User login
Neurology Reviews covers innovative and emerging news in neurology and neuroscience every month, with a focus on practical approaches to treating Parkinson's disease, epilepsy, headache, stroke, multiple sclerosis, Alzheimer's disease, and other neurologic disorders.
PML
Progressive multifocal leukoencephalopathy
Rituxan
The leading independent newspaper covering neurology news and commentary.
Editor’s note
2023 is indeed a noteworthy year. As you will read in this issue, it marks the 40th anniversary of the landmark Orphan Drug Act and the formation of the National Organization for Rare Disorders (NORD). 2023 also marks the 30th anniversary of Neurology Reviews, the parent publication of the Rare Neurological Disease Special Report. While Neurology Reviews covers rare disease news throughout the year (see our Rare Disease Roundup in this issue), it is in our annual supplement where our rare disease news coverage and our partnership with NORD truly shines.

In this issue we take pride in taking a deeper look at some of the rare neurological diseases that have made headlines as well as the therapeutic advances and research breakthroughs that continue to benefit patients and the rare disease community as a whole. While I would prefer to humbly serve the rare disease community through our news coverage and educational efforts, I would be remiss if I didn’t mention that our 2022 Rare Neurological Disease Special Report won a Silver Regional Award in the category of annual supplement in the American Society of Business Publication Editors (Azbee) yearly competition. With that moment of bragging aside, I invite you to read this year’s issue, and I thank you for the success that this supplement has enjoyed since it launched in 2015.
Glenn S. Williams,
2023 is indeed a noteworthy year. As you will read in this issue, it marks the 40th anniversary of the landmark Orphan Drug Act and the formation of the National Organization for Rare Disorders (NORD). 2023 also marks the 30th anniversary of Neurology Reviews, the parent publication of the Rare Neurological Disease Special Report. While Neurology Reviews covers rare disease news throughout the year (see our Rare Disease Roundup in this issue), it is in our annual supplement where our rare disease news coverage and our partnership with NORD truly shines.
In this issue we take pride in taking a deeper look at some of the rare neurological diseases that have made headlines as well as the therapeutic advances and research breakthroughs that continue to benefit patients and the rare disease community as a whole. While I would prefer to humbly serve the rare disease community through our news coverage and educational efforts, I would be remiss if I didn’t mention that our 2022 Rare Neurological Disease Special Report won a Silver Regional Award in the category of annual supplement in the American Society of Business Publication Editors (Azbee) yearly competition. With that moment of bragging aside, I invite you to read this year’s issue, and I thank you for the success that this supplement has enjoyed since it launched in 2015.
Glenn S. Williams,
2023 is indeed a noteworthy year. As you will read in this issue, it marks the 40th anniversary of the landmark Orphan Drug Act and the formation of the National Organization for Rare Disorders (NORD). 2023 also marks the 30th anniversary of Neurology Reviews, the parent publication of the Rare Neurological Disease Special Report. While Neurology Reviews covers rare disease news throughout the year (see our Rare Disease Roundup in this issue), it is in our annual supplement where our rare disease news coverage and our partnership with NORD truly shines.
In this issue we take pride in taking a deeper look at some of the rare neurological diseases that have made headlines as well as the therapeutic advances and research breakthroughs that continue to benefit patients and the rare disease community as a whole. While I would prefer to humbly serve the rare disease community through our news coverage and educational efforts, I would be remiss if I didn’t mention that our 2022 Rare Neurological Disease Special Report won a Silver Regional Award in the category of annual supplement in the American Society of Business Publication Editors (Azbee) yearly competition. With that moment of bragging aside, I invite you to read this year’s issue, and I thank you for the success that this supplement has enjoyed since it launched in 2015.
Glenn S. Williams,
2023 Rare Neurological Disease Special Report

INTRODUCTIONS
Editor’s note
By Glenn S. Williams
2023 is indeed a noteworthy year. As you will read in this issue, it marks the 40th anniversary of the landmark Orphan Drug Act (ODA) and the formation of the National Organization for Rare Disorders. 2023 also marks the 30th anniversary of Neurology Reviews, the parent publication of the Rare Neurological Disease Special Report.
A note from NORD
By Edward Neilan, MD, PhD
The coalition of rare disease advocates who sparked rare disease advocacy and convinced lawmakers to pass the ODA in 1983 established NORD that same year to provide an ongoing, unified voice for the needs of the rare disease community.
Rare disease roundup
A look back at some of the 2023 rare disease headlines from Neurology Reviews.
CLINICAL REVIEWS
The Orphan Drug Act and NORD at their 40th anniversary: Dramatic achievements and ongoing innovation
By Batya Swift Yasgur, MA, MSW
The movement whose face is ODA and NORD continues to build its legacy. Next? Progress in rare disease care will require an all-in approach to solving a looming and massive public health challenge.
Emerging therapies in Duchenne and facioscapulohumeral muscular dystrophy
By Frieda Wiley, PharmD
Newly approved and investigational therapies, and enhanced diagnostics, are sparking optimism about treating MD – especially Duchenne and facioscapulohumeral types.
Has prompt diagnosis of amyotrophic lateral sclerosis become urgent?
By Ted Bosworth
Optimism is high about improving the survival and care of ALS patients. Neurologists who don’t specialize in ALS can add to the positivity by endorsing a role in speedier diagnostic pathways.
A new chapter for research on treating Huntington’s disease
By Jennie Smith
Setbacks in trials of protein-lowering therapies – mostly over their safety – mask a story of rapid advances and a more recently discovered treatment pathway that also offers promise for other diseases.
The dawning age of therapy for Friedreich ataxia
By Neil Osterweil
The first therapy to target the underlying pathology of Friedreich ataxia was approved in 2023. Other drug and genetic therapies are in the pipeline.
Stiff person syndrome: When a rare disorder hits the headlines
By Kate Johnson
Awareness of this disorder is increasing, but clinicians are challenged to apply the proper workup to avoid wrong turns in identifying affected patients.
Advances in testing and therapeutics are improving the lives of patients with Fabry disease
By Lorraine L. Janeczko, MPH
Thanks to robust research efforts, treatment options are expanding and patients are getting their diagnosis earlier – often, when they are presymptomatic and treatment has greater potential for enhancing quality of life.
Guillain-Barré syndrome: Honing treatment strategies
By John Jesitus
Classic subtypes of Guillain-Barré syndrome are varying manifestations of a shared disease process, novel insights into the disease indicate. This understanding is yielding new treatment strategies.
INTRODUCTIONS
Editor’s note
By Glenn S. Williams
2023 is indeed a noteworthy year. As you will read in this issue, it marks the 40th anniversary of the landmark Orphan Drug Act (ODA) and the formation of the National Organization for Rare Disorders. 2023 also marks the 30th anniversary of Neurology Reviews, the parent publication of the Rare Neurological Disease Special Report.
A note from NORD
By Edward Neilan, MD, PhD
The coalition of rare disease advocates who sparked rare disease advocacy and convinced lawmakers to pass the ODA in 1983 established NORD that same year to provide an ongoing, unified voice for the needs of the rare disease community.
Rare disease roundup
A look back at some of the 2023 rare disease headlines from Neurology Reviews.
CLINICAL REVIEWS
The Orphan Drug Act and NORD at their 40th anniversary: Dramatic achievements and ongoing innovation
By Batya Swift Yasgur, MA, MSW
The movement whose face is ODA and NORD continues to build its legacy. Next? Progress in rare disease care will require an all-in approach to solving a looming and massive public health challenge.
Emerging therapies in Duchenne and facioscapulohumeral muscular dystrophy
By Frieda Wiley, PharmD
Newly approved and investigational therapies, and enhanced diagnostics, are sparking optimism about treating MD – especially Duchenne and facioscapulohumeral types.
Has prompt diagnosis of amyotrophic lateral sclerosis become urgent?
By Ted Bosworth
Optimism is high about improving the survival and care of ALS patients. Neurologists who don’t specialize in ALS can add to the positivity by endorsing a role in speedier diagnostic pathways.
A new chapter for research on treating Huntington’s disease
By Jennie Smith
Setbacks in trials of protein-lowering therapies – mostly over their safety – mask a story of rapid advances and a more recently discovered treatment pathway that also offers promise for other diseases.
The dawning age of therapy for Friedreich ataxia
By Neil Osterweil
The first therapy to target the underlying pathology of Friedreich ataxia was approved in 2023. Other drug and genetic therapies are in the pipeline.
Stiff person syndrome: When a rare disorder hits the headlines
By Kate Johnson
Awareness of this disorder is increasing, but clinicians are challenged to apply the proper workup to avoid wrong turns in identifying affected patients.
Advances in testing and therapeutics are improving the lives of patients with Fabry disease
By Lorraine L. Janeczko, MPH
Thanks to robust research efforts, treatment options are expanding and patients are getting their diagnosis earlier – often, when they are presymptomatic and treatment has greater potential for enhancing quality of life.
Guillain-Barré syndrome: Honing treatment strategies
By John Jesitus
Classic subtypes of Guillain-Barré syndrome are varying manifestations of a shared disease process, novel insights into the disease indicate. This understanding is yielding new treatment strategies.
INTRODUCTIONS
Editor’s note
By Glenn S. Williams
2023 is indeed a noteworthy year. As you will read in this issue, it marks the 40th anniversary of the landmark Orphan Drug Act (ODA) and the formation of the National Organization for Rare Disorders. 2023 also marks the 30th anniversary of Neurology Reviews, the parent publication of the Rare Neurological Disease Special Report.
A note from NORD
By Edward Neilan, MD, PhD
The coalition of rare disease advocates who sparked rare disease advocacy and convinced lawmakers to pass the ODA in 1983 established NORD that same year to provide an ongoing, unified voice for the needs of the rare disease community.
Rare disease roundup
A look back at some of the 2023 rare disease headlines from Neurology Reviews.
CLINICAL REVIEWS
The Orphan Drug Act and NORD at their 40th anniversary: Dramatic achievements and ongoing innovation
By Batya Swift Yasgur, MA, MSW
The movement whose face is ODA and NORD continues to build its legacy. Next? Progress in rare disease care will require an all-in approach to solving a looming and massive public health challenge.
Emerging therapies in Duchenne and facioscapulohumeral muscular dystrophy
By Frieda Wiley, PharmD
Newly approved and investigational therapies, and enhanced diagnostics, are sparking optimism about treating MD – especially Duchenne and facioscapulohumeral types.
Has prompt diagnosis of amyotrophic lateral sclerosis become urgent?
By Ted Bosworth
Optimism is high about improving the survival and care of ALS patients. Neurologists who don’t specialize in ALS can add to the positivity by endorsing a role in speedier diagnostic pathways.
A new chapter for research on treating Huntington’s disease
By Jennie Smith
Setbacks in trials of protein-lowering therapies – mostly over their safety – mask a story of rapid advances and a more recently discovered treatment pathway that also offers promise for other diseases.
The dawning age of therapy for Friedreich ataxia
By Neil Osterweil
The first therapy to target the underlying pathology of Friedreich ataxia was approved in 2023. Other drug and genetic therapies are in the pipeline.
Stiff person syndrome: When a rare disorder hits the headlines
By Kate Johnson
Awareness of this disorder is increasing, but clinicians are challenged to apply the proper workup to avoid wrong turns in identifying affected patients.
Advances in testing and therapeutics are improving the lives of patients with Fabry disease
By Lorraine L. Janeczko, MPH
Thanks to robust research efforts, treatment options are expanding and patients are getting their diagnosis earlier – often, when they are presymptomatic and treatment has greater potential for enhancing quality of life.
Guillain-Barré syndrome: Honing treatment strategies
By John Jesitus
Classic subtypes of Guillain-Barré syndrome are varying manifestations of a shared disease process, novel insights into the disease indicate. This understanding is yielding new treatment strategies.

Has prompt diagnosis of amyotrophic lateral sclerosis become urgent?
Amyotrophic lateral sclerosis (ALS) falls easily into the Food and Drug Administration definition of “rare disease.” With an estimated prevalence in the United States of fewer than 20,000 cases,1 ALS sits comfortably below the cutoff of 200,000 cases that serves to define a disease as “rare.”
After a recent steep climb, there are something on the order of 50 therapies, across more than 10 drug classes, in clinical trials for the treatment of ALS.2 This bounty represents exciting progress toward the development of targeted therapies for a characteristically fatal disease.
That headway is coupled with a sobering limitation, however: Relatively few ALS patients are being enrolled.
The knotty problem with therapeutic trials for ALS
“Trials are generally designed for patients with adequate functional reserve and predicted survival, to ensure that a signal of benefit can be seen,” said Nicholas John Maragakis, MD, director of the ALS Clinical Trials Unit at Johns Hopkins University, Baltimore. “Many of my patients are too severely affected at presentation.”

Dr. Maragakis hasn’t calculated the precise percentage of patients he is enrolling in one of the many available trials available at the Johns Hopkins center. He estimates that it is less than 20%, however.
That percentage is comparable to what is reported by Stephen Scelsa, MD, and Daniel J. Macgowan, MD, who share much of the ALS caseload in a dedicated, comprehensive ALS center at Mount Sinai Beth Israel, New York. Both are on the faculty at the Icahn School of Medicine at Mount Sinai.
“The considerable delay in the diagnosis of ALS remains a challenge,” Dr. Scelsa acknowledges. Like Dr. Maragakis, he reports that, by the time patients develop symptoms that make referral to a comprehensive ALS center like Mount Sinai Beth Israel appropriate, many no longer meet eligibility criteria for most experimental treatments.
Some therapeutic targets in clinical trials, such as neuroinflammation, offer potential benefit even in advancing disease, but it is prevention that is usually the goal of experimental ALS therapies. This approach is associated with far more promise than attempting to reverse existing neurologic damage, which might not be possible, according to both Dr. Scelsa and Dr. Macgowan.
“The clinical trials are typically looking for patients with less than 2 years since the onset of symptoms and at least 60% of predicted respiratory function,” Dr. Macgowan said.
Because of these or other similarly restrictive criteria, coupled with common delays before patients arrive at a center where trials are available, “the window for clinical research closes very quickly,” Dr. Macgowan added, and “the band of patients who are eligible is relatively narrow.”
