randomized, controlled trial and without demonstration of improved outcomes. A couple of well-conducted observational trials might be sufficient to convince physicians to introduce PADN as well,” he said. If such studies associated PADN with “improvements in hemodynamics, exercise capacity, and patient-reported outcomes, it might be sufficient.”
Currently, Dr. Hoeper is most concerned about obtaining further evidence of safety, which he characterized as a “major issue.”
If a multicenter trial is conducted “the primary endpoint should be focused on clinical events,” according to Dr. Romanov, who was asked to comment on the next steps in validating PADN for the treatment of CTEPH-associated pulmonary hypertension persisting after endarterectomy.
“The mortality rate during 1-year long-term follow-up is not so high, but heart failure progression is a problem. So in my view, the primary endpoint should be a composite of death and heart failure hospitalization,” he said. He called for follow-up duration of 2-3 years.
Jonathan Steinberg, MD, director of cardiac clinical trials and education, Summit Medical Group, Montclair, N.J., also called a trial with hard endpoints, such as death, the ideal.
In the meantime, hemodynamic and functional measures “are still quite valuable and move the ball forward for this intervention,” he said in an interview. Senior author of this trial and principle investigator of the recent ERADICATE-AF trial, which evaluated renal denervation in preventing recurrence of atrial fibrillation (JAMA. 2020;323:248-55), Dr. Steinberg predicted, “I do indeed suspect we will see trials that are more accomplishable [than a large-scale, randomized, controlled trial] in the not too distant future.”
Dr. Romanov received funding from Biosense Webster. Dr. Hoeper has received fees for lectures and/or consultations from Acceleron, Actelion, Bayer, Janssen, Merck Sharp & Dohme, and Pfizer.
SOURCE: Romanov A et al. J Am Coll Cardiol. 2020 Aug 17;76:916-26.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Machine learning shows ability to predict diastolic dysfunction with ECG
A machine-learning model that uses readily available clinical and electrocardiography data may have the potential to identify left ventricular (LV) diastolic dysfunction, a key biomarker in predicting heart failure, without echocardiography, but a workable clinical platform is still far off, a team of North American researchers reported.
“This cost-effective strategy may be a valuable first clinical step for assessing the presence of LV dysfunction and may potentially aid in the early diagnosis and management of heart failure patients,” Nobuyuki Kagiyama, MD, PhD, of West Virginia University, Morgantown, and colleagues, wrote in the Journal of the American Academy of Cardiology.
The researchers reported on a multicenter, prospective study that evaluated 1,202 patients from three centers in the United States and one in Canada. To develop machine-learning models, the study pooled 814 patients from the U.S. institutions as an internal cohort. They were then randomly divided into a training set and an internal test set on an 80:20 basis (651 and 163). The 388 Canadian patients were reserved as an external set to test the model.
All patients had 12-lead ECG and simultaneous body surface signal-processed ECG (spECG) along with comprehensive two-dimensional Doppler ECG on the same day.
How the model works
The machine-learning model estimated echocardiographic LV relaxation velocities (e’) values using traditional ECG and spECG features. The model also took into account 10 basic clinical features: age; sex; systolic and diastolic blood pressure; and comorbid conditions such as cerebrovascular and cardiovascular disease, diabetes, hypertension, dyslipidemia, and chronic kidney disease.
Patient characteristics were starkly different between the internal (United States) and external (Canadian) cohorts, with the latter being 10 years older on average (65 vs. 44; P < .001), predominantly male (58.2% vs. 47.3%; P < .001) and with significantly lower rates of coronary artery disease (1.8% vs. 21.1%; P < .001), although average blood pressure was similar between the two groups.
The study used area under the curve (AUC) to calculate the predictability of the machine-learning estimated e’ values versus the guideline-based reduced e’, finding close correlation between the internal (AUC, 0.83; sensitivity, 78%; specificity, 77%; negative predictive value, 73%; and positive predictive value, 82%) and external test sets (AUC, 0.84; sensitivity, 90%; specificity, 61%; NPV, 81%; and PPV, 77%).
Similar variations between the two cohorts were reported for global LV diastolic dysfunction and reduced LV ejection fraction.
The final model used 18 features in all, including 3 clinical features (age, dyslipidemia, and hypertension), 7 scores from spECG features, and 8 from traditional ECG features.
Interpreting the results
Dr. Kagiyama and colleagues noted that, because impaired myocardial relaxation is an early sign of cardiac tissue deterioration, screening for it can aid in early detection of subclinical LVDD and earlier treatment for hypertension and diabetes. But they acknowledged that further studies are needed.
In an invited editorial, Khurram Nasir, MD, MPH, MSc, of Houston Methodist DeBakey Heart and Vascular Center and Rohan Khera, MD, MS, of Yale University, New Haven, Conn., wrote that the machine-learning model has a way to go.
They noted that the 73%-77% accuracy of the model in identifying diastolic dysfunction impedes its imminent use. “Although we are excited about the prospects of such developments, we hold out for better evidence for their actual use,” they wrote, adding that the algorithms have limited use in the clinic because most patients already get “definitive testing” if they need it.
Developing a machine-learning model that obviates the need for ECG for evaluating LV diastolic dysfunction seems dubious at this time, said Luigi Di Biase, MD, PhD, section head of electrophysiology and director of arrhythmia services at Montefiore Medical Center and professor at Albert Einstein College of Medicine, both in New York. “The echo is not a difficult test. It’s the most proven usable tool that we have in cardiology because it’s easy to reproduce, low cost, and noninvasive – so we have all that we want in medicine.”
But machine learning does have potential, added Dr. Di Biase, who’s also a member of the American College of Cardiology’s Electrophysiology Section Leadership Council. “If this application could predict the people that would develop diastolic dysfunction that leads to heart failure – because an echo at that time may be negative but there may be other features that tell me this patient will develop disease – then it would have a much different clinical impact.”