At Hennepin Healthcare in Minneapolis, which, like Johns Hopkins and Mount Sinai, offers an advanced multidisciplinary approach to ALS care in a dedicated clinic, the problem of late referrals is no different. Samuel Maiser, MD, chair of neurology, does attempt to counter this delay by moving quickly.
“I almost always offer a therapeutic trial to a patient with early-stage ALS,” he said. He does so earlier, rather than later, and explains: “I do not want to delay that conversation, because any delay might reduce the chance for getting into a trial.”
The generalist can make a difference in therapeutic success
The proliferation of clinical trials has made early diagnosis of ALS urgent. However, the experts interviewed for this article agreed: Accelerating the time to diagnosis is more dependent on the general neurologist or primary care physician than on the ALS specialist. ALS is a diagnosis of exclusion, but there is now very little delay in reaching a probable diagnosis at a dedicated center.
Yet neurodegenerative complaints in early-stage ALS are often nonspecific and mild; confidence in making a potential diagnosis of ALS is limited among primary care clinicians and general neurologists, who almost always see these patients first. Usually, the problem is not failure to include ALS in the differential diagnosis but hesitation in being candid when there is still doubt.
General neurologists, in particular, Dr. Maragakis said, “are often highly suspicious of a diagnosis of ALS very early on but are concerned about using this term until the clinical signs are more compelling.”
This is understandable. There is reluctance to deliver bad news when confidence in the diagnosis is limited. But the experts agreed: Delayed diagnosis is not in the patient’s interest now that there is at least the potential for entering a trial supported by a scientific rationale for benefit.
“Waiting for 100% certainty – this could actually harm our patients,” Dr. Maiser said. The tendency to avoid delivering bad news, he said, “is human nature, and it is not easy to tell people that ALS is the potential cause, but it’s important for early treatment.”
Some evidence suggests that the incidence of ALS is increasing3 but this is not necessarily evident at the clinical level. “It is not my impression that the incidence of ALS is increasing,” Dr. Macgowan said, “so much as I think we are getting better at making the diagnosis.”
Where we stand: Pathophysiology, diagnosis, treatment
Pathophysiology. ALS is characterized by muscle denervation.4 In the great majority of cases, the disease represents a proteinopathy involving loss of the TDP-43 protein from nuclei. However, pathological heterogeneity means that other pathophysiological mechanisms – mediated by oxidative stress, mitochondrial dysfunction, and neurotoxicity related to excessive stimulation of postsynaptic glutamate receptors – can participate.2,5,6

Approximately 10% of patients have a known gene associated with ALS.7 The rest have what is considered sporadic ALS, although some experts estimate that heritability will eventually be confirmed in 50% or more of cases that have been given the “sporadic” label.8,9 More than 30 genes have been linked to ALS in genomewide association studies. Among patients whose disease carries a known familial link, four genes – SOD1, TARDBP, FUS, and C9orf72 – account for approximately 70% of cases.2
Diagnosis. Genetic testing in patients with suspected or confirmed ALS is the standard of care at most, if not all, comprehensive ALS treatment centers, according to the four experts interviewed by Neurology Reviews 2023 Rare Neurological Disease Special Report for this article. Such testing was routine for years because of its potential for helping researchers to understand subtypes of disease; today, testing has assumed even greater practical value with recent approval of the first ALS gene therapy: Tofersen (Qalsody, Biogen), licensed in 2023, is an antisense oligonucleotide therapy that targets SOD1 mRNA to reduce production of the SOD1 protein, a mediator of disease progression.
“Genetic testing has been useful for telling us something about the disease and its prognosis,” Dr. Maragakis said, “but an approved gene therapy means it can have a direct effect on treatment.”
ALS therapeutics. Other gene therapies are in development. Gene signatures are likely to provide even more opportunities for clinical trials in the future.
Following three loading doses of tofersen at 14-day intervals, the maintenance regimen, administered intrathecally by lumbar puncture, is every 28 days. In the phase 3 trial, tofersen reduced levels of SOD1 protein and neurofilament light chain, a biomarker of axonal injury.10 Tofersen is appropriate only in patients with SOD1-associated ALS; the drug’s favorable clinical impact, including a positive effect, if any, on survival has not been demonstrated. Extension studies are underway.
Tofersen joins three other FDA-approved ALS therapies:
• Riluzole, an oral drug available since 1995 that slows disease progression by blocking glutamate.
• Edaravone, an antioxidant approved in 2017, administered orally or intravenously.
• An orally administered combination of sodium phenylbutyrate and taurursodiol marketed as Relyvrio and formerly known as AMX0035, that was introduced in 2022.
“We offer riluzole, which is safe in combination with other therapies, to most patients,” said Dr. Scelsa, who noted that treatment trials often test experimental drugs on top of riluzole. He moves to edaravone or Relyvrio, which are far more expensive, selectively. Tofersen, which is also expensive, is reserved for patients with SOD1-associated disease; however, not all eligible patients opt for this therapy after reviewing its benefits and risks.
“There is not yet a guarantee that tofersen will improve outcomes, and it requires intrathecal injections for life,”
Dr. Maiser said. “Some patients, particularly my older patients, have said, ‘No thank you,’ based on the available data.”
Dr. Macgowan pointed out that lumbar puncture repeated indefinitely can be “challenging.” He, too, discusses all available treatment options with every patient, including riluzole, which he agreed is associated with a meaningful benefit, particularly when started early.
Because of the safety of riluzole, Dr. Maragakis takes early treatment a step further. For neurologists who have a high level of suspicion of ALS in a given patient, “my advice would be to treat aggressively from the get-go. Even if not 100% certain of the diagnosis, I would start them on riluzole while waiting for confirmation.” Like the other experts interviewed here, he acknowledged that referral to a busy comprehensive ALS center often takes time, making it reasonable to initiate treatment when suspicion is high.
On the front lines, “the neurologist can tell the patient that ALS is just one of several potential explanations for symptoms but there is concern,” said Dr. Maragakis, proposing a strategy to introduce the possibility of ALS and start treatment that might slow disease while waiting for confirmation of the diagnosis. “My biggest concern is that no one is making that call,” he said, trying to address at least one reason for the current delay in making referrals.
Comprehensive care at specialty centers
Whenever possible, ALS is a disease best managed at a center that offers comprehensive management, including multidisciplinary care. On this point, the four experts agreed.
“Tertiary-care centers for ALS serve a critical purpose,”
Dr. Maiser said. For a disease that affects nearly every aspect of life, the skills of a multidisciplinary support staff offer an “opportunity to stay in front of the disease” for as long as possible. Teamwork often leads to “outside-of-the-box thinking” for helping patients and families cope with the range of disabilities that undermine the patient’s quality of life.
Details of ALS management matter. At Mount Sinai and Hennepin Healthcare, and at Johns Hopkins, where demand recently led to the opening of a second ALS clinic, the ALS center is set up to address the full spectrum of needs. Staff members have multiple skills so that they can work together to make patients comfortable and prepare them for what is inevitably progression – even if the rate of that progression varies.
All these centers incorporate a rational, thorough discussion of end-of-life options in a palliative care approach that targets optimized quality of life. One goal is to prepare patients to consider and be prepared to make decisions when it is time for tracheostomy, percutaneous endoscopic gastrostomy, and other life support options that are not always well tolerated. The goal? Avoiding unnecessary anguish during end-stage disease when impaired respiratory function – the primary cause of ALS-related death – no longer sustains unassisted survival.
“I am concerned for the many ALS patients without access to this type of comprehensive care,” Dr. Macgowan said.
Like the other experts here, he emphasized that the demands of ALS care can be “overwhelming” outside a comprehensive care setting – for the patient, their family, and individual providers.
Looking ahead
There are many reasons to be optimistic about improving the survival and care of patients with ALS. Besides therapies in clinical trials, Dr. Scelsa explained, there is the potential role for monitoring neurofilament light changes, a biomarker of neurodegeneration, in patients who are at risk of ALS.
Dr. Maragakis offered an analogy to the gene therapy onasemnogene abeparvovec, which can prevent the associated neurodegeneration of spinal muscular atrophy if initiated before symptoms appear. He said that, in ALS, neurofilament light changes or other biomarkers might offer an opportunity to halt the progression of disease before it starts – if one or more therapies in development prove workable.
In the meantime, neurologists who do not specialize in ALS should be thinking about how they can participate in speedier diagnostic pathways.
“There are a number of therapies that look promising,” Dr. Maiser told Rare Neurological Disease Special Report. He singled out strategies to degrade TDP-43 or prevent it from forming. If these treatments are found effective, it’s expected that they would be of value in sporadic ALS, the most common form. Again, though, “the challenge is getting patients on this therapy at the earliest stages of disease.”
Dr. Maragakis discloses equity ownership/stock options with Braintrust Bio and Akava; he is a patent holder with Johns Hopkins [ALS] and has received grant/research/clinical trial support from Apellis Pharma, Biogen Idec, Cytokinetics, Helixmith, Calico, Sanofi, Department of Defense ALSRP, Maryland Stem Cell Research Fund, Massachusetts General Hospital, Medicinova, and NINDS. He serves as consultant or advisory board member for Amylyx; Cytokinetics, Roche, Healey Center, Nura Bio, Northeast ALS Consortium, Akava, Inflammx, and Secretome. Dr. Scelsa did not report any conflicts of interest. Dr. Macgowan and Dr. Maiser have no relevant conflicts of interest to disclose.
References
1. Mehta P et al. Prevalence of amyotrophic lateral sclerosis in the United States using established and novel methodologies, 2017. Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(1-2):108-16. doi: 10.1080/21678421.2022.2059380.
2. Mead RJ et al. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov. 2023;22(3):185-212. doi: 10.1038/s41573-022-00612-2.
3. Longinetti E and Fang F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr Opin Neurol. 2019;32(5):771-6. doi: 10.1097/WCO.0000000000000730.
4. van den Bos MAJ et al. Pathophysiology and diagnosis of ALS: Insights from advances in neurophysiological techniques. Int J Mol Sci. 2019;20(11):2818. doi: 10.3390/ijms20112818.
5. Neumann M et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130-3. doi: 10.1126/science.1134108.
6. Ling S-C et al. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416-38. doi: 10.1016/j.neuron.2013.07.033.
7. Ranganathan R et al. Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Front Neurosci. 2020;14:684. doi: 10.3389/fnins.2020.00684.
8. Ryan M et al. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2019;76(11):1367-74. doi: 10.1001/jamaneurol.2019.2044.
9. van Rheenen W et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet. 2021;53(12):1636-48. doi: 10.1038/s41588-021-00973-1.
10. Miller TM et al; VALOR and OLE Working Group. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099-110. doi: 10.1056/NEJMoa2204705.
Amyotrophic lateral sclerosis (ALS) falls easily into the Food and Drug Administration definition of “rare disease.” With an estimated prevalence in the United States of fewer than 20,000 cases,1 ALS sits comfortably below the cutoff of 200,000 cases that serves to define a disease as “rare.”
After a recent steep climb, there are something on the order of 50 therapies, across more than 10 drug classes, in clinical trials for the treatment of ALS.2 This bounty represents exciting progress toward the development of targeted therapies for a characteristically fatal disease.
That headway is coupled with a sobering limitation, however: Relatively few ALS patients are being enrolled.
The knotty problem with therapeutic trials for ALS
“Trials are generally designed for patients with adequate functional reserve and predicted survival, to ensure that a signal of benefit can be seen,” said Nicholas John Maragakis, MD, director of the ALS Clinical Trials Unit at Johns Hopkins University, Baltimore. “Many of my patients are too severely affected at presentation.”
Dr. Maragakis hasn’t calculated the precise percentage of patients he is enrolling in one of the many available trials available at the Johns Hopkins center. He estimates that it is less than 20%, however.
That percentage is comparable to what is reported by Stephen Scelsa, MD, and Daniel J. Macgowan, MD, who share much of the ALS caseload in a dedicated, comprehensive ALS center at Mount Sinai Beth Israel, New York. Both are on the faculty at the Icahn School of Medicine at Mount Sinai.
“The considerable delay in the diagnosis of ALS remains a challenge,” Dr. Scelsa acknowledges. Like Dr. Maragakis, he reports that, by the time patients develop symptoms that make referral to a comprehensive ALS center like Mount Sinai Beth Israel appropriate, many no longer meet eligibility criteria for most experimental treatments.
Some therapeutic targets in clinical trials, such as neuroinflammation, offer potential benefit even in advancing disease, but it is prevention that is usually the goal of experimental ALS therapies. This approach is associated with far more promise than attempting to reverse existing neurologic damage, which might not be possible, according to both Dr. Scelsa and Dr. Macgowan.
“The clinical trials are typically looking for patients with less than 2 years since the onset of symptoms and at least 60% of predicted respiratory function,” Dr. Macgowan said.
Because of these or other similarly restrictive criteria, coupled with common delays before patients arrive at a center where trials are available, “the window for clinical research closes very quickly,” Dr. Macgowan added, and “the band of patients who are eligible is relatively narrow.”
At Hennepin Healthcare in Minneapolis, which, like Johns Hopkins and Mount Sinai, offers an advanced multidisciplinary approach to ALS care in a dedicated clinic, the problem of late referrals is no different. Samuel Maiser, MD, chair of neurology, does attempt to counter this delay by moving quickly.
“I almost always offer a therapeutic trial to a patient with early-stage ALS,” he said. He does so earlier, rather than later, and explains: “I do not want to delay that conversation, because any delay might reduce the chance for getting into a trial.”
The generalist can make a difference in therapeutic success
The proliferation of clinical trials has made early diagnosis of ALS urgent. However, the experts interviewed for this article agreed: Accelerating the time to diagnosis is more dependent on the general neurologist or primary care physician than on the ALS specialist. ALS is a diagnosis of exclusion, but there is now very little delay in reaching a probable diagnosis at a dedicated center.
Yet neurodegenerative complaints in early-stage ALS are often nonspecific and mild; confidence in making a potential diagnosis of ALS is limited among primary care clinicians and general neurologists, who almost always see these patients first. Usually, the problem is not failure to include ALS in the differential diagnosis but hesitation in being candid when there is still doubt.
General neurologists, in particular, Dr. Maragakis said, “are often highly suspicious of a diagnosis of ALS very early on but are concerned about using this term until the clinical signs are more compelling.”
This is understandable. There is reluctance to deliver bad news when confidence in the diagnosis is limited. But the experts agreed: Delayed diagnosis is not in the patient’s interest now that there is at least the potential for entering a trial supported by a scientific rationale for benefit.
“Waiting for 100% certainty – this could actually harm our patients,” Dr. Maiser said. The tendency to avoid delivering bad news, he said, “is human nature, and it is not easy to tell people that ALS is the potential cause, but it’s important for early treatment.”
Some evidence suggests that the incidence of ALS is increasing3 but this is not necessarily evident at the clinical level. “It is not my impression that the incidence of ALS is increasing,” Dr. Macgowan said, “so much as I think we are getting better at making the diagnosis.”
Where we stand: Pathophysiology, diagnosis, treatment
Pathophysiology. ALS is characterized by muscle denervation.4 In the great majority of cases, the disease represents a proteinopathy involving loss of the TDP-43 protein from nuclei. However, pathological heterogeneity means that other pathophysiological mechanisms – mediated by oxidative stress, mitochondrial dysfunction, and neurotoxicity related to excessive stimulation of postsynaptic glutamate receptors – can participate.2,5,6
Approximately 10% of patients have a known gene associated with ALS.7 The rest have what is considered sporadic ALS, although some experts estimate that heritability will eventually be confirmed in 50% or more of cases that have been given the “sporadic” label.8,9 More than 30 genes have been linked to ALS in genomewide association studies. Among patients whose disease carries a known familial link, four genes – SOD1, TARDBP, FUS, and C9orf72 – account for approximately 70% of cases.2
Diagnosis. Genetic testing in patients with suspected or confirmed ALS is the standard of care at most, if not all, comprehensive ALS treatment centers, according to the four experts interviewed by Neurology Reviews 2023 Rare Neurological Disease Special Report for this article. Such testing was routine for years because of its potential for helping researchers to understand subtypes of disease; today, testing has assumed even greater practical value with recent approval of the first ALS gene therapy: Tofersen (Qalsody, Biogen), licensed in 2023, is an antisense oligonucleotide therapy that targets SOD1 mRNA to reduce production of the SOD1 protein, a mediator of disease progression.
“Genetic testing has been useful for telling us something about the disease and its prognosis,” Dr. Maragakis said, “but an approved gene therapy means it can have a direct effect on treatment.”
ALS therapeutics. Other gene therapies are in development. Gene signatures are likely to provide even more opportunities for clinical trials in the future.
Following three loading doses of tofersen at 14-day intervals, the maintenance regimen, administered intrathecally by lumbar puncture, is every 28 days. In the phase 3 trial, tofersen reduced levels of SOD1 protein and neurofilament light chain, a biomarker of axonal injury.10 Tofersen is appropriate only in patients with SOD1-associated ALS; the drug’s favorable clinical impact, including a positive effect, if any, on survival has not been demonstrated. Extension studies are underway.
Tofersen joins three other FDA-approved ALS therapies:
• Riluzole, an oral drug available since 1995 that slows disease progression by blocking glutamate.
• Edaravone, an antioxidant approved in 2017, administered orally or intravenously.
• An orally administered combination of sodium phenylbutyrate and taurursodiol marketed as Relyvrio and formerly known as AMX0035, that was introduced in 2022.
“We offer riluzole, which is safe in combination with other therapies, to most patients,” said Dr. Scelsa, who noted that treatment trials often test experimental drugs on top of riluzole. He moves to edaravone or Relyvrio, which are far more expensive, selectively. Tofersen, which is also expensive, is reserved for patients with SOD1-associated disease; however, not all eligible patients opt for this therapy after reviewing its benefits and risks.
“There is not yet a guarantee that tofersen will improve outcomes, and it requires intrathecal injections for life,”
Dr. Maiser said. “Some patients, particularly my older patients, have said, ‘No thank you,’ based on the available data.”
Dr. Macgowan pointed out that lumbar puncture repeated indefinitely can be “challenging.” He, too, discusses all available treatment options with every patient, including riluzole, which he agreed is associated with a meaningful benefit, particularly when started early.
Because of the safety of riluzole, Dr. Maragakis takes early treatment a step further. For neurologists who have a high level of suspicion of ALS in a given patient, “my advice would be to treat aggressively from the get-go. Even if not 100% certain of the diagnosis, I would start them on riluzole while waiting for confirmation.” Like the other experts interviewed here, he acknowledged that referral to a busy comprehensive ALS center often takes time, making it reasonable to initiate treatment when suspicion is high.
On the front lines, “the neurologist can tell the patient that ALS is just one of several potential explanations for symptoms but there is concern,” said Dr. Maragakis, proposing a strategy to introduce the possibility of ALS and start treatment that might slow disease while waiting for confirmation of the diagnosis. “My biggest concern is that no one is making that call,” he said, trying to address at least one reason for the current delay in making referrals.
Comprehensive care at specialty centers
Whenever possible, ALS is a disease best managed at a center that offers comprehensive management, including multidisciplinary care. On this point, the four experts agreed.
“Tertiary-care centers for ALS serve a critical purpose,”
Dr. Maiser said. For a disease that affects nearly every aspect of life, the skills of a multidisciplinary support staff offer an “opportunity to stay in front of the disease” for as long as possible. Teamwork often leads to “outside-of-the-box thinking” for helping patients and families cope with the range of disabilities that undermine the patient’s quality of life.
Details of ALS management matter. At Mount Sinai and Hennepin Healthcare, and at Johns Hopkins, where demand recently led to the opening of a second ALS clinic, the ALS center is set up to address the full spectrum of needs. Staff members have multiple skills so that they can work together to make patients comfortable and prepare them for what is inevitably progression – even if the rate of that progression varies.
All these centers incorporate a rational, thorough discussion of end-of-life options in a palliative care approach that targets optimized quality of life. One goal is to prepare patients to consider and be prepared to make decisions when it is time for tracheostomy, percutaneous endoscopic gastrostomy, and other life support options that are not always well tolerated. The goal? Avoiding unnecessary anguish during end-stage disease when impaired respiratory function – the primary cause of ALS-related death – no longer sustains unassisted survival.
“I am concerned for the many ALS patients without access to this type of comprehensive care,” Dr. Macgowan said.
Like the other experts here, he emphasized that the demands of ALS care can be “overwhelming” outside a comprehensive care setting – for the patient, their family, and individual providers.
Looking ahead
There are many reasons to be optimistic about improving the survival and care of patients with ALS. Besides therapies in clinical trials, Dr. Scelsa explained, there is the potential role for monitoring neurofilament light changes, a biomarker of neurodegeneration, in patients who are at risk of ALS.
Dr. Maragakis offered an analogy to the gene therapy onasemnogene abeparvovec, which can prevent the associated neurodegeneration of spinal muscular atrophy if initiated before symptoms appear. He said that, in ALS, neurofilament light changes or other biomarkers might offer an opportunity to halt the progression of disease before it starts – if one or more therapies in development prove workable.
In the meantime, neurologists who do not specialize in ALS should be thinking about how they can participate in speedier diagnostic pathways.
“There are a number of therapies that look promising,” Dr. Maiser told Rare Neurological Disease Special Report. He singled out strategies to degrade TDP-43 or prevent it from forming. If these treatments are found effective, it’s expected that they would be of value in sporadic ALS, the most common form. Again, though, “the challenge is getting patients on this therapy at the earliest stages of disease.”
Dr. Maragakis discloses equity ownership/stock options with Braintrust Bio and Akava; he is a patent holder with Johns Hopkins [ALS] and has received grant/research/clinical trial support from Apellis Pharma, Biogen Idec, Cytokinetics, Helixmith, Calico, Sanofi, Department of Defense ALSRP, Maryland Stem Cell Research Fund, Massachusetts General Hospital, Medicinova, and NINDS. He serves as consultant or advisory board member for Amylyx; Cytokinetics, Roche, Healey Center, Nura Bio, Northeast ALS Consortium, Akava, Inflammx, and Secretome. Dr. Scelsa did not report any conflicts of interest. Dr. Macgowan and Dr. Maiser have no relevant conflicts of interest to disclose.
References
1. Mehta P et al. Prevalence of amyotrophic lateral sclerosis in the United States using established and novel methodologies, 2017. Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(1-2):108-16. doi: 10.1080/21678421.2022.2059380.
2. Mead RJ et al. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov. 2023;22(3):185-212. doi: 10.1038/s41573-022-00612-2.
3. Longinetti E and Fang F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr Opin Neurol. 2019;32(5):771-6. doi: 10.1097/WCO.0000000000000730.
4. van den Bos MAJ et al. Pathophysiology and diagnosis of ALS: Insights from advances in neurophysiological techniques. Int J Mol Sci. 2019;20(11):2818. doi: 10.3390/ijms20112818.
5. Neumann M et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130-3. doi: 10.1126/science.1134108.
6. Ling S-C et al. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416-38. doi: 10.1016/j.neuron.2013.07.033.
7. Ranganathan R et al. Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Front Neurosci. 2020;14:684. doi: 10.3389/fnins.2020.00684.
8. Ryan M et al. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2019;76(11):1367-74. doi: 10.1001/jamaneurol.2019.2044.
9. van Rheenen W et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet. 2021;53(12):1636-48. doi: 10.1038/s41588-021-00973-1.
10. Miller TM et al; VALOR and OLE Working Group. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099-110. doi: 10.1056/NEJMoa2204705.
Amyotrophic lateral sclerosis (ALS) falls easily into the Food and Drug Administration definition of “rare disease.” With an estimated prevalence in the United States of fewer than 20,000 cases,1 ALS sits comfortably below the cutoff of 200,000 cases that serves to define a disease as “rare.”
After a recent steep climb, there are something on the order of 50 therapies, across more than 10 drug classes, in clinical trials for the treatment of ALS.2 This bounty represents exciting progress toward the development of targeted therapies for a characteristically fatal disease.
That headway is coupled with a sobering limitation, however: Relatively few ALS patients are being enrolled.
The knotty problem with therapeutic trials for ALS
“Trials are generally designed for patients with adequate functional reserve and predicted survival, to ensure that a signal of benefit can be seen,” said Nicholas John Maragakis, MD, director of the ALS Clinical Trials Unit at Johns Hopkins University, Baltimore. “Many of my patients are too severely affected at presentation.”
Dr. Maragakis hasn’t calculated the precise percentage of patients he is enrolling in one of the many available trials available at the Johns Hopkins center. He estimates that it is less than 20%, however.
That percentage is comparable to what is reported by Stephen Scelsa, MD, and Daniel J. Macgowan, MD, who share much of the ALS caseload in a dedicated, comprehensive ALS center at Mount Sinai Beth Israel, New York. Both are on the faculty at the Icahn School of Medicine at Mount Sinai.
“The considerable delay in the diagnosis of ALS remains a challenge,” Dr. Scelsa acknowledges. Like Dr. Maragakis, he reports that, by the time patients develop symptoms that make referral to a comprehensive ALS center like Mount Sinai Beth Israel appropriate, many no longer meet eligibility criteria for most experimental treatments.
Some therapeutic targets in clinical trials, such as neuroinflammation, offer potential benefit even in advancing disease, but it is prevention that is usually the goal of experimental ALS therapies. This approach is associated with far more promise than attempting to reverse existing neurologic damage, which might not be possible, according to both Dr. Scelsa and Dr. Macgowan.
“The clinical trials are typically looking for patients with less than 2 years since the onset of symptoms and at least 60% of predicted respiratory function,” Dr. Macgowan said.
Because of these or other similarly restrictive criteria, coupled with common delays before patients arrive at a center where trials are available, “the window for clinical research closes very quickly,” Dr. Macgowan added, and “the band of patients who are eligible is relatively narrow.”
At Hennepin Healthcare in Minneapolis, which, like Johns Hopkins and Mount Sinai, offers an advanced multidisciplinary approach to ALS care in a dedicated clinic, the problem of late referrals is no different. Samuel Maiser, MD, chair of neurology, does attempt to counter this delay by moving quickly.
“I almost always offer a therapeutic trial to a patient with early-stage ALS,” he said. He does so earlier, rather than later, and explains: “I do not want to delay that conversation, because any delay might reduce the chance for getting into a trial.”
The generalist can make a difference in therapeutic success
The proliferation of clinical trials has made early diagnosis of ALS urgent. However, the experts interviewed for this article agreed: Accelerating the time to diagnosis is more dependent on the general neurologist or primary care physician than on the ALS specialist. ALS is a diagnosis of exclusion, but there is now very little delay in reaching a probable diagnosis at a dedicated center.
Yet neurodegenerative complaints in early-stage ALS are often nonspecific and mild; confidence in making a potential diagnosis of ALS is limited among primary care clinicians and general neurologists, who almost always see these patients first. Usually, the problem is not failure to include ALS in the differential diagnosis but hesitation in being candid when there is still doubt.
General neurologists, in particular, Dr. Maragakis said, “are often highly suspicious of a diagnosis of ALS very early on but are concerned about using this term until the clinical signs are more compelling.”
This is understandable. There is reluctance to deliver bad news when confidence in the diagnosis is limited. But the experts agreed: Delayed diagnosis is not in the patient’s interest now that there is at least the potential for entering a trial supported by a scientific rationale for benefit.
“Waiting for 100% certainty – this could actually harm our patients,” Dr. Maiser said. The tendency to avoid delivering bad news, he said, “is human nature, and it is not easy to tell people that ALS is the potential cause, but it’s important for early treatment.”
Some evidence suggests that the incidence of ALS is increasing3 but this is not necessarily evident at the clinical level. “It is not my impression that the incidence of ALS is increasing,” Dr. Macgowan said, “so much as I think we are getting better at making the diagnosis.”