The National Science Foundation provided funding for the study. Heart Test Laboratories, doing business as Heart Sciences, provided funding and spECG devices. Dr. Kagiyama reported receiving a research grant from Hitachi Healthcare. A coauthor disclosed financial relationships with Heart Sciences, Ultronics, and Kencor Health.
Dr. Nasir, Dr. Khera, and Dr. Di Biase have no relevant financial relationships to disclose.
SOURCE: Kagiyama N et al. J Am Coll Cardiol. 2020;76:930-41.
A machine-learning model that uses readily available clinical and electrocardiography data may have the potential to identify left ventricular (LV) diastolic dysfunction, a key biomarker in predicting heart failure, without echocardiography, but a workable clinical platform is still far off, a team of North American researchers reported.
“This cost-effective strategy may be a valuable first clinical step for assessing the presence of LV dysfunction and may potentially aid in the early diagnosis and management of heart failure patients,” Nobuyuki Kagiyama, MD, PhD, of West Virginia University, Morgantown, and colleagues, wrote in the Journal of the American Academy of Cardiology.
The researchers reported on a multicenter, prospective study that evaluated 1,202 patients from three centers in the United States and one in Canada. To develop machine-learning models, the study pooled 814 patients from the U.S. institutions as an internal cohort. They were then randomly divided into a training set and an internal test set on an 80:20 basis (651 and 163). The 388 Canadian patients were reserved as an external set to test the model.
All patients had 12-lead ECG and simultaneous body surface signal-processed ECG (spECG) along with comprehensive two-dimensional Doppler ECG on the same day.
How the model works
The machine-learning model estimated echocardiographic LV relaxation velocities (e’) values using traditional ECG and spECG features. The model also took into account 10 basic clinical features: age; sex; systolic and diastolic blood pressure; and comorbid conditions such as cerebrovascular and cardiovascular disease, diabetes, hypertension, dyslipidemia, and chronic kidney disease.
Patient characteristics were starkly different between the internal (United States) and external (Canadian) cohorts, with the latter being 10 years older on average (65 vs. 44; P < .001), predominantly male (58.2% vs. 47.3%; P < .001) and with significantly lower rates of coronary artery disease (1.8% vs. 21.1%; P < .001), although average blood pressure was similar between the two groups.
The study used area under the curve (AUC) to calculate the predictability of the machine-learning estimated e’ values versus the guideline-based reduced e’, finding close correlation between the internal (AUC, 0.83; sensitivity, 78%; specificity, 77%; negative predictive value, 73%; and positive predictive value, 82%) and external test sets (AUC, 0.84; sensitivity, 90%; specificity, 61%; NPV, 81%; and PPV, 77%).
Similar variations between the two cohorts were reported for global LV diastolic dysfunction and reduced LV ejection fraction.
The final model used 18 features in all, including 3 clinical features (age, dyslipidemia, and hypertension), 7 scores from spECG features, and 8 from traditional ECG features.
Interpreting the results
Dr. Kagiyama and colleagues noted that, because impaired myocardial relaxation is an early sign of cardiac tissue deterioration, screening for it can aid in early detection of subclinical LVDD and earlier treatment for hypertension and diabetes. But they acknowledged that further studies are needed.
In an invited editorial, Khurram Nasir, MD, MPH, MSc, of Houston Methodist DeBakey Heart and Vascular Center and Rohan Khera, MD, MS, of Yale University, New Haven, Conn., wrote that the machine-learning model has a way to go.
They noted that the 73%-77% accuracy of the model in identifying diastolic dysfunction impedes its imminent use. “Although we are excited about the prospects of such developments, we hold out for better evidence for their actual use,” they wrote, adding that the algorithms have limited use in the clinic because most patients already get “definitive testing” if they need it.
Developing a machine-learning model that obviates the need for ECG for evaluating LV diastolic dysfunction seems dubious at this time, said Luigi Di Biase, MD, PhD, section head of electrophysiology and director of arrhythmia services at Montefiore Medical Center and professor at Albert Einstein College of Medicine, both in New York. “The echo is not a difficult test. It’s the most proven usable tool that we have in cardiology because it’s easy to reproduce, low cost, and noninvasive – so we have all that we want in medicine.”
But machine learning does have potential, added Dr. Di Biase, who’s also a member of the American College of Cardiology’s Electrophysiology Section Leadership Council. “If this application could predict the people that would develop diastolic dysfunction that leads to heart failure – because an echo at that time may be negative but there may be other features that tell me this patient will develop disease – then it would have a much different clinical impact.”
The National Science Foundation provided funding for the study. Heart Test Laboratories, doing business as Heart Sciences, provided funding and spECG devices. Dr. Kagiyama reported receiving a research grant from Hitachi Healthcare. A coauthor disclosed financial relationships with Heart Sciences, Ultronics, and Kencor Health.
Dr. Nasir, Dr. Khera, and Dr. Di Biase have no relevant financial relationships to disclose.
SOURCE: Kagiyama N et al. J Am Coll Cardiol. 2020;76:930-41.
A machine-learning model that uses readily available clinical and electrocardiography data may have the potential to identify left ventricular (LV) diastolic dysfunction, a key biomarker in predicting heart failure, without echocardiography, but a workable clinical platform is still far off, a team of North American researchers reported.
“This cost-effective strategy may be a valuable first clinical step for assessing the presence of LV dysfunction and may potentially aid in the early diagnosis and management of heart failure patients,” Nobuyuki Kagiyama, MD, PhD, of West Virginia University, Morgantown, and colleagues, wrote in the Journal of the American Academy of Cardiology.
The researchers reported on a multicenter, prospective study that evaluated 1,202 patients from three centers in the United States and one in Canada. To develop machine-learning models, the study pooled 814 patients from the U.S. institutions as an internal cohort. They were then randomly divided into a training set and an internal test set on an 80:20 basis (651 and 163). The 388 Canadian patients were reserved as an external set to test the model.
All patients had 12-lead ECG and simultaneous body surface signal-processed ECG (spECG) along with comprehensive two-dimensional Doppler ECG on the same day.
How the model works
The machine-learning model estimated echocardiographic LV relaxation velocities (e’) values using traditional ECG and spECG features. The model also took into account 10 basic clinical features: age; sex; systolic and diastolic blood pressure; and comorbid conditions such as cerebrovascular and cardiovascular disease, diabetes, hypertension, dyslipidemia, and chronic kidney disease.
Patient characteristics were starkly different between the internal (United States) and external (Canadian) cohorts, with the latter being 10 years older on average (65 vs. 44; P < .001), predominantly male (58.2% vs. 47.3%; P < .001) and with significantly lower rates of coronary artery disease (1.8% vs. 21.1%; P < .001), although average blood pressure was similar between the two groups.
The study used area under the curve (AUC) to calculate the predictability of the machine-learning estimated e’ values versus the guideline-based reduced e’, finding close correlation between the internal (AUC, 0.83; sensitivity, 78%; specificity, 77%; negative predictive value, 73%; and positive predictive value, 82%) and external test sets (AUC, 0.84; sensitivity, 90%; specificity, 61%; NPV, 81%; and PPV, 77%).
Similar variations between the two cohorts were reported for global LV diastolic dysfunction and reduced LV ejection fraction.
The final model used 18 features in all, including 3 clinical features (age, dyslipidemia, and hypertension), 7 scores from spECG features, and 8 from traditional ECG features.
Interpreting the results
Dr. Kagiyama and colleagues noted that, because impaired myocardial relaxation is an early sign of cardiac tissue deterioration, screening for it can aid in early detection of subclinical LVDD and earlier treatment for hypertension and diabetes. But they acknowledged that further studies are needed.
In an invited editorial, Khurram Nasir, MD, MPH, MSc, of Houston Methodist DeBakey Heart and Vascular Center and Rohan Khera, MD, MS, of Yale University, New Haven, Conn., wrote that the machine-learning model has a way to go.
They noted that the 73%-77% accuracy of the model in identifying diastolic dysfunction impedes its imminent use. “Although we are excited about the prospects of such developments, we hold out for better evidence for their actual use,” they wrote, adding that the algorithms have limited use in the clinic because most patients already get “definitive testing” if they need it.
Developing a machine-learning model that obviates the need for ECG for evaluating LV diastolic dysfunction seems dubious at this time, said Luigi Di Biase, MD, PhD, section head of electrophysiology and director of arrhythmia services at Montefiore Medical Center and professor at Albert Einstein College of Medicine, both in New York. “The echo is not a difficult test. It’s the most proven usable tool that we have in cardiology because it’s easy to reproduce, low cost, and noninvasive – so we have all that we want in medicine.”
But machine learning does have potential, added Dr. Di Biase, who’s also a member of the American College of Cardiology’s Electrophysiology Section Leadership Council. “If this application could predict the people that would develop diastolic dysfunction that leads to heart failure – because an echo at that time may be negative but there may be other features that tell me this patient will develop disease – then it would have a much different clinical impact.”
The National Science Foundation provided funding for the study. Heart Test Laboratories, doing business as Heart Sciences, provided funding and spECG devices. Dr. Kagiyama reported receiving a research grant from Hitachi Healthcare. A coauthor disclosed financial relationships with Heart Sciences, Ultronics, and Kencor Health.
Dr. Nasir, Dr. Khera, and Dr. Di Biase have no relevant financial relationships to disclose.
SOURCE: Kagiyama N et al. J Am Coll Cardiol. 2020;76:930-41.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Ulcerated hand lesion

While the eroded scabbed area in a raised lesion with pearly borders raised the suspicion of basal cell carcinoma (BCC), a superficial shave biopsy revealed that this was sclerosing basosquamous carcinoma.
Basosquamous carcinoma is an uncommon form of BCC. It features nests/clusters of basaloid cells arising from the basal layer of the epidermis (a hallmark of BCC), as well as keratinization, which is seen in squamous cell carcinoma (SCC). Basosquamous carcinoma occurs in the same areas as more common types of nonmelanoma skin cancers (NMSCs)—that is, sun-exposed areas. Not surprisingly, then, the highest risk areas for basosquamous carcinoma are the face, hands, arms, scalp (in those without hair), and back. People with occupational or intentional sun exposure through tanning have even higher rates of NMSC than the average population; patients using tanning booths exposing their entire bodies warrant more extensive physical exams.
Basosquamous carcinoma is one of the subtypes of BCC that is associated with a higher risk of local invasion, recurrence, and metastasis. Treatment with Mohs micrographic surgery (MMS) reduces the risk of recurrence to 4% (from 12% to 51%). Even with MMS, basosquamous carcinoma can be particularly challenging. It may require additional excision stages, as well as a larger than anticipated excision size at the time of surgery.
In this case, the patient was referred for MMS due to the high-risk nature of this lesion.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Garcia C, Poletti E, Crowson AN. Basosquamous carcinoma. J Am Acad Dermatol. 2009;60:137-143.
While the eroded scabbed area in a raised lesion with pearly borders raised the suspicion of basal cell carcinoma (BCC), a superficial shave biopsy revealed that this was sclerosing basosquamous carcinoma.
Basosquamous carcinoma is an uncommon form of BCC. It features nests/clusters of basaloid cells arising from the basal layer of the epidermis (a hallmark of BCC), as well as keratinization, which is seen in squamous cell carcinoma (SCC). Basosquamous carcinoma occurs in the same areas as more common types of nonmelanoma skin cancers (NMSCs)—that is, sun-exposed areas. Not surprisingly, then, the highest risk areas for basosquamous carcinoma are the face, hands, arms, scalp (in those without hair), and back. People with occupational or intentional sun exposure through tanning have even higher rates of NMSC than the average population; patients using tanning booths exposing their entire bodies warrant more extensive physical exams.