Where we stand: Pathophysiology, diagnosis, treatment
Pathophysiology. ALS is characterized by muscle denervation.4 In the great majority of cases, the disease represents a proteinopathy involving loss of the TDP-43 protein from nuclei. However, pathological heterogeneity means that other pathophysiological mechanisms – mediated by oxidative stress, mitochondrial dysfunction, and neurotoxicity related to excessive stimulation of postsynaptic glutamate receptors – can participate.2,5,6
Approximately 10% of patients have a known gene associated with ALS.7 The rest have what is considered sporadic ALS, although some experts estimate that heritability will eventually be confirmed in 50% or more of cases that have been given the “sporadic” label.8,9 More than 30 genes have been linked to ALS in genomewide association studies. Among patients whose disease carries a known familial link, four genes – SOD1, TARDBP, FUS, and C9orf72 – account for approximately 70% of cases.2
Diagnosis. Genetic testing in patients with suspected or confirmed ALS is the standard of care at most, if not all, comprehensive ALS treatment centers, according to the four experts interviewed by Neurology Reviews 2023 Rare Neurological Disease Special Report for this article. Such testing was routine for years because of its potential for helping researchers to understand subtypes of disease; today, testing has assumed even greater practical value with recent approval of the first ALS gene therapy: Tofersen (Qalsody, Biogen), licensed in 2023, is an antisense oligonucleotide therapy that targets SOD1 mRNA to reduce production of the SOD1 protein, a mediator of disease progression.
“Genetic testing has been useful for telling us something about the disease and its prognosis,” Dr. Maragakis said, “but an approved gene therapy means it can have a direct effect on treatment.”
ALS therapeutics. Other gene therapies are in development. Gene signatures are likely to provide even more opportunities for clinical trials in the future.
Following three loading doses of tofersen at 14-day intervals, the maintenance regimen, administered intrathecally by lumbar puncture, is every 28 days. In the phase 3 trial, tofersen reduced levels of SOD1 protein and neurofilament light chain, a biomarker of axonal injury.10 Tofersen is appropriate only in patients with SOD1-associated ALS; the drug’s favorable clinical impact, including a positive effect, if any, on survival has not been demonstrated. Extension studies are underway.
Tofersen joins three other FDA-approved ALS therapies:
• Riluzole, an oral drug available since 1995 that slows disease progression by blocking glutamate.
• Edaravone, an antioxidant approved in 2017, administered orally or intravenously.
• An orally administered combination of sodium phenylbutyrate and taurursodiol marketed as Relyvrio and formerly known as AMX0035, that was introduced in 2022.
“We offer riluzole, which is safe in combination with other therapies, to most patients,” said Dr. Scelsa, who noted that treatment trials often test experimental drugs on top of riluzole. He moves to edaravone or Relyvrio, which are far more expensive, selectively. Tofersen, which is also expensive, is reserved for patients with SOD1-associated disease; however, not all eligible patients opt for this therapy after reviewing its benefits and risks.
“There is not yet a guarantee that tofersen will improve outcomes, and it requires intrathecal injections for life,”
Dr. Maiser said. “Some patients, particularly my older patients, have said, ‘No thank you,’ based on the available data.”
Dr. Macgowan pointed out that lumbar puncture repeated indefinitely can be “challenging.” He, too, discusses all available treatment options with every patient, including riluzole, which he agreed is associated with a meaningful benefit, particularly when started early.
Because of the safety of riluzole, Dr. Maragakis takes early treatment a step further. For neurologists who have a high level of suspicion of ALS in a given patient, “my advice would be to treat aggressively from the get-go. Even if not 100% certain of the diagnosis, I would start them on riluzole while waiting for confirmation.” Like the other experts interviewed here, he acknowledged that referral to a busy comprehensive ALS center often takes time, making it reasonable to initiate treatment when suspicion is high.
On the front lines, “the neurologist can tell the patient that ALS is just one of several potential explanations for symptoms but there is concern,” said Dr. Maragakis, proposing a strategy to introduce the possibility of ALS and start treatment that might slow disease while waiting for confirmation of the diagnosis. “My biggest concern is that no one is making that call,” he said, trying to address at least one reason for the current delay in making referrals.
Comprehensive care at specialty centers
Whenever possible, ALS is a disease best managed at a center that offers comprehensive management, including multidisciplinary care. On this point, the four experts agreed.
“Tertiary-care centers for ALS serve a critical purpose,”
Dr. Maiser said. For a disease that affects nearly every aspect of life, the skills of a multidisciplinary support staff offer an “opportunity to stay in front of the disease” for as long as possible. Teamwork often leads to “outside-of-the-box thinking” for helping patients and families cope with the range of disabilities that undermine the patient’s quality of life.
Details of ALS management matter. At Mount Sinai and Hennepin Healthcare, and at Johns Hopkins, where demand recently led to the opening of a second ALS clinic, the ALS center is set up to address the full spectrum of needs. Staff members have multiple skills so that they can work together to make patients comfortable and prepare them for what is inevitably progression – even if the rate of that progression varies.
All these centers incorporate a rational, thorough discussion of end-of-life options in a palliative care approach that targets optimized quality of life. One goal is to prepare patients to consider and be prepared to make decisions when it is time for tracheostomy, percutaneous endoscopic gastrostomy, and other life support options that are not always well tolerated. The goal? Avoiding unnecessary anguish during end-stage disease when impaired respiratory function – the primary cause of ALS-related death – no longer sustains unassisted survival.
“I am concerned for the many ALS patients without access to this type of comprehensive care,” Dr. Macgowan said.
Like the other experts here, he emphasized that the demands of ALS care can be “overwhelming” outside a comprehensive care setting – for the patient, their family, and individual providers.
Looking ahead
There are many reasons to be optimistic about improving the survival and care of patients with ALS. Besides therapies in clinical trials, Dr. Scelsa explained, there is the potential role for monitoring neurofilament light changes, a biomarker of neurodegeneration, in patients who are at risk of ALS.
Dr. Maragakis offered an analogy to the gene therapy onasemnogene abeparvovec, which can prevent the associated neurodegeneration of spinal muscular atrophy if initiated before symptoms appear. He said that, in ALS, neurofilament light changes or other biomarkers might offer an opportunity to halt the progression of disease before it starts – if one or more therapies in development prove workable.
In the meantime, neurologists who do not specialize in ALS should be thinking about how they can participate in speedier diagnostic pathways.
“There are a number of therapies that look promising,” Dr. Maiser told Rare Neurological Disease Special Report. He singled out strategies to degrade TDP-43 or prevent it from forming. If these treatments are found effective, it’s expected that they would be of value in sporadic ALS, the most common form. Again, though, “the challenge is getting patients on this therapy at the earliest stages of disease.”
Dr. Maragakis discloses equity ownership/stock options with Braintrust Bio and Akava; he is a patent holder with Johns Hopkins [ALS] and has received grant/research/clinical trial support from Apellis Pharma, Biogen Idec, Cytokinetics, Helixmith, Calico, Sanofi, Department of Defense ALSRP, Maryland Stem Cell Research Fund, Massachusetts General Hospital, Medicinova, and NINDS. He serves as consultant or advisory board member for Amylyx; Cytokinetics, Roche, Healey Center, Nura Bio, Northeast ALS Consortium, Akava, Inflammx, and Secretome. Dr. Scelsa did not report any conflicts of interest. Dr. Macgowan and Dr. Maiser have no relevant conflicts of interest to disclose.
References
1. Mehta P et al. Prevalence of amyotrophic lateral sclerosis in the United States using established and novel methodologies, 2017. Amyotroph Lateral Scler Frontotemporal Degener. 2023;24(1-2):108-16. doi: 10.1080/21678421.2022.2059380.
2. Mead RJ et al. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov. 2023;22(3):185-212. doi: 10.1038/s41573-022-00612-2.
3. Longinetti E and Fang F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr Opin Neurol. 2019;32(5):771-6. doi: 10.1097/WCO.0000000000000730.
4. van den Bos MAJ et al. Pathophysiology and diagnosis of ALS: Insights from advances in neurophysiological techniques. Int J Mol Sci. 2019;20(11):2818. doi: 10.3390/ijms20112818.
5. Neumann M et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130-3. doi: 10.1126/science.1134108.
6. Ling S-C et al. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416-38. doi: 10.1016/j.neuron.2013.07.033.
7. Ranganathan R et al. Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Front Neurosci. 2020;14:684. doi: 10.3389/fnins.2020.00684.
8. Ryan M et al. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2019;76(11):1367-74. doi: 10.1001/jamaneurol.2019.2044.
9. van Rheenen W et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet. 2021;53(12):1636-48. doi: 10.1038/s41588-021-00973-1.
10. Miller TM et al; VALOR and OLE Working Group. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099-110. doi: 10.1056/NEJMoa2204705.
Emerging therapies in Duchenne and facioscapulohumeral muscular dystrophy
“There have been so many breakthroughs recently on the side of genetically targeted treatment [for muscular dystrophy] that supports muscle better,” said John F. Brandsema, MD, a child neurologist and section head at Children’s Hospital of Philadelphia, in an interview with Neurology Reviews 2023 Rare Neurological Disease Special Report. “We’re starting to see clinical response to some things that have been in trials – after decades of banging our heads on the wall trying new therapies, only to see them fail. I think it’s about reframing Duchenne muscular dystrophy [DMD] and facioscapulohumeral muscular dystrophy [FSHD] as treatable by target therapy because previously, they were treated with supportive care.”
DMD: Current and emerging therapies

DMD is caused by a mutation in the dystrophin gene on the X chromosome that inhibits production of dystrophin, a protein that shields muscles from injury during contraction. Dystrophin deficiency prevents muscle recovery, resulting in muscle-cell death and, ultimately, loss of function due to muscle degeneration.
FDA-approved exon-skipping therapies. Treatment modalities for what has historically been an incurable, lifespan-shortening disease involved supportive care that addresses symptoms, not the underlying cause. Consequently, many patients with DMD live only into their 20s and 30s. The tide began to turn in 2016, however, when the U.S. Food and Drug Administration granted accelerated approval for eteplirsen, an exon 51–skipping treatment that was the first RNA-based therapy for DMD to target the underlying cause. Additional exon-skipping therapies followed, including casimersen, which skips exon 45, and golodirsen and viltolarsen, which skip exon 53.
AOC 1044: Novel exon-skipping. In April 2023, the FDA granted orphan-drug designation to the experimental drug antibody oligonucleotide conjugate (AOC) 1044 that skips exon 44. A small interfering RNA (siRNA), AOC 1044 works in patients who have a mutation amenable to exon 44 skipping (a disease type known as DMD44) by delivering phosphorodiamidate morpholino to skeletal muscle and heart tissue that skips exon 44. The process allows for dystrophin production, thereby preventing degradation of muscle tissue.
The orphan drug status of AOC 1044 made it available to the population of patients enrolled in the EXPLORE44 Phase 1/2 trial. However, studies demonstrating effectiveness of the drug – with the hope of, ultimately, providing widespread access to AOC 1044 – are still underway. In one of those studies, investigators expect to enroll approximately 40 healthy volunteers and 24 DMD44 patients 7-27 years of age.3 The study will evaluate the effects of exon skipping and dystrophin protein levels in participants who have DMD44.
Delandistrogene moxeparvovec. Oct. 27, 2021, marked the inception of the phase 3 Multinational, Randomized, Double-Blind, Placebo-Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Subjects With Duchenne Muscular Dystrophy (EMBARK). The trial is evaluating the safety and efficacy of the gene-therapy agent delandistrogene moxeparvovec in ambulatory boys who were 4 to less than 8 years of age at randomization. The 126 boys enrolled in the trial met the criteria of (1) a diagnosis of DMD confirmed by documented clinical findings and previous genetic testing and (2) a pathogenic frameshift mutation stop codon located between exons 18 and 79 (inclusive), except for a mutation fully contained within exon 45.
Additional inclusion criteria were (1) the ability to cooperate with motor-assessment testing and (2) receiving a steady daily dose of oral corticosteroid for 12 weeks or longer prior to screening, and (3) the expectation of maintaining the study dosage throughout screening. Boys who had previously received gene therapy, investigational medication, or any treatment that could have amplified dystrophin expression within the time limit specified by the protocol were ineligible to participate. Boys were excluded from the study if they presented with any other illness, medical condition, or need for chronic drug treatment.
Exon-skipping therapies in trials. Various biotech and pharmaceutical companies have initiated clinical trials to explore the potential of additional exon-skipping therapies for the DMD population:
ENTR-601-44 is another exon 44–skipping therapy in the pipeline.
On Aug. 22, 2023, the FDA approved delandistrogene moxeparvovec-rokl, a recombinant gene therapy utilizing an adenovirus vector. The product is indicated for ambulatory patients with DMD 4-5 years of age who have a confirmed mutation of the dystrophin gene.
Dyne Therapeutics is actively recruiting participants to investigate Dyne 251, its exon 51–skipping therapy.
Trials are in the works by BioMarin Pharmaceutical for its next-generation peptide-conjugated phosphorodiamidate morpholino oligomers (PPMO) for skipping exon 51.
Despite the prospects of such therapy, therapeutic targeting of exon 44 addresses only patients with DMD44, who account for approximately 10% of the DMD population. Disease involving the most prevalent site of a dystrophin gene mutation, exon 51, affects 13% of the DMD population. This leaves the majority of patients with DMD without gene therapy. Yet Dr. Brandsema is optimistic nevertheless.
“We were just failing over and over again with DMD treatment, but there is some hope now,” Dr. Brandsema said. “Also, FSHD is right on the cusp of having new therapies approaching.”
FSHD: Emerging therapies
The third more common type of muscular dystrophy is not a life-threatening condition. FSHD affects approximately 4 of every 100,000 people.1 An autosomal-dominant condition, FSHD is ultimately caused by inappropriate expression of the DUX4 protein product – a consequence of a complex genetic activity involving DUX4, its chromosomal locus, and the number of repeats of a microsatellite called D4Z4.4 The disease usually starts in proximal regions of the face (that is, surrounding the eyes and mouth), before spreading to muscular groups of the limbs – most prominently, muscles of the scapulae and humeri. Symptoms usually appear in these places initially, but the condition can affect any part of the body. Fifty percent of FSHD patients experience loss of high-frequency hearing and present with retinovasculopathy. Like DMD, FSHD varies in severity, with some forms presenting at birth.
AOC 1020-CS1 is an example of a new FSHD treatment under investigation. The phase 1/2 FORTITUDE trial is a randomized, double-blind, placebo-controlled study exploring the safety, tolerability, pharmacokinetics, pharmacodynamics, and potential efficacy of single- and multiple-dose AOC 1020-CS1 therapy in FSHD.5 The trial began in April 2023; estimated completion date is September 2025.
As with many rare diseases, however, following patients and capturing data that fully narrate their story remains challenging in both DMD and FSHD. Although clinical trials undoubtedly offer hope of expanding treatment options and additional insights into disease-state management, the often insidious, complex nature of some rare diseases, such as DMD and FSHD, presents some limitations.