Basosquamous carcinoma is one of the subtypes of BCC that is associated with a higher risk of local invasion, recurrence, and metastasis. Treatment with Mohs micrographic surgery (MMS) reduces the risk of recurrence to 4% (from 12% to 51%). Even with MMS, basosquamous carcinoma can be particularly challenging. It may require additional excision stages, as well as a larger than anticipated excision size at the time of surgery.
In this case, the patient was referred for MMS due to the high-risk nature of this lesion.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
While the eroded scabbed area in a raised lesion with pearly borders raised the suspicion of basal cell carcinoma (BCC), a superficial shave biopsy revealed that this was sclerosing basosquamous carcinoma.
Basosquamous carcinoma is an uncommon form of BCC. It features nests/clusters of basaloid cells arising from the basal layer of the epidermis (a hallmark of BCC), as well as keratinization, which is seen in squamous cell carcinoma (SCC). Basosquamous carcinoma occurs in the same areas as more common types of nonmelanoma skin cancers (NMSCs)—that is, sun-exposed areas. Not surprisingly, then, the highest risk areas for basosquamous carcinoma are the face, hands, arms, scalp (in those without hair), and back. People with occupational or intentional sun exposure through tanning have even higher rates of NMSC than the average population; patients using tanning booths exposing their entire bodies warrant more extensive physical exams.
Basosquamous carcinoma is one of the subtypes of BCC that is associated with a higher risk of local invasion, recurrence, and metastasis. Treatment with Mohs micrographic surgery (MMS) reduces the risk of recurrence to 4% (from 12% to 51%). Even with MMS, basosquamous carcinoma can be particularly challenging. It may require additional excision stages, as well as a larger than anticipated excision size at the time of surgery.
In this case, the patient was referred for MMS due to the high-risk nature of this lesion.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Garcia C, Poletti E, Crowson AN. Basosquamous carcinoma. J Am Acad Dermatol. 2009;60:137-143.
Garcia C, Poletti E, Crowson AN. Basosquamous carcinoma. J Am Acad Dermatol. 2009;60:137-143.

Canakinumab (Ilaris) tapering tested in systemic JIA trial
A trial that tested two ways of tapering canakinumab (Ilaris) monotherapy in children with systemic juvenile idiopathic arthritis (sJIA) showed that both approaches might be feasible, but the lack of a control arm means that more data are needed before putting either into routine clinical practice.

Pierre Quartier, MD, and associates reported in Arthritis & Rheumatology. That’s with the proviso that children are taking canakinumab at the recommended dose of 4 mg/kg every 4 weeks and achieve clinical remission before any tapering is started.
The researchers found that 70%-80% of children who were in complete clinical remission (CR) maintained this for 6 months after the first dose-tapering step had been taken. However, at least one-fifth of study participants experienced a disease flare during treatment withdrawal, and only one-third were able to discontinue treatment altogether, which suggests continued treatment is needed.
“The results are a step in the right direction,” commented Athimaleipet Ramanan, MBBS, a consultant pediatric rheumatologist who was not involved in the study. They “offer the first vision of whether we can actually taper a medication” in sJIA because “until now we have not had any evidence for this,” he added.
“What we really need to know, which the study doesn’t tell you, is: Is decreasing the dose better than stopping?” said Dr. Ramanan, of the Bristol (England) Royal Hospital for Children.
Another thing that is important to know is: Does reducing the dose lead to the development of anti-drug antibodies? he said.

“There were some concerns that, when you give less of a monoclonal antibody, you might get more neutralizing anti-drug antibodies or you might make a drug more immunogenic,” he said. These data perhaps suggest that this isn’t the case because only one child on one occasion had detectable non-neutralizing anti-drug antibodies during the entire study, and that was 11 weeks after the last dose of canakinumab had been given.
Study results and design
Canakinumab is a monoclonal antibody that inhibits interleukin-1 (IL-1) that has been approved in the United States and Europe for the treatment of sJIA since 2013. Reducing a child’s exposure to canakinumab once their disease is under control is an attractive proposition given the treat-to-target approach used increasingly throughout modern rheumatologic practice. It could also help reduce the cost of what is an expensive treatment, compared with other available options for sJIA such as the interleukin-6 inhibitor tocilizumab (Actemra) or another IL-1 inhibitor anakinra (Kineret), which is not FDA-approved for use in sJIA and requires weekly injections.
“We don’t want to the disease to reappear, to flare, but we also don’t want to overuse treatments in these patients,” explained Dr. Quartier of Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris in an interview.
The study he coconducted, given the acronym B-SPECIFIC-4 Patients, was an open-label study that consisted of two parts. In part 1, 182 children were given subcutaneous canakinumab 4 mg/kg every 4 weeks. In part 2, 76 children who were in complete CR after canakinumab treatment were randomized to one of two tapering strategies. In one arm, the dose of canakinumab was reduced from 4 mg/kg to 2 mg/kg given every 4 weeks before eventually discontinuing treatment, and in other arm the duration between dosing was increased by 4-week intervals, from 4 to 8 weeks, then 12 weeks, then discontinuation.
If children were taking glucocorticoids or methotrexate, the treating physicians were encouraged to stop these medications if possible, with 34%-39% and 42%-59% of children, respectively, being able to do so. The rate depended on whether children had received canakinumab before entering the study because some had been recruited from a long-term extension study while others were naive to the biologic; all had inactive disease at study entry.
This was the first study to evaluate the effectiveness of canakinumab in enabling the discontinuation of methotrexate, but the primary objective was to assess whether more than 40% of randomized patients in either discontinuation arm remained in CR for 24 weeks after the first step of discontinuation. This was achieved in 71% of children who were in the dose-reduction arm and in 84% of children in the dose-prolongation arm (P ≤ .0001 for each arm vs. 40%).
Prior exposure to canakinumab did not seem to affect the maintenance of CR, but it was also found that more children who maintained CR at their second but not their first attempt at tapering were in CR than those who were still in CR at the first step (76% and 89% in the dose-reduction and dose-prolongation arms, respectively).
Among the two dosing regimens, failure occurred in 18% of children in the dose-reduction arm at the first step, 10% at the second, and 8% at the third, whereas 2.7% of children in the dose-prolongation arm experienced regimen failure at the first step, 6.1% at the second, and 15% at the third.
No substantial difference between the two tapering approaches was observed because the study was not powered to look at this. There was also no control arm, such as a group continuing treatment while the other groups tapered, or as Dr. Ramanan had pointed out, stopping canakinumab altogether.
“As long as the treatment was continued, even at very low dosage, most patients remained in inactive disease,” Dr. Quartier said. He added, however, that only a minority of patients could stop treatment completely. Treatment should not be stopped abruptly, he advised, because this was associated with a substantial number of disease flares that needed treatment to be reinstated.
These findings suggest that “a certain level of sustained inhibition of the IL-1 pathway seems important for the maintenance of CR in most sJIA patients,” Dr. Quartier and coinvestigators wrote in their article.
They added: “We believe that these results are relevant for clinical practice, particularly for designing personalized tapering strategies that can allow an adequate control of disease while minimizing the side effects of certain medications, notably glucocorticoids.”
The study was funded by Novartis in collaboration with the Pediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group. Dr. Quartier was an investigator for the trial and has received research and consultancy fees from Novartis, among other pharmaceutical companies. Two of his coauthors are employees of Novartis. Dr. Ramanan has acted as an investigator in prior canakinumab trials and has received consultancy fees from Novartis and multiple other companies.
SOURCE: Quartier P et al. Arthritis Rheumatol. 2020 Aug 11. doi: 10.1002/art.41488.
A trial that tested two ways of tapering canakinumab (Ilaris) monotherapy in children with systemic juvenile idiopathic arthritis (sJIA) showed that both approaches might be feasible, but the lack of a control arm means that more data are needed before putting either into routine clinical practice.
Pierre Quartier, MD, and associates reported in Arthritis & Rheumatology. That’s with the proviso that children are taking canakinumab at the recommended dose of 4 mg/kg every 4 weeks and achieve clinical remission before any tapering is started.
The researchers found that 70%-80% of children who were in complete clinical remission (CR) maintained this for 6 months after the first dose-tapering step had been taken. However, at least one-fifth of study participants experienced a disease flare during treatment withdrawal, and only one-third were able to discontinue treatment altogether, which suggests continued treatment is needed.
“The results are a step in the right direction,” commented Athimaleipet Ramanan, MBBS, a consultant pediatric rheumatologist who was not involved in the study. They “offer the first vision of whether we can actually taper a medication” in sJIA because “until now we have not had any evidence for this,” he added.
“What we really need to know, which the study doesn’t tell you, is: Is decreasing the dose better than stopping?” said Dr. Ramanan, of the Bristol (England) Royal Hospital for Children.
Another thing that is important to know is: Does reducing the dose lead to the development of anti-drug antibodies? he said.
“There were some concerns that, when you give less of a monoclonal antibody, you might get more neutralizing anti-drug antibodies or you might make a drug more immunogenic,” he said. These data perhaps suggest that this isn’t the case because only one child on one occasion had detectable non-neutralizing anti-drug antibodies during the entire study, and that was 11 weeks after the last dose of canakinumab had been given.
Study results and design
Canakinumab is a monoclonal antibody that inhibits interleukin-1 (IL-1) that has been approved in the United States and Europe for the treatment of sJIA since 2013. Reducing a child’s exposure to canakinumab once their disease is under control is an attractive proposition given the treat-to-target approach used increasingly throughout modern rheumatologic practice. It could also help reduce the cost of what is an expensive treatment, compared with other available options for sJIA such as the interleukin-6 inhibitor tocilizumab (Actemra) or another IL-1 inhibitor anakinra (Kineret), which is not FDA-approved for use in sJIA and requires weekly injections.
“We don’t want to the disease to reappear, to flare, but we also don’t want to overuse treatments in these patients,” explained Dr. Quartier of Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris in an interview.
The study he coconducted, given the acronym B-SPECIFIC-4 Patients, was an open-label study that consisted of two parts. In part 1, 182 children were given subcutaneous canakinumab 4 mg/kg every 4 weeks. In part 2, 76 children who were in complete CR after canakinumab treatment were randomized to one of two tapering strategies. In one arm, the dose of canakinumab was reduced from 4 mg/kg to 2 mg/kg given every 4 weeks before eventually discontinuing treatment, and in other arm the duration between dosing was increased by 4-week intervals, from 4 to 8 weeks, then 12 weeks, then discontinuation.
If children were taking glucocorticoids or methotrexate, the treating physicians were encouraged to stop these medications if possible, with 34%-39% and 42%-59% of children, respectively, being able to do so. The rate depended on whether children had received canakinumab before entering the study because some had been recruited from a long-term extension study while others were naive to the biologic; all had inactive disease at study entry.
This was the first study to evaluate the effectiveness of canakinumab in enabling the discontinuation of methotrexate, but the primary objective was to assess whether more than 40% of randomized patients in either discontinuation arm remained in CR for 24 weeks after the first step of discontinuation. This was achieved in 71% of children who were in the dose-reduction arm and in 84% of children in the dose-prolongation arm (P ≤ .0001 for each arm vs. 40%).
Prior exposure to canakinumab did not seem to affect the maintenance of CR, but it was also found that more children who maintained CR at their second but not their first attempt at tapering were in CR than those who were still in CR at the first step (76% and 89% in the dose-reduction and dose-prolongation arms, respectively).
Among the two dosing regimens, failure occurred in 18% of children in the dose-reduction arm at the first step, 10% at the second, and 8% at the third, whereas 2.7% of children in the dose-prolongation arm experienced regimen failure at the first step, 6.1% at the second, and 15% at the third.
No substantial difference between the two tapering approaches was observed because the study was not powered to look at this. There was also no control arm, such as a group continuing treatment while the other groups tapered, or as Dr. Ramanan had pointed out, stopping canakinumab altogether.