“Patients are hard to measure,” Dr. Brandsema explained, “because they’re so variable at baseline in history and progress in a different [slower] way than timelines are set up in our system to study drugs.”
Neonatal screening and early diagnosis: Imperative for improving outcomes
Neonatal screening helps with early detection and treatment. Prompt diagnosis does not necessarily prolong a DMD patient’s life, but it can enhance their quality of life.

DNA diagnostics. A critical component of the path to treatment is DNA diagnostics. According to Barry J. Byrne, MD, PhD, chief medical advisor of the Muscular Dystrophy Association, the Human Genome Project conducted by the National Institutes of Health helped make DNA tests affordable; such tests run about $800 today. However, given continuous advancements in sequencing, Dr. Byrne said that whole-exome sequencing for $100 is within reach.
In terms of accessibility, some nations – Canada is an example – include testing as part of national health care services. In the United States, coverage for testing varies by health insurance plan. In addition, some plans have favored rapid diagnostic testing, and the overall cost is often individualized to the patient.
Early diagnosis and supportive care. Early diagnosis can certainly help improve DMD patients’ quality of life; supportive care provides some benefit. Dr. Byrne stressed the importance of managing extraskeletal clinical manifestations in this patient population. A critical area is initiating cardiovascular treatment immediately following diagnosis, even if the patient does not exhibit cardiovascular symptoms.
“Cardiac manifestations are actually the cause of mortality in DMD, and most boys with DMD should begin cardiovascular treatment shortly after diagnosis,” Dr. Byrne told Neurology Reviews 2023 Rare Neurological Disease Special Report. “The message to neurologists is that these patients can benefit from early cardiovascular treatment because we can prevent the complications of DMD-related heart failure until much later in life.”
Historically, clinicians used echocardiography as the mainstay tool to assess cardiovascular function; however, more and more clinicians are turning to magnetic resonance imaging for such investigation. Dr. Byrne, a cardiologist, explained that magnetic resonance imaging identifies cardiovascular dysfunction at earlier stages than echocardiography can. In addition, although DMD patients frequently experience fatigue, Dr. Byrne cautions neurologists that fatigue is usually related to muscle weakness, not necessarily heart failure.
DMD therapies carry a hefty price
Right now, the projected price range of AOC 1044 is $3.2 million to $3.4 million. Akin to the case with onasemnogene abeparvovec-xioi (Zolgensma) for spinal muscular atrophy, the world’s first gene therapy and first seven-figure drug, the manufacturer of AOC 1044 based pricing on the anticipated cost of treating a DMD44 patient throughout the lifespan, according to Dr. Byrne.
Delandistrogene moxeparvovec might come with an even higher price tag. A cost-effectiveness analysis study priced the therapy at $5 million. In a presentation to investors, the manufacturer projected the price in the range of $5 million to $13 million.6,7
‘It takes a village’: Comprehensive care requires a multidisciplinary team
Dr. Brandsema and Dr. Byrne agree: Optimizing outcomes requires ongoing coordinated and collaborative efforts of an interdisciplinary team of health care providers for the duration of DMD and FSHD patients’ lifespan.
A neurologist by training, Dr. Brandsema recognizes the importance of interdisciplinary collaboration in caring for patients with DMD, given the multiorgan manifestations of the disease.
“We have some hope with DMD, and FSHD is right on the cusp of having new therapies approaching ... It is important to recognize that interdisciplinary follow-up and optimized standard of care are important after dosing.”
“I think many patients living with neurological disorders have multiple providers they rely on for care,” Dr. Byrne said, “but cardiovascular and pulmonary care are important because both are affected in the case of DMD – not so much in FSHD.”
Ultimately, advancements in therapy and care give patients living with these disorders, and their caregivers, a renewed sense of hope – hope that their life will be improved by breakthrough therapies that have been approved or will arrive soon.
Dr. Brandsema discloses he is a consultant for Alexion, Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech/Roche, Janssen, Marathon, Momenta, NS Pharma, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is a speaker for AveXis and Biogen, a medical advisory council member for Cure SMA, and a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Catabasis, CSL Behring, Cytokinetics, Fibrogen, Genentech/Roche, Ionis, Lilly, Janssen, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Dr. Byrne has no relevant financial disclosures.
References
1. Centers for Disease Control and Prevention. What is muscular dystrophy? Updated Nov. 21, 2022. Accessed Sept. 3, 2023. https://www.cdc.gov/ncbddd/musculardystrophy/facts.html.
2. FDA approves first gene therapy for treatment of certain patients with Duchenne muscular dystrophy. U.S. Food and Drug Administration. Press release. June 22, 2023. Accessed Sept. 3, 2023. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-certain-patients-duchenne-muscular-dystrophy.
3. Study of AOC 1044 in healthy adult volunteers and participants with Duchenne muscular dystrophy (DMD) mutations amenable to exon 44 skipping (EXPLORE44). ClinicalTrials.gov Identifier: NCT05670730. Updated April 4, 2023. Accessed Sep. 3, 2023. https://www.clinicaltrials.gov/study/NCT05670730?cond=DMD&intr=AOC%201044&rank=1.
4. Statland JM, Tawil R. Facioscapulohumeral muscular dystrophy. Continuum (Minneap. Minn). 2016;22(6, Muscle and Neuromuscular Junction Disorders):1916-31. doi: 10.1212/CON.0000000000000399.
5. Phase 1/2 study of AOC 1020 in adults with facioscapulohumeral muscular dystrophy (FSHD) (FORTITUDE). ClinicalTrials.gov Identifier: NCT05747924. Updated Aug. 9, 2023. Accessed Sept. 3, 2023. https://clinicaltrials.gov/study/NCT05747924?term=fORTITUDE&cond=Facioscapulohumeral%20Muscular%20Dystrophy&rank=1.
6. Klimchak AC, Sedita LE, Rodino-Klapac LR, et al. Assessing the value of delandistrogene moxeparvovec (SRP-9001) gene therapy in patients with Duchenne muscular dystrophy in the United States. J Mark Access Health Policy. 2023;11(1):2216518. doi: 10.1080/20016689.2023.2216518.
7. Ingram D. [Investor relations presentation.] Sarepta Therapeutics website. June 22, 2023. Accessed Sept. 3, 2023. https://investorrelations.sarepta.com/static-files/7216948c-f688-4024-922e-39761bc7a984.
“There have been so many breakthroughs recently on the side of genetically targeted treatment [for muscular dystrophy] that supports muscle better,” said John F. Brandsema, MD, a child neurologist and section head at Children’s Hospital of Philadelphia, in an interview with Neurology Reviews 2023 Rare Neurological Disease Special Report. “We’re starting to see clinical response to some things that have been in trials – after decades of banging our heads on the wall trying new therapies, only to see them fail. I think it’s about reframing Duchenne muscular dystrophy [DMD] and facioscapulohumeral muscular dystrophy [FSHD] as treatable by target therapy because previously, they were treated with supportive care.”
DMD: Current and emerging therapies
DMD is caused by a mutation in the dystrophin gene on the X chromosome that inhibits production of dystrophin, a protein that shields muscles from injury during contraction. Dystrophin deficiency prevents muscle recovery, resulting in muscle-cell death and, ultimately, loss of function due to muscle degeneration.
FDA-approved exon-skipping therapies. Treatment modalities for what has historically been an incurable, lifespan-shortening disease involved supportive care that addresses symptoms, not the underlying cause. Consequently, many patients with DMD live only into their 20s and 30s. The tide began to turn in 2016, however, when the U.S. Food and Drug Administration granted accelerated approval for eteplirsen, an exon 51–skipping treatment that was the first RNA-based therapy for DMD to target the underlying cause. Additional exon-skipping therapies followed, including casimersen, which skips exon 45, and golodirsen and viltolarsen, which skip exon 53.
AOC 1044: Novel exon-skipping. In April 2023, the FDA granted orphan-drug designation to the experimental drug antibody oligonucleotide conjugate (AOC) 1044 that skips exon 44. A small interfering RNA (siRNA), AOC 1044 works in patients who have a mutation amenable to exon 44 skipping (a disease type known as DMD44) by delivering phosphorodiamidate morpholino to skeletal muscle and heart tissue that skips exon 44. The process allows for dystrophin production, thereby preventing degradation of muscle tissue.
The orphan drug status of AOC 1044 made it available to the population of patients enrolled in the EXPLORE44 Phase 1/2 trial. However, studies demonstrating effectiveness of the drug – with the hope of, ultimately, providing widespread access to AOC 1044 – are still underway. In one of those studies, investigators expect to enroll approximately 40 healthy volunteers and 24 DMD44 patients 7-27 years of age.3 The study will evaluate the effects of exon skipping and dystrophin protein levels in participants who have DMD44.
Delandistrogene moxeparvovec. Oct. 27, 2021, marked the inception of the phase 3 Multinational, Randomized, Double-Blind, Placebo-Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Subjects With Duchenne Muscular Dystrophy (EMBARK). The trial is evaluating the safety and efficacy of the gene-therapy agent delandistrogene moxeparvovec in ambulatory boys who were 4 to less than 8 years of age at randomization. The 126 boys enrolled in the trial met the criteria of (1) a diagnosis of DMD confirmed by documented clinical findings and previous genetic testing and (2) a pathogenic frameshift mutation stop codon located between exons 18 and 79 (inclusive), except for a mutation fully contained within exon 45.
Additional inclusion criteria were (1) the ability to cooperate with motor-assessment testing and (2) receiving a steady daily dose of oral corticosteroid for 12 weeks or longer prior to screening, and (3) the expectation of maintaining the study dosage throughout screening. Boys who had previously received gene therapy, investigational medication, or any treatment that could have amplified dystrophin expression within the time limit specified by the protocol were ineligible to participate. Boys were excluded from the study if they presented with any other illness, medical condition, or need for chronic drug treatment.
Exon-skipping therapies in trials. Various biotech and pharmaceutical companies have initiated clinical trials to explore the potential of additional exon-skipping therapies for the DMD population:
ENTR-601-44 is another exon 44–skipping therapy in the pipeline.
On Aug. 22, 2023, the FDA approved delandistrogene moxeparvovec-rokl, a recombinant gene therapy utilizing an adenovirus vector. The product is indicated for ambulatory patients with DMD 4-5 years of age who have a confirmed mutation of the dystrophin gene.
Dyne Therapeutics is actively recruiting participants to investigate Dyne 251, its exon 51–skipping therapy.
Trials are in the works by BioMarin Pharmaceutical for its next-generation peptide-conjugated phosphorodiamidate morpholino oligomers (PPMO) for skipping exon 51.
Despite the prospects of such therapy, therapeutic targeting of exon 44 addresses only patients with DMD44, who account for approximately 10% of the DMD population. Disease involving the most prevalent site of a dystrophin gene mutation, exon 51, affects 13% of the DMD population. This leaves the majority of patients with DMD without gene therapy. Yet Dr. Brandsema is optimistic nevertheless.
“We were just failing over and over again with DMD treatment, but there is some hope now,” Dr. Brandsema said. “Also, FSHD is right on the cusp of having new therapies approaching.”
FSHD: Emerging therapies
The third more common type of muscular dystrophy is not a life-threatening condition. FSHD affects approximately 4 of every 100,000 people.1 An autosomal-dominant condition, FSHD is ultimately caused by inappropriate expression of the DUX4 protein product – a consequence of a complex genetic activity involving DUX4, its chromosomal locus, and the number of repeats of a microsatellite called D4Z4.4 The disease usually starts in proximal regions of the face (that is, surrounding the eyes and mouth), before spreading to muscular groups of the limbs – most prominently, muscles of the scapulae and humeri. Symptoms usually appear in these places initially, but the condition can affect any part of the body. Fifty percent of FSHD patients experience loss of high-frequency hearing and present with retinovasculopathy. Like DMD, FSHD varies in severity, with some forms presenting at birth.
AOC 1020-CS1 is an example of a new FSHD treatment under investigation. The phase 1/2 FORTITUDE trial is a randomized, double-blind, placebo-controlled study exploring the safety, tolerability, pharmacokinetics, pharmacodynamics, and potential efficacy of single- and multiple-dose AOC 1020-CS1 therapy in FSHD.5 The trial began in April 2023; estimated completion date is September 2025.
As with many rare diseases, however, following patients and capturing data that fully narrate their story remains challenging in both DMD and FSHD. Although clinical trials undoubtedly offer hope of expanding treatment options and additional insights into disease-state management, the often insidious, complex nature of some rare diseases, such as DMD and FSHD, presents some limitations.
“Patients are hard to measure,” Dr. Brandsema explained, “because they’re so variable at baseline in history and progress in a different [slower] way than timelines are set up in our system to study drugs.”
Neonatal screening and early diagnosis: Imperative for improving outcomes
Neonatal screening helps with early detection and treatment. Prompt diagnosis does not necessarily prolong a DMD patient’s life, but it can enhance their quality of life.
DNA diagnostics. A critical component of the path to treatment is DNA diagnostics. According to Barry J. Byrne, MD, PhD, chief medical advisor of the Muscular Dystrophy Association, the Human Genome Project conducted by the National Institutes of Health helped make DNA tests affordable; such tests run about $800 today. However, given continuous advancements in sequencing, Dr. Byrne said that whole-exome sequencing for $100 is within reach.
In terms of accessibility, some nations – Canada is an example – include testing as part of national health care services. In the United States, coverage for testing varies by health insurance plan. In addition, some plans have favored rapid diagnostic testing, and the overall cost is often individualized to the patient.
Early diagnosis and supportive care. Early diagnosis can certainly help improve DMD patients’ quality of life; supportive care provides some benefit. Dr. Byrne stressed the importance of managing extraskeletal clinical manifestations in this patient population. A critical area is initiating cardiovascular treatment immediately following diagnosis, even if the patient does not exhibit cardiovascular symptoms.
“Cardiac manifestations are actually the cause of mortality in DMD, and most boys with DMD should begin cardiovascular treatment shortly after diagnosis,” Dr. Byrne told Neurology Reviews 2023 Rare Neurological Disease Special Report. “The message to neurologists is that these patients can benefit from early cardiovascular treatment because we can prevent the complications of DMD-related heart failure until much later in life.”