“As long as the treatment was continued, even at very low dosage, most patients remained in inactive disease,” Dr. Quartier said. He added, however, that only a minority of patients could stop treatment completely. Treatment should not be stopped abruptly, he advised, because this was associated with a substantial number of disease flares that needed treatment to be reinstated.
These findings suggest that “a certain level of sustained inhibition of the IL-1 pathway seems important for the maintenance of CR in most sJIA patients,” Dr. Quartier and coinvestigators wrote in their article.
They added: “We believe that these results are relevant for clinical practice, particularly for designing personalized tapering strategies that can allow an adequate control of disease while minimizing the side effects of certain medications, notably glucocorticoids.”
The study was funded by Novartis in collaboration with the Pediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group. Dr. Quartier was an investigator for the trial and has received research and consultancy fees from Novartis, among other pharmaceutical companies. Two of his coauthors are employees of Novartis. Dr. Ramanan has acted as an investigator in prior canakinumab trials and has received consultancy fees from Novartis and multiple other companies.
SOURCE: Quartier P et al. Arthritis Rheumatol. 2020 Aug 11. doi: 10.1002/art.41488.
A trial that tested two ways of tapering canakinumab (Ilaris) monotherapy in children with systemic juvenile idiopathic arthritis (sJIA) showed that both approaches might be feasible, but the lack of a control arm means that more data are needed before putting either into routine clinical practice.
Pierre Quartier, MD, and associates reported in Arthritis & Rheumatology. That’s with the proviso that children are taking canakinumab at the recommended dose of 4 mg/kg every 4 weeks and achieve clinical remission before any tapering is started.
The researchers found that 70%-80% of children who were in complete clinical remission (CR) maintained this for 6 months after the first dose-tapering step had been taken. However, at least one-fifth of study participants experienced a disease flare during treatment withdrawal, and only one-third were able to discontinue treatment altogether, which suggests continued treatment is needed.
“The results are a step in the right direction,” commented Athimaleipet Ramanan, MBBS, a consultant pediatric rheumatologist who was not involved in the study. They “offer the first vision of whether we can actually taper a medication” in sJIA because “until now we have not had any evidence for this,” he added.
“What we really need to know, which the study doesn’t tell you, is: Is decreasing the dose better than stopping?” said Dr. Ramanan, of the Bristol (England) Royal Hospital for Children.
Another thing that is important to know is: Does reducing the dose lead to the development of anti-drug antibodies? he said.
“There were some concerns that, when you give less of a monoclonal antibody, you might get more neutralizing anti-drug antibodies or you might make a drug more immunogenic,” he said. These data perhaps suggest that this isn’t the case because only one child on one occasion had detectable non-neutralizing anti-drug antibodies during the entire study, and that was 11 weeks after the last dose of canakinumab had been given.
Study results and design
Canakinumab is a monoclonal antibody that inhibits interleukin-1 (IL-1) that has been approved in the United States and Europe for the treatment of sJIA since 2013. Reducing a child’s exposure to canakinumab once their disease is under control is an attractive proposition given the treat-to-target approach used increasingly throughout modern rheumatologic practice. It could also help reduce the cost of what is an expensive treatment, compared with other available options for sJIA such as the interleukin-6 inhibitor tocilizumab (Actemra) or another IL-1 inhibitor anakinra (Kineret), which is not FDA-approved for use in sJIA and requires weekly injections.
“We don’t want to the disease to reappear, to flare, but we also don’t want to overuse treatments in these patients,” explained Dr. Quartier of Hôpital Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris in an interview.
The study he coconducted, given the acronym B-SPECIFIC-4 Patients, was an open-label study that consisted of two parts. In part 1, 182 children were given subcutaneous canakinumab 4 mg/kg every 4 weeks. In part 2, 76 children who were in complete CR after canakinumab treatment were randomized to one of two tapering strategies. In one arm, the dose of canakinumab was reduced from 4 mg/kg to 2 mg/kg given every 4 weeks before eventually discontinuing treatment, and in other arm the duration between dosing was increased by 4-week intervals, from 4 to 8 weeks, then 12 weeks, then discontinuation.
If children were taking glucocorticoids or methotrexate, the treating physicians were encouraged to stop these medications if possible, with 34%-39% and 42%-59% of children, respectively, being able to do so. The rate depended on whether children had received canakinumab before entering the study because some had been recruited from a long-term extension study while others were naive to the biologic; all had inactive disease at study entry.
This was the first study to evaluate the effectiveness of canakinumab in enabling the discontinuation of methotrexate, but the primary objective was to assess whether more than 40% of randomized patients in either discontinuation arm remained in CR for 24 weeks after the first step of discontinuation. This was achieved in 71% of children who were in the dose-reduction arm and in 84% of children in the dose-prolongation arm (P ≤ .0001 for each arm vs. 40%).
Prior exposure to canakinumab did not seem to affect the maintenance of CR, but it was also found that more children who maintained CR at their second but not their first attempt at tapering were in CR than those who were still in CR at the first step (76% and 89% in the dose-reduction and dose-prolongation arms, respectively).
Among the two dosing regimens, failure occurred in 18% of children in the dose-reduction arm at the first step, 10% at the second, and 8% at the third, whereas 2.7% of children in the dose-prolongation arm experienced regimen failure at the first step, 6.1% at the second, and 15% at the third.
No substantial difference between the two tapering approaches was observed because the study was not powered to look at this. There was also no control arm, such as a group continuing treatment while the other groups tapered, or as Dr. Ramanan had pointed out, stopping canakinumab altogether.
“As long as the treatment was continued, even at very low dosage, most patients remained in inactive disease,” Dr. Quartier said. He added, however, that only a minority of patients could stop treatment completely. Treatment should not be stopped abruptly, he advised, because this was associated with a substantial number of disease flares that needed treatment to be reinstated.
These findings suggest that “a certain level of sustained inhibition of the IL-1 pathway seems important for the maintenance of CR in most sJIA patients,” Dr. Quartier and coinvestigators wrote in their article.
They added: “We believe that these results are relevant for clinical practice, particularly for designing personalized tapering strategies that can allow an adequate control of disease while minimizing the side effects of certain medications, notably glucocorticoids.”
The study was funded by Novartis in collaboration with the Pediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group. Dr. Quartier was an investigator for the trial and has received research and consultancy fees from Novartis, among other pharmaceutical companies. Two of his coauthors are employees of Novartis. Dr. Ramanan has acted as an investigator in prior canakinumab trials and has received consultancy fees from Novartis and multiple other companies.
SOURCE: Quartier P et al. Arthritis Rheumatol. 2020 Aug 11. doi: 10.1002/art.41488.
FROM ARTHRITIS & RHEUMATOLOGY
Novel botulinum toxin type A earns high marks for forehead lines
, Jeremy B. Green, MD, said at the virtual annual meeting of the American Academy of Dermatology.
When the study is completed, conclusions can be reached about the investigational product’s durability of benefit for treatment of dynamic forehead lines, which are notoriously challenging to treat. However, much is already known about the product’s durability for treatment of glabellar lines, as demonstrated in SAKURA 1 and SAKURA 2, two pivotal, phase 3, multicenter, randomized, double-blind, placebo-controlled trials totaling 609 patients.
In SAKURA 1 and 2, glabellar line severity didn’t return to baseline until a median of 28 and 26 weeks after injection. In contrast, as the study authors noted, the majority of patients whose glabellar lines are treated with the currently available botulinum toxin type A products are no longer responders by 3-4 months after treatment. Since surveys indicate most patients receive repeated injections every 5-6 months, that means they’re walking around with suboptimal results for the last 2-3 months before their next treatment session (Plast Reconstr Surg. 2020 Jan;145[1]:45-58).
This investigational neuromodulator, known as DaxibotulinumtoxinA for Injection, or DAXI, is composed of a highly purified 150-KDa botulinum toxin type A coupled with a proprietary stabilizing peptide. The product is formulated without human serum albumin and, once reconstituted, is stable at room temperature.
Dr. Green, a dermatologist in private practice in Coral Gables, Fla., reported on 61 participants in the phase 2 study, all with moderate or severe forehead lines and glabellar lines as assessed by both investigators and patients on structured scales. The patients’ glabellar lines were treated with 40 U of DAXI at baseline. Then 2 weeks later, their dynamic forehead lines were treated with either 12 U, 18 U, 24 U, or 30 U of DAXI. This sequential treatment recapitulates the approach widely used in clinical practice, he noted.
At baseline, two-thirds of patients had severe forehead lines at maximum eyebrow elevation as determined by Investigator Global Assessment – Forehead Wrinkle Severity and Patient Forehead Wrinkle Severity. The other third of participants had moderate forehead lines.
The primary endpoint was the presence of no or mild forehead lines by investigator assessment 4 weeks after treatment. This was achieved in 86% of patients who received 12 U of DAXI, 87% who recieved 18 U, 94% who received 24 U, and 100% of those who received 30 U.
“There appears to be a dose-dependent response, but this hasn’t yet been statistically analyzed,” Dr. Green said.
By patient assessment, there were no or only mild forehead lines at 4 weeks in 57% of those who received the lowest dose of DAXI, with rates of 80%, 100%, and 93% in those who received 18 U, 24 U, and 30 U.
At week 4, 57% of patients who got 12 U of DAXI pronounced themselves “satisfied” or “very satisfied” with DAXI therapy, as did 73%, 100%, and 93% of those who got the higher doses.
The treatment-related adverse events consisted of a smattering of cases of edema, erythema, or headache, similar to what’s described in the product labeling of all the neuromodulators.
Revance Therapeutics has applied to the Food and Drug Administration for marketing approval of DAXI for the treatment of glabellar lines. A regulatory decision is expected in late November. The company is also developing DAXI for the treatment of variety of neurologic and musculoskeletal conditions, including poststroke upper limb spasticity.
In an interview, Dr. Green said he was favorably impressed with DAXI’s durability for amelioration of forehead lines in the patients he personally treated in the ongoing phase 2 study, although there was no head-to-head comparison with other neuromodulators in the trial. He’s not aware of any planned phase 3 trial aimed at obtaining a forehead line indication.
“Of course, all four of the neuromodulators currently approved in the U.S. have glabellar line indications, but all are also used off-label in other locations, so I would imagine that DAXI will be used similarly if and when it is FDA-approved,” the dermatologist added.
He reported serving as a paid investigator for Revance.
, Jeremy B. Green, MD, said at the virtual annual meeting of the American Academy of Dermatology.
When the study is completed, conclusions can be reached about the investigational product’s durability of benefit for treatment of dynamic forehead lines, which are notoriously challenging to treat. However, much is already known about the product’s durability for treatment of glabellar lines, as demonstrated in SAKURA 1 and SAKURA 2, two pivotal, phase 3, multicenter, randomized, double-blind, placebo-controlled trials totaling 609 patients.
In SAKURA 1 and 2, glabellar line severity didn’t return to baseline until a median of 28 and 26 weeks after injection. In contrast, as the study authors noted, the majority of patients whose glabellar lines are treated with the currently available botulinum toxin type A products are no longer responders by 3-4 months after treatment. Since surveys indicate most patients receive repeated injections every 5-6 months, that means they’re walking around with suboptimal results for the last 2-3 months before their next treatment session (Plast Reconstr Surg. 2020 Jan;145[1]:45-58).
This investigational neuromodulator, known as DaxibotulinumtoxinA for Injection, or DAXI, is composed of a highly purified 150-KDa botulinum toxin type A coupled with a proprietary stabilizing peptide. The product is formulated without human serum albumin and, once reconstituted, is stable at room temperature.