Historically, clinicians used echocardiography as the mainstay tool to assess cardiovascular function; however, more and more clinicians are turning to magnetic resonance imaging for such investigation. Dr. Byrne, a cardiologist, explained that magnetic resonance imaging identifies cardiovascular dysfunction at earlier stages than echocardiography can. In addition, although DMD patients frequently experience fatigue, Dr. Byrne cautions neurologists that fatigue is usually related to muscle weakness, not necessarily heart failure.
DMD therapies carry a hefty price
Right now, the projected price range of AOC 1044 is $3.2 million to $3.4 million. Akin to the case with onasemnogene abeparvovec-xioi (Zolgensma) for spinal muscular atrophy, the world’s first gene therapy and first seven-figure drug, the manufacturer of AOC 1044 based pricing on the anticipated cost of treating a DMD44 patient throughout the lifespan, according to Dr. Byrne.
Delandistrogene moxeparvovec might come with an even higher price tag. A cost-effectiveness analysis study priced the therapy at $5 million. In a presentation to investors, the manufacturer projected the price in the range of $5 million to $13 million.6,7
‘It takes a village’: Comprehensive care requires a multidisciplinary team
Dr. Brandsema and Dr. Byrne agree: Optimizing outcomes requires ongoing coordinated and collaborative efforts of an interdisciplinary team of health care providers for the duration of DMD and FSHD patients’ lifespan.
A neurologist by training, Dr. Brandsema recognizes the importance of interdisciplinary collaboration in caring for patients with DMD, given the multiorgan manifestations of the disease.
“We have some hope with DMD, and FSHD is right on the cusp of having new therapies approaching ... It is important to recognize that interdisciplinary follow-up and optimized standard of care are important after dosing.”
“I think many patients living with neurological disorders have multiple providers they rely on for care,” Dr. Byrne said, “but cardiovascular and pulmonary care are important because both are affected in the case of DMD – not so much in FSHD.”
Ultimately, advancements in therapy and care give patients living with these disorders, and their caregivers, a renewed sense of hope – hope that their life will be improved by breakthrough therapies that have been approved or will arrive soon.
Dr. Brandsema discloses he is a consultant for Alexion, Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech/Roche, Janssen, Marathon, Momenta, NS Pharma, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is a speaker for AveXis and Biogen, a medical advisory council member for Cure SMA, and a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Catabasis, CSL Behring, Cytokinetics, Fibrogen, Genentech/Roche, Ionis, Lilly, Janssen, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Dr. Byrne has no relevant financial disclosures.
References
1. Centers for Disease Control and Prevention. What is muscular dystrophy? Updated Nov. 21, 2022. Accessed Sept. 3, 2023. https://www.cdc.gov/ncbddd/musculardystrophy/facts.html.
2. FDA approves first gene therapy for treatment of certain patients with Duchenne muscular dystrophy. U.S. Food and Drug Administration. Press release. June 22, 2023. Accessed Sept. 3, 2023. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-certain-patients-duchenne-muscular-dystrophy.
3. Study of AOC 1044 in healthy adult volunteers and participants with Duchenne muscular dystrophy (DMD) mutations amenable to exon 44 skipping (EXPLORE44). ClinicalTrials.gov Identifier: NCT05670730. Updated April 4, 2023. Accessed Sep. 3, 2023. https://www.clinicaltrials.gov/study/NCT05670730?cond=DMD&intr=AOC%201044&rank=1.
4. Statland JM, Tawil R. Facioscapulohumeral muscular dystrophy. Continuum (Minneap. Minn). 2016;22(6, Muscle and Neuromuscular Junction Disorders):1916-31. doi: 10.1212/CON.0000000000000399.
5. Phase 1/2 study of AOC 1020 in adults with facioscapulohumeral muscular dystrophy (FSHD) (FORTITUDE). ClinicalTrials.gov Identifier: NCT05747924. Updated Aug. 9, 2023. Accessed Sept. 3, 2023. https://clinicaltrials.gov/study/NCT05747924?term=fORTITUDE&cond=Facioscapulohumeral%20Muscular%20Dystrophy&rank=1.
6. Klimchak AC, Sedita LE, Rodino-Klapac LR, et al. Assessing the value of delandistrogene moxeparvovec (SRP-9001) gene therapy in patients with Duchenne muscular dystrophy in the United States. J Mark Access Health Policy. 2023;11(1):2216518. doi: 10.1080/20016689.2023.2216518.
7. Ingram D. [Investor relations presentation.] Sarepta Therapeutics website. June 22, 2023. Accessed Sept. 3, 2023. https://investorrelations.sarepta.com/static-files/7216948c-f688-4024-922e-39761bc7a984.
“There have been so many breakthroughs recently on the side of genetically targeted treatment [for muscular dystrophy] that supports muscle better,” said John F. Brandsema, MD, a child neurologist and section head at Children’s Hospital of Philadelphia, in an interview with Neurology Reviews 2023 Rare Neurological Disease Special Report. “We’re starting to see clinical response to some things that have been in trials – after decades of banging our heads on the wall trying new therapies, only to see them fail. I think it’s about reframing Duchenne muscular dystrophy [DMD] and facioscapulohumeral muscular dystrophy [FSHD] as treatable by target therapy because previously, they were treated with supportive care.”
DMD: Current and emerging therapies
DMD is caused by a mutation in the dystrophin gene on the X chromosome that inhibits production of dystrophin, a protein that shields muscles from injury during contraction. Dystrophin deficiency prevents muscle recovery, resulting in muscle-cell death and, ultimately, loss of function due to muscle degeneration.
FDA-approved exon-skipping therapies. Treatment modalities for what has historically been an incurable, lifespan-shortening disease involved supportive care that addresses symptoms, not the underlying cause. Consequently, many patients with DMD live only into their 20s and 30s. The tide began to turn in 2016, however, when the U.S. Food and Drug Administration granted accelerated approval for eteplirsen, an exon 51–skipping treatment that was the first RNA-based therapy for DMD to target the underlying cause. Additional exon-skipping therapies followed, including casimersen, which skips exon 45, and golodirsen and viltolarsen, which skip exon 53.
AOC 1044: Novel exon-skipping. In April 2023, the FDA granted orphan-drug designation to the experimental drug antibody oligonucleotide conjugate (AOC) 1044 that skips exon 44. A small interfering RNA (siRNA), AOC 1044 works in patients who have a mutation amenable to exon 44 skipping (a disease type known as DMD44) by delivering phosphorodiamidate morpholino to skeletal muscle and heart tissue that skips exon 44. The process allows for dystrophin production, thereby preventing degradation of muscle tissue.
The orphan drug status of AOC 1044 made it available to the population of patients enrolled in the EXPLORE44 Phase 1/2 trial. However, studies demonstrating effectiveness of the drug – with the hope of, ultimately, providing widespread access to AOC 1044 – are still underway. In one of those studies, investigators expect to enroll approximately 40 healthy volunteers and 24 DMD44 patients 7-27 years of age.3 The study will evaluate the effects of exon skipping and dystrophin protein levels in participants who have DMD44.
Delandistrogene moxeparvovec. Oct. 27, 2021, marked the inception of the phase 3 Multinational, Randomized, Double-Blind, Placebo-Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Subjects With Duchenne Muscular Dystrophy (EMBARK). The trial is evaluating the safety and efficacy of the gene-therapy agent delandistrogene moxeparvovec in ambulatory boys who were 4 to less than 8 years of age at randomization. The 126 boys enrolled in the trial met the criteria of (1) a diagnosis of DMD confirmed by documented clinical findings and previous genetic testing and (2) a pathogenic frameshift mutation stop codon located between exons 18 and 79 (inclusive), except for a mutation fully contained within exon 45.
Additional inclusion criteria were (1) the ability to cooperate with motor-assessment testing and (2) receiving a steady daily dose of oral corticosteroid for 12 weeks or longer prior to screening, and (3) the expectation of maintaining the study dosage throughout screening. Boys who had previously received gene therapy, investigational medication, or any treatment that could have amplified dystrophin expression within the time limit specified by the protocol were ineligible to participate. Boys were excluded from the study if they presented with any other illness, medical condition, or need for chronic drug treatment.
Exon-skipping therapies in trials. Various biotech and pharmaceutical companies have initiated clinical trials to explore the potential of additional exon-skipping therapies for the DMD population:
ENTR-601-44 is another exon 44–skipping therapy in the pipeline.
On Aug. 22, 2023, the FDA approved delandistrogene moxeparvovec-rokl, a recombinant gene therapy utilizing an adenovirus vector. The product is indicated for ambulatory patients with DMD 4-5 years of age who have a confirmed mutation of the dystrophin gene.
Dyne Therapeutics is actively recruiting participants to investigate Dyne 251, its exon 51–skipping therapy.
Trials are in the works by BioMarin Pharmaceutical for its next-generation peptide-conjugated phosphorodiamidate morpholino oligomers (PPMO) for skipping exon 51.
Despite the prospects of such therapy, therapeutic targeting of exon 44 addresses only patients with DMD44, who account for approximately 10% of the DMD population. Disease involving the most prevalent site of a dystrophin gene mutation, exon 51, affects 13% of the DMD population. This leaves the majority of patients with DMD without gene therapy. Yet Dr. Brandsema is optimistic nevertheless.
“We were just failing over and over again with DMD treatment, but there is some hope now,” Dr. Brandsema said. “Also, FSHD is right on the cusp of having new therapies approaching.”
FSHD: Emerging therapies
The third more common type of muscular dystrophy is not a life-threatening condition. FSHD affects approximately 4 of every 100,000 people.1 An autosomal-dominant condition, FSHD is ultimately caused by inappropriate expression of the DUX4 protein product – a consequence of a complex genetic activity involving DUX4, its chromosomal locus, and the number of repeats of a microsatellite called D4Z4.4 The disease usually starts in proximal regions of the face (that is, surrounding the eyes and mouth), before spreading to muscular groups of the limbs – most prominently, muscles of the scapulae and humeri. Symptoms usually appear in these places initially, but the condition can affect any part of the body. Fifty percent of FSHD patients experience loss of high-frequency hearing and present with retinovasculopathy. Like DMD, FSHD varies in severity, with some forms presenting at birth.
AOC 1020-CS1 is an example of a new FSHD treatment under investigation. The phase 1/2 FORTITUDE trial is a randomized, double-blind, placebo-controlled study exploring the safety, tolerability, pharmacokinetics, pharmacodynamics, and potential efficacy of single- and multiple-dose AOC 1020-CS1 therapy in FSHD.5 The trial began in April 2023; estimated completion date is September 2025.
As with many rare diseases, however, following patients and capturing data that fully narrate their story remains challenging in both DMD and FSHD. Although clinical trials undoubtedly offer hope of expanding treatment options and additional insights into disease-state management, the often insidious, complex nature of some rare diseases, such as DMD and FSHD, presents some limitations.
“Patients are hard to measure,” Dr. Brandsema explained, “because they’re so variable at baseline in history and progress in a different [slower] way than timelines are set up in our system to study drugs.”
Neonatal screening and early diagnosis: Imperative for improving outcomes
Neonatal screening helps with early detection and treatment. Prompt diagnosis does not necessarily prolong a DMD patient’s life, but it can enhance their quality of life.
DNA diagnostics. A critical component of the path to treatment is DNA diagnostics. According to Barry J. Byrne, MD, PhD, chief medical advisor of the Muscular Dystrophy Association, the Human Genome Project conducted by the National Institutes of Health helped make DNA tests affordable; such tests run about $800 today. However, given continuous advancements in sequencing, Dr. Byrne said that whole-exome sequencing for $100 is within reach.
In terms of accessibility, some nations – Canada is an example – include testing as part of national health care services. In the United States, coverage for testing varies by health insurance plan. In addition, some plans have favored rapid diagnostic testing, and the overall cost is often individualized to the patient.
Early diagnosis and supportive care. Early diagnosis can certainly help improve DMD patients’ quality of life; supportive care provides some benefit. Dr. Byrne stressed the importance of managing extraskeletal clinical manifestations in this patient population. A critical area is initiating cardiovascular treatment immediately following diagnosis, even if the patient does not exhibit cardiovascular symptoms.
“Cardiac manifestations are actually the cause of mortality in DMD, and most boys with DMD should begin cardiovascular treatment shortly after diagnosis,” Dr. Byrne told Neurology Reviews 2023 Rare Neurological Disease Special Report. “The message to neurologists is that these patients can benefit from early cardiovascular treatment because we can prevent the complications of DMD-related heart failure until much later in life.”
Historically, clinicians used echocardiography as the mainstay tool to assess cardiovascular function; however, more and more clinicians are turning to magnetic resonance imaging for such investigation. Dr. Byrne, a cardiologist, explained that magnetic resonance imaging identifies cardiovascular dysfunction at earlier stages than echocardiography can. In addition, although DMD patients frequently experience fatigue, Dr. Byrne cautions neurologists that fatigue is usually related to muscle weakness, not necessarily heart failure.
DMD therapies carry a hefty price
Right now, the projected price range of AOC 1044 is $3.2 million to $3.4 million. Akin to the case with onasemnogene abeparvovec-xioi (Zolgensma) for spinal muscular atrophy, the world’s first gene therapy and first seven-figure drug, the manufacturer of AOC 1044 based pricing on the anticipated cost of treating a DMD44 patient throughout the lifespan, according to Dr. Byrne.
Delandistrogene moxeparvovec might come with an even higher price tag. A cost-effectiveness analysis study priced the therapy at $5 million. In a presentation to investors, the manufacturer projected the price in the range of $5 million to $13 million.6,7
‘It takes a village’: Comprehensive care requires a multidisciplinary team
Dr. Brandsema and Dr. Byrne agree: Optimizing outcomes requires ongoing coordinated and collaborative efforts of an interdisciplinary team of health care providers for the duration of DMD and FSHD patients’ lifespan.
A neurologist by training, Dr. Brandsema recognizes the importance of interdisciplinary collaboration in caring for patients with DMD, given the multiorgan manifestations of the disease.
“We have some hope with DMD, and FSHD is right on the cusp of having new therapies approaching ... It is important to recognize that interdisciplinary follow-up and optimized standard of care are important after dosing.”
“I think many patients living with neurological disorders have multiple providers they rely on for care,” Dr. Byrne said, “but cardiovascular and pulmonary care are important because both are affected in the case of DMD – not so much in FSHD.”
Ultimately, advancements in therapy and care give patients living with these disorders, and their caregivers, a renewed sense of hope – hope that their life will be improved by breakthrough therapies that have been approved or will arrive soon.
Dr. Brandsema discloses he is a consultant for Alexion, Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech/Roche, Janssen, Marathon, Momenta, NS Pharma, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is a speaker for AveXis and Biogen, a medical advisory council member for Cure SMA, and a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Catabasis, CSL Behring, Cytokinetics, Fibrogen, Genentech/Roche, Ionis, Lilly, Janssen, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Dr. Byrne has no relevant financial disclosures.
References
1. Centers for Disease Control and Prevention. What is muscular dystrophy? Updated Nov. 21, 2022. Accessed Sept. 3, 2023. https://www.cdc.gov/ncbddd/musculardystrophy/facts.html.
2. FDA approves first gene therapy for treatment of certain patients with Duchenne muscular dystrophy. U.S. Food and Drug Administration. Press release. June 22, 2023. Accessed Sept. 3, 2023. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-certain-patients-duchenne-muscular-dystrophy.
3. Study of AOC 1044 in healthy adult volunteers and participants with Duchenne muscular dystrophy (DMD) mutations amenable to exon 44 skipping (EXPLORE44). ClinicalTrials.gov Identifier: NCT05670730. Updated April 4, 2023. Accessed Sep. 3, 2023. https://www.clinicaltrials.gov/study/NCT05670730?cond=DMD&intr=AOC%201044&rank=1.
4. Statland JM, Tawil R. Facioscapulohumeral muscular dystrophy. Continuum (Minneap. Minn). 2016;22(6, Muscle and Neuromuscular Junction Disorders):1916-31. doi: 10.1212/CON.0000000000000399.
5. Phase 1/2 study of AOC 1020 in adults with facioscapulohumeral muscular dystrophy (FSHD) (FORTITUDE). ClinicalTrials.gov Identifier: NCT05747924. Updated Aug. 9, 2023. Accessed Sept. 3, 2023. https://clinicaltrials.gov/study/NCT05747924?term=fORTITUDE&cond=Facioscapulohumeral%20Muscular%20Dystrophy&rank=1.
6. Klimchak AC, Sedita LE, Rodino-Klapac LR, et al. Assessing the value of delandistrogene moxeparvovec (SRP-9001) gene therapy in patients with Duchenne muscular dystrophy in the United States. J Mark Access Health Policy. 2023;11(1):2216518. doi: 10.1080/20016689.2023.2216518.
7. Ingram D. [Investor relations presentation.] Sarepta Therapeutics website. June 22, 2023. Accessed Sept. 3, 2023. https://investorrelations.sarepta.com/static-files/7216948c-f688-4024-922e-39761bc7a984.

Pragmatic solutions to ‘catastrophic’ global stroke burden
Deaths and disability because of stroke are expected to rise alarmingly over the next 30 years, with almost 10 million stroke deaths forecast annually by 2050, according to a new report from the World Stroke Organization–Lancet Neurology Commission Stroke Collaboration Group.
“This highlights the need for urgent measures to reduce stroke burden worldwide, with an emphasis on low- and middle-income countries,” the report authors stated.
These measures include an increase in trained health care workers who can implement effective primary prevention strategies, including the early detection and adequate management of hypertension.
On the basis of a review of evidence-based guidelines, recent surveys, and in-depth interviews with stroke experts around the world, the WSO–Lancet Neurology Commission made evidence-based pragmatic recommendations to reduce the global burden of stroke, including measures to improve surveillance, prevention, acute care, and rehabilitation.
The report was announced on Oct. 10 by WSO President, Sheila Martins, MD, at the World Stroke Conference in Toronto. The report was also published online in The Lancet Neurology.
“Stroke care has changed a lot in the last few years,” said Dr. Martins, who is chief of neurology and neurosurgery at Hospital Moinhos de Vento, Porto Alegre, Brazil, and founder and president of the Brazilian Stroke Network. “We know what we need to do to reduce the global burden of stroke, and high-income countries are making progress in that regard. But the situation in low- and middle-income countries is catastrophic, with mortality rates of up to 80% in individuals who have had a stroke in some countries. There is a very large gap between knowledge and implementation.”
Dr. Martins said that the commission is offering potential innovative suggestions on how to change this reality.
“While we have the knowledge on the strategies needed to reduce stroke burden, the mechanisms needed to implement this knowledge will be different in different countries and cultures. Our commission includes several representatives from low- and middle-income countries, and we will be working with local stakeholders in these countries to try and implement our recommendations,” Dr. Martins explained.
Stroke mortality and disability is on the rise
In the report, the authors pointed out that the global burden of stroke is “huge.” In 2020, stroke was the second leading cause of death (6.6 million deaths) and the third leading cause of disability – responsible for 143 million disability-adjusted life-years – after neonatal disorders and ischemic heart disease. Stroke is also a leading cause of depression and dementia.
The absolute number of people affected by stroke, which includes those who die or remain disabled, has almost doubled in the past 30 years, the report authors noted. Most of the contemporary stroke burden is in low- and middle-income countries, and the burden of disability after a stroke is increasing at a faster pace in low- and middle-income countries than in high-income countries. Alarmingly, the incidence of stroke is increasing in young and middle-aged people globally.
The commission forecasts the burden of stroke from 2020 to 2050, with projections estimating that stroke mortality will increase by 50% to 9.7 million and disability-adjusted life-years growing to over 189.3 million by 2050.
“Stroke exerts an enormous toll on the world’s population, leading to the death and permanent disability of millions of people each year, and costing billions of dollars,” said Valery L. Feigin, MD, of Auckland (New Zealand) University of Technology, and commission cochair. “Precisely forecasting the health and economic impacts of stroke decades into the future is inherently challenging given the levels of uncertainty involved, but these estimates are indicative of the ever-increasing burden we will see in the years ahead unless urgent, effective action is taken.”
The report authors explained that multiple factors contribute to the high burden of stroke in low- and middle-income countries, including undetected and uncontrolled hypertension; lack of easily accessible, high-quality health services; insufficient attention to and investment in prevention, air pollution; population growth; unhealthy lifestyles (for example, poor diet, smoking, sedentary lifestyle, obesity); an earlier age of stroke onset and greater proportion of hemorrhagic strokes than in high-income countries; and the burden of infectious diseases resulting in competition for limited health care resources.
The enormous financial cost of stroke
The total cost of stroke (both direct treatment and rehabilitation costs and indirect costs due to loss of income) is estimated to rise from $891 billion per year in 2017 to as much as $2.31 trillion by 2050. “These substantial increases in the costs associated with stroke will cause distressing financial circumstances for many communities and national health systems,” the authors said.
However, this increase can be avoided because stroke is highly preventable and treatable, they stressed. “These unsustainable trends in burden and costs of stroke underline the importance of identifying interventions to prevent and manage stroke more effectively.”
The Commission pointed out that population-wide primary prevention across the lifespan is extremely cost effective. It has been estimated that for every $1 spent on the prevention of stroke and cardiovascular disease, there is a more than $10 return on investment.
Additionally, primary prevention efforts directed at stroke would probably yield large gains because of the secondary effects of reducing the risk for heart disease, type 2 diabetes, dementia, and some types of cancer that share common risk factors, the authors noted.
“One of the most common problems in implementing stroke prevention and care recommendations is the lack of funding. Our commission recommends introducing legislative regulations and taxations of unhealthy products (such as salt, alcohol, sugary drinks, trans fats) by each and every government in the world,” Dr. Feigin said.
“Such taxation would not only reduce consumption of these products – and therefore lead to the reduction of burden from stroke and major other noncommunicable diseases – but also generate a large revenue sufficient to fund not only prevention programs and services for stroke and other major disorders, but also reduce poverty, inequality in health service provision, and improve wellbeing of the population,” he added.
Recommendations
The commission authors made the following recommendations for key priorities to reduce the burden of stroke:
Surveillance and prevention
- Incorporate stroke events and risk factor surveillance into national stroke action plans.
- Establish a system for population-wide primary and secondary stroke prevention, with emphasis on lifestyle modification for people at any level of risk of stroke and cardiovascular disease.
- Primary and secondary stroke prevention services should be freely accessible and supported by universal health coverage, with access to affordable drugs for management of hypertension, dyslipidemia, diabetes, and clotting disorders.
- Governments must allocate a fixed proportion of their annual health care funding for prevention of stroke and related noncommunicable diseases. This funding could come from taxation of tobacco, salt, alcohol, and sugar.
- Raise public awareness and take action to encourage a healthy lifestyle and prevent stroke via population-wide deployment of digital technologies with simple, inexpensive screening for cardiovascular disease and modifiable risk factors.
- Establish protocol-based shifting of tasks from highly trained health care professionals to supervised paramedical health care workers, to facilitate population-wide primary stroke prevention interventions across rural and urban settings.
Acute care
- Prioritize effective planning of acute stroke care services; capacity building, training, and certification of a multidisciplinary workforce; provision of evidence-based equipment and affordable medicines; and adequate resource allocation at national and regional levels.
- Establish regional networks and protocol-driven services, including community-wide awareness campaigns for early recognition of a stroke, regionally coordinated prehospital services, telemedicine networks, and stroke centers that can triage and treat all cases of acute stroke, and facilitate timely access to reperfusion therapy.
- Integrate acute care networks into the four pillars of the stroke “quadrangle” of resources, including surveillance, prevention, and rehabilitation services, by involving all relevant stakeholders (that is, communities, policy makers, nongovernmental organizations, national and regional stroke organizations, and public and private health care providers) in the stroke care continuum.
Rehabilitation
- Establish multidisciplinary rehabilitation services and adapt evidence-based recommendations to the local context, including the training, support, and supervision of community health care workers and caregivers to assist in long-term care.
- Invest in research to generate innovative low-cost interventions, in public awareness to improve demand for rehabilitation services, and in advocacy to mobilize resources for multidisciplinary rehabilitation.
- Promote the training of stroke rehabilitation professionals. Use digital portals to improve training and to extend the use of assessment tools – such as the Modified Rankin Scale and the U.S. National Institutes of Health Stroke Scale – and quality of life measures to assess functional impairment and monitor recovery.
The commission concluded that, “overall, if the recommendations of this Commission are implemented, the burden of stroke will be reduced substantially ... which will improve brain health and overall wellbeing worldwide.”
Dr. Martins said that the WSO is committed to supporting and accelerating the implementation of these recommendations globally through the WSO Implementation Task Force, with stroke experts to advise the establishment of stroke prevention and care and to contribute with educational programs, and through Global Stroke Alliance meetings facilitating the discussions between stroke experts and policy makers, giving technical support to governments to elaborate national plans for stroke and to include stroke care in universal health coverage packages.
The Commission received funding from the WSO, Bill and Melinda Gates Foundation, Health Research Council of New Zealand, and National Health & Medical Research Council of Australia and was supported by the NIH.
A version of this article first appeared on Medscape.com.
Deaths and disability because of stroke are expected to rise alarmingly over the next 30 years, with almost 10 million stroke deaths forecast annually by 2050, according to a new report from the World Stroke Organization–Lancet Neurology Commission Stroke Collaboration Group.
“This highlights the need for urgent measures to reduce stroke burden worldwide, with an emphasis on low- and middle-income countries,” the report authors stated.
These measures include an increase in trained health care workers who can implement effective primary prevention strategies, including the early detection and adequate management of hypertension.
On the basis of a review of evidence-based guidelines, recent surveys, and in-depth interviews with stroke experts around the world, the WSO–Lancet Neurology Commission made evidence-based pragmatic recommendations to reduce the global burden of stroke, including measures to improve surveillance, prevention, acute care, and rehabilitation.
The report was announced on Oct. 10 by WSO President, Sheila Martins, MD, at the World Stroke Conference in Toronto. The report was also published online in The Lancet Neurology.
“Stroke care has changed a lot in the last few years,” said Dr. Martins, who is chief of neurology and neurosurgery at Hospital Moinhos de Vento, Porto Alegre, Brazil, and founder and president of the Brazilian Stroke Network. “We know what we need to do to reduce the global burden of stroke, and high-income countries are making progress in that regard. But the situation in low- and middle-income countries is catastrophic, with mortality rates of up to 80% in individuals who have had a stroke in some countries. There is a very large gap between knowledge and implementation.”
Dr. Martins said that the commission is offering potential innovative suggestions on how to change this reality.
“While we have the knowledge on the strategies needed to reduce stroke burden, the mechanisms needed to implement this knowledge will be different in different countries and cultures. Our commission includes several representatives from low- and middle-income countries, and we will be working with local stakeholders in these countries to try and implement our recommendations,” Dr. Martins explained.
Stroke mortality and disability is on the rise
In the report, the authors pointed out that the global burden of stroke is “huge.” In 2020, stroke was the second leading cause of death (6.6 million deaths) and the third leading cause of disability – responsible for 143 million disability-adjusted life-years – after neonatal disorders and ischemic heart disease. Stroke is also a leading cause of depression and dementia.
The absolute number of people affected by stroke, which includes those who die or remain disabled, has almost doubled in the past 30 years, the report authors noted. Most of the contemporary stroke burden is in low- and middle-income countries, and the burden of disability after a stroke is increasing at a faster pace in low- and middle-income countries than in high-income countries. Alarmingly, the incidence of stroke is increasing in young and middle-aged people globally.
The commission forecasts the burden of stroke from 2020 to 2050, with projections estimating that stroke mortality will increase by 50% to 9.7 million and disability-adjusted life-years growing to over 189.3 million by 2050.
“Stroke exerts an enormous toll on the world’s population, leading to the death and permanent disability of millions of people each year, and costing billions of dollars,” said Valery L. Feigin, MD, of Auckland (New Zealand) University of Technology, and commission cochair. “Precisely forecasting the health and economic impacts of stroke decades into the future is inherently challenging given the levels of uncertainty involved, but these estimates are indicative of the ever-increasing burden we will see in the years ahead unless urgent, effective action is taken.”
The report authors explained that multiple factors contribute to the high burden of stroke in low- and middle-income countries, including undetected and uncontrolled hypertension; lack of easily accessible, high-quality health services; insufficient attention to and investment in prevention, air pollution; population growth; unhealthy lifestyles (for example, poor diet, smoking, sedentary lifestyle, obesity); an earlier age of stroke onset and greater proportion of hemorrhagic strokes than in high-income countries; and the burden of infectious diseases resulting in competition for limited health care resources.
The enormous financial cost of stroke
The total cost of stroke (both direct treatment and rehabilitation costs and indirect costs due to loss of income) is estimated to rise from $891 billion per year in 2017 to as much as $2.31 trillion by 2050. “These substantial increases in the costs associated with stroke will cause distressing financial circumstances for many communities and national health systems,” the authors said.
However, this increase can be avoided because stroke is highly preventable and treatable, they stressed. “These unsustainable trends in burden and costs of stroke underline the importance of identifying interventions to prevent and manage stroke more effectively.”
The Commission pointed out that population-wide primary prevention across the lifespan is extremely cost effective. It has been estimated that for every $1 spent on the prevention of stroke and cardiovascular disease, there is a more than $10 return on investment.
Additionally, primary prevention efforts directed at stroke would probably yield large gains because of the secondary effects of reducing the risk for heart disease, type 2 diabetes, dementia, and some types of cancer that share common risk factors, the authors noted.
“One of the most common problems in implementing stroke prevention and care recommendations is the lack of funding. Our commission recommends introducing legislative regulations and taxations of unhealthy products (such as salt, alcohol, sugary drinks, trans fats) by each and every government in the world,” Dr. Feigin said.
“Such taxation would not only reduce consumption of these products – and therefore lead to the reduction of burden from stroke and major other noncommunicable diseases – but also generate a large revenue sufficient to fund not only prevention programs and services for stroke and other major disorders, but also reduce poverty, inequality in health service provision, and improve wellbeing of the population,” he added.
Recommendations
The commission authors made the following recommendations for key priorities to reduce the burden of stroke:
Surveillance and prevention
- Incorporate stroke events and risk factor surveillance into national stroke action plans.
- Establish a system for population-wide primary and secondary stroke prevention, with emphasis on lifestyle modification for people at any level of risk of stroke and cardiovascular disease.
- Primary and secondary stroke prevention services should be freely accessible and supported by universal health coverage, with access to affordable drugs for management of hypertension, dyslipidemia, diabetes, and clotting disorders.
- Governments must allocate a fixed proportion of their annual health care funding for prevention of stroke and related noncommunicable diseases. This funding could come from taxation of tobacco, salt, alcohol, and sugar.
- Raise public awareness and take action to encourage a healthy lifestyle and prevent stroke via population-wide deployment of digital technologies with simple, inexpensive screening for cardiovascular disease and modifiable risk factors.
- Establish protocol-based shifting of tasks from highly trained health care professionals to supervised paramedical health care workers, to facilitate population-wide primary stroke prevention interventions across rural and urban settings.
Acute care
- Prioritize effective planning of acute stroke care services; capacity building, training, and certification of a multidisciplinary workforce; provision of evidence-based equipment and affordable medicines; and adequate resource allocation at national and regional levels.
- Establish regional networks and protocol-driven services, including community-wide awareness campaigns for early recognition of a stroke, regionally coordinated prehospital services, telemedicine networks, and stroke centers that can triage and treat all cases of acute stroke, and facilitate timely access to reperfusion therapy.
- Integrate acute care networks into the four pillars of the stroke “quadrangle” of resources, including surveillance, prevention, and rehabilitation services, by involving all relevant stakeholders (that is, communities, policy makers, nongovernmental organizations, national and regional stroke organizations, and public and private health care providers) in the stroke care continuum.
Rehabilitation
- Establish multidisciplinary rehabilitation services and adapt evidence-based recommendations to the local context, including the training, support, and supervision of community health care workers and caregivers to assist in long-term care.
- Invest in research to generate innovative low-cost interventions, in public awareness to improve demand for rehabilitation services, and in advocacy to mobilize resources for multidisciplinary rehabilitation.
- Promote the training of stroke rehabilitation professionals. Use digital portals to improve training and to extend the use of assessment tools – such as the Modified Rankin Scale and the U.S. National Institutes of Health Stroke Scale – and quality of life measures to assess functional impairment and monitor recovery.
The commission concluded that, “overall, if the recommendations of this Commission are implemented, the burden of stroke will be reduced substantially ... which will improve brain health and overall wellbeing worldwide.”
Dr. Martins said that the WSO is committed to supporting and accelerating the implementation of these recommendations globally through the WSO Implementation Task Force, with stroke experts to advise the establishment of stroke prevention and care and to contribute with educational programs, and through Global Stroke Alliance meetings facilitating the discussions between stroke experts and policy makers, giving technical support to governments to elaborate national plans for stroke and to include stroke care in universal health coverage packages.
The Commission received funding from the WSO, Bill and Melinda Gates Foundation, Health Research Council of New Zealand, and National Health & Medical Research Council of Australia and was supported by the NIH.
A version of this article first appeared on Medscape.com.
Deaths and disability because of stroke are expected to rise alarmingly over the next 30 years, with almost 10 million stroke deaths forecast annually by 2050, according to a new report from the World Stroke Organization–Lancet Neurology Commission Stroke Collaboration Group.
“This highlights the need for urgent measures to reduce stroke burden worldwide, with an emphasis on low- and middle-income countries,” the report authors stated.
These measures include an increase in trained health care workers who can implement effective primary prevention strategies, including the early detection and adequate management of hypertension.
On the basis of a review of evidence-based guidelines, recent surveys, and in-depth interviews with stroke experts around the world, the WSO–Lancet Neurology Commission made evidence-based pragmatic recommendations to reduce the global burden of stroke, including measures to improve surveillance, prevention, acute care, and rehabilitation.
The report was announced on Oct. 10 by WSO President, Sheila Martins, MD, at the World Stroke Conference in Toronto. The report was also published online in The Lancet Neurology.
“Stroke care has changed a lot in the last few years,” said Dr. Martins, who is chief of neurology and neurosurgery at Hospital Moinhos de Vento, Porto Alegre, Brazil, and founder and president of the Brazilian Stroke Network. “We know what we need to do to reduce the global burden of stroke, and high-income countries are making progress in that regard. But the situation in low- and middle-income countries is catastrophic, with mortality rates of up to 80% in individuals who have had a stroke in some countries. There is a very large gap between knowledge and implementation.”
Dr. Martins said that the commission is offering potential innovative suggestions on how to change this reality.
“While we have the knowledge on the strategies needed to reduce stroke burden, the mechanisms needed to implement this knowledge will be different in different countries and cultures. Our commission includes several representatives from low- and middle-income countries, and we will be working with local stakeholders in these countries to try and implement our recommendations,” Dr. Martins explained.
Stroke mortality and disability is on the rise
In the report, the authors pointed out that the global burden of stroke is “huge.” In 2020, stroke was the second leading cause of death (6.6 million deaths) and the third leading cause of disability – responsible for 143 million disability-adjusted life-years – after neonatal disorders and ischemic heart disease. Stroke is also a leading cause of depression and dementia.
The absolute number of people affected by stroke, which includes those who die or remain disabled, has almost doubled in the past 30 years, the report authors noted. Most of the contemporary stroke burden is in low- and middle-income countries, and the burden of disability after a stroke is increasing at a faster pace in low- and middle-income countries than in high-income countries. Alarmingly, the incidence of stroke is increasing in young and middle-aged people globally.
The commission forecasts the burden of stroke from 2020 to 2050, with projections estimating that stroke mortality will increase by 50% to 9.7 million and disability-adjusted life-years growing to over 189.3 million by 2050.
“Stroke exerts an enormous toll on the world’s population, leading to the death and permanent disability of millions of people each year, and costing billions of dollars,” said Valery L. Feigin, MD, of Auckland (New Zealand) University of Technology, and commission cochair. “Precisely forecasting the health and economic impacts of stroke decades into the future is inherently challenging given the levels of uncertainty involved, but these estimates are indicative of the ever-increasing burden we will see in the years ahead unless urgent, effective action is taken.”
The report authors explained that multiple factors contribute to the high burden of stroke in low- and middle-income countries, including undetected and uncontrolled hypertension; lack of easily accessible, high-quality health services; insufficient attention to and investment in prevention, air pollution; population growth; unhealthy lifestyles (for example, poor diet, smoking, sedentary lifestyle, obesity); an earlier age of stroke onset and greater proportion of hemorrhagic strokes than in high-income countries; and the burden of infectious diseases resulting in competition for limited health care resources.
The enormous financial cost of stroke
The total cost of stroke (both direct treatment and rehabilitation costs and indirect costs due to loss of income) is estimated to rise from $891 billion per year in 2017 to as much as $2.31 trillion by 2050. “These substantial increases in the costs associated with stroke will cause distressing financial circumstances for many communities and national health systems,” the authors said.
However, this increase can be avoided because stroke is highly preventable and treatable, they stressed. “These unsustainable trends in burden and costs of stroke underline the importance of identifying interventions to prevent and manage stroke more effectively.”
The Commission pointed out that population-wide primary prevention across the lifespan is extremely cost effective. It has been estimated that for every $1 spent on the prevention of stroke and cardiovascular disease, there is a more than $10 return on investment.
Additionally, primary prevention efforts directed at stroke would probably yield large gains because of the secondary effects of reducing the risk for heart disease, type 2 diabetes, dementia, and some types of cancer that share common risk factors, the authors noted.
“One of the most common problems in implementing stroke prevention and care recommendations is the lack of funding. Our commission recommends introducing legislative regulations and taxations of unhealthy products (such as salt, alcohol, sugary drinks, trans fats) by each and every government in the world,” Dr. Feigin said.
“Such taxation would not only reduce consumption of these products – and therefore lead to the reduction of burden from stroke and major other noncommunicable diseases – but also generate a large revenue sufficient to fund not only prevention programs and services for stroke and other major disorders, but also reduce poverty, inequality in health service provision, and improve wellbeing of the population,” he added.
Recommendations
The commission authors made the following recommendations for key priorities to reduce the burden of stroke:
Surveillance and prevention
- Incorporate stroke events and risk factor surveillance into national stroke action plans.
- Establish a system for population-wide primary and secondary stroke prevention, with emphasis on lifestyle modification for people at any level of risk of stroke and cardiovascular disease.
- Primary and secondary stroke prevention services should be freely accessible and supported by universal health coverage, with access to affordable drugs for management of hypertension, dyslipidemia, diabetes, and clotting disorders.
- Governments must allocate a fixed proportion of their annual health care funding for prevention of stroke and related noncommunicable diseases. This funding could come from taxation of tobacco, salt, alcohol, and sugar.
- Raise public awareness and take action to encourage a healthy lifestyle and prevent stroke via population-wide deployment of digital technologies with simple, inexpensive screening for cardiovascular disease and modifiable risk factors.
- Establish protocol-based shifting of tasks from highly trained health care professionals to supervised paramedical health care workers, to facilitate population-wide primary stroke prevention interventions across rural and urban settings.
Acute care
- Prioritize effective planning of acute stroke care services; capacity building, training, and certification of a multidisciplinary workforce; provision of evidence-based equipment and affordable medicines; and adequate resource allocation at national and regional levels.
- Establish regional networks and protocol-driven services, including community-wide awareness campaigns for early recognition of a stroke, regionally coordinated prehospital services, telemedicine networks, and stroke centers that can triage and treat all cases of acute stroke, and facilitate timely access to reperfusion therapy.
- Integrate acute care networks into the four pillars of the stroke “quadrangle” of resources, including surveillance, prevention, and rehabilitation services, by involving all relevant stakeholders (that is, communities, policy makers, nongovernmental organizations, national and regional stroke organizations, and public and private health care providers) in the stroke care continuum.
Rehabilitation
- Establish multidisciplinary rehabilitation services and adapt evidence-based recommendations to the local context, including the training, support, and supervision of community health care workers and caregivers to assist in long-term care.
- Invest in research to generate innovative low-cost interventions, in public awareness to improve demand for rehabilitation services, and in advocacy to mobilize resources for multidisciplinary rehabilitation.
- Promote the training of stroke rehabilitation professionals. Use digital portals to improve training and to extend the use of assessment tools – such as the Modified Rankin Scale and the U.S. National Institutes of Health Stroke Scale – and quality of life measures to assess functional impairment and monitor recovery.
The commission concluded that, “overall, if the recommendations of this Commission are implemented, the burden of stroke will be reduced substantially ... which will improve brain health and overall wellbeing worldwide.”
Dr. Martins said that the WSO is committed to supporting and accelerating the implementation of these recommendations globally through the WSO Implementation Task Force, with stroke experts to advise the establishment of stroke prevention and care and to contribute with educational programs, and through Global Stroke Alliance meetings facilitating the discussions between stroke experts and policy makers, giving technical support to governments to elaborate national plans for stroke and to include stroke care in universal health coverage packages.
The Commission received funding from the WSO, Bill and Melinda Gates Foundation, Health Research Council of New Zealand, and National Health & Medical Research Council of Australia and was supported by the NIH.
A version of this article first appeared on Medscape.com.
FROM THE LANCET NEUROLOGY
New guidelines for determining brain death released
The consensus practice guideline on brain death, also known as death by neurologic criteria (BD/DNC), was developed by a panel of 20 experts from different specialties, institutions, and medical societies.
As with previous guidelines, the updated version stipulates that brain death should be declared when a patient with a known cause of catastrophic brain injury has permanent loss of function of the brain, including the brain stem, which results in coma, brain stem areflexia, and apnea in the setting of an adequate stimulus.
But the updated version also clarifies questions on neurological examinations and apnea testing and offers new guidance on pre-evaluation targets for blood pressure and body temperature and evaluating brain death in patients who are pregnant, are on extracorporeal membrane oxygenation, or have an injury to the base of the brain.
Also, for the first time, the guidance clarifies that clinicians don’t need to obtain consent before performing a brain death evaluation, unless institutional policy, state laws, or regulations stipulate otherwise.
“The 2023 guidelines will be considered the standard of care in the U.S.,” lead author David M. Greer, MD, chair and chief of neurology, Boston University, and chief of neurology, Boston Medical Center, said in an interview. “Each hospital in the U.S. is responsible for its own policy for BD/DNC determination, and our hope