Dr. Green, a dermatologist in private practice in Coral Gables, Fla., reported on 61 participants in the phase 2 study, all with moderate or severe forehead lines and glabellar lines as assessed by both investigators and patients on structured scales. The patients’ glabellar lines were treated with 40 U of DAXI at baseline. Then 2 weeks later, their dynamic forehead lines were treated with either 12 U, 18 U, 24 U, or 30 U of DAXI. This sequential treatment recapitulates the approach widely used in clinical practice, he noted.
At baseline, two-thirds of patients had severe forehead lines at maximum eyebrow elevation as determined by Investigator Global Assessment – Forehead Wrinkle Severity and Patient Forehead Wrinkle Severity. The other third of participants had moderate forehead lines.
The primary endpoint was the presence of no or mild forehead lines by investigator assessment 4 weeks after treatment. This was achieved in 86% of patients who received 12 U of DAXI, 87% who recieved 18 U, 94% who received 24 U, and 100% of those who received 30 U.
“There appears to be a dose-dependent response, but this hasn’t yet been statistically analyzed,” Dr. Green said.
By patient assessment, there were no or only mild forehead lines at 4 weeks in 57% of those who received the lowest dose of DAXI, with rates of 80%, 100%, and 93% in those who received 18 U, 24 U, and 30 U.
At week 4, 57% of patients who got 12 U of DAXI pronounced themselves “satisfied” or “very satisfied” with DAXI therapy, as did 73%, 100%, and 93% of those who got the higher doses.
The treatment-related adverse events consisted of a smattering of cases of edema, erythema, or headache, similar to what’s described in the product labeling of all the neuromodulators.
Revance Therapeutics has applied to the Food and Drug Administration for marketing approval of DAXI for the treatment of glabellar lines. A regulatory decision is expected in late November. The company is also developing DAXI for the treatment of variety of neurologic and musculoskeletal conditions, including poststroke upper limb spasticity.
In an interview, Dr. Green said he was favorably impressed with DAXI’s durability for amelioration of forehead lines in the patients he personally treated in the ongoing phase 2 study, although there was no head-to-head comparison with other neuromodulators in the trial. He’s not aware of any planned phase 3 trial aimed at obtaining a forehead line indication.
“Of course, all four of the neuromodulators currently approved in the U.S. have glabellar line indications, but all are also used off-label in other locations, so I would imagine that DAXI will be used similarly if and when it is FDA-approved,” the dermatologist added.
He reported serving as a paid investigator for Revance.
, Jeremy B. Green, MD, said at the virtual annual meeting of the American Academy of Dermatology.
When the study is completed, conclusions can be reached about the investigational product’s durability of benefit for treatment of dynamic forehead lines, which are notoriously challenging to treat. However, much is already known about the product’s durability for treatment of glabellar lines, as demonstrated in SAKURA 1 and SAKURA 2, two pivotal, phase 3, multicenter, randomized, double-blind, placebo-controlled trials totaling 609 patients.
In SAKURA 1 and 2, glabellar line severity didn’t return to baseline until a median of 28 and 26 weeks after injection. In contrast, as the study authors noted, the majority of patients whose glabellar lines are treated with the currently available botulinum toxin type A products are no longer responders by 3-4 months after treatment. Since surveys indicate most patients receive repeated injections every 5-6 months, that means they’re walking around with suboptimal results for the last 2-3 months before their next treatment session (Plast Reconstr Surg. 2020 Jan;145[1]:45-58).
This investigational neuromodulator, known as DaxibotulinumtoxinA for Injection, or DAXI, is composed of a highly purified 150-KDa botulinum toxin type A coupled with a proprietary stabilizing peptide. The product is formulated without human serum albumin and, once reconstituted, is stable at room temperature.
Dr. Green, a dermatologist in private practice in Coral Gables, Fla., reported on 61 participants in the phase 2 study, all with moderate or severe forehead lines and glabellar lines as assessed by both investigators and patients on structured scales. The patients’ glabellar lines were treated with 40 U of DAXI at baseline. Then 2 weeks later, their dynamic forehead lines were treated with either 12 U, 18 U, 24 U, or 30 U of DAXI. This sequential treatment recapitulates the approach widely used in clinical practice, he noted.
At baseline, two-thirds of patients had severe forehead lines at maximum eyebrow elevation as determined by Investigator Global Assessment – Forehead Wrinkle Severity and Patient Forehead Wrinkle Severity. The other third of participants had moderate forehead lines.
The primary endpoint was the presence of no or mild forehead lines by investigator assessment 4 weeks after treatment. This was achieved in 86% of patients who received 12 U of DAXI, 87% who recieved 18 U, 94% who received 24 U, and 100% of those who received 30 U.
“There appears to be a dose-dependent response, but this hasn’t yet been statistically analyzed,” Dr. Green said.
By patient assessment, there were no or only mild forehead lines at 4 weeks in 57% of those who received the lowest dose of DAXI, with rates of 80%, 100%, and 93% in those who received 18 U, 24 U, and 30 U.
At week 4, 57% of patients who got 12 U of DAXI pronounced themselves “satisfied” or “very satisfied” with DAXI therapy, as did 73%, 100%, and 93% of those who got the higher doses.
The treatment-related adverse events consisted of a smattering of cases of edema, erythema, or headache, similar to what’s described in the product labeling of all the neuromodulators.
Revance Therapeutics has applied to the Food and Drug Administration for marketing approval of DAXI for the treatment of glabellar lines. A regulatory decision is expected in late November. The company is also developing DAXI for the treatment of variety of neurologic and musculoskeletal conditions, including poststroke upper limb spasticity.
In an interview, Dr. Green said he was favorably impressed with DAXI’s durability for amelioration of forehead lines in the patients he personally treated in the ongoing phase 2 study, although there was no head-to-head comparison with other neuromodulators in the trial. He’s not aware of any planned phase 3 trial aimed at obtaining a forehead line indication.
“Of course, all four of the neuromodulators currently approved in the U.S. have glabellar line indications, but all are also used off-label in other locations, so I would imagine that DAXI will be used similarly if and when it is FDA-approved,” the dermatologist added.
He reported serving as a paid investigator for Revance.
FROM AAD 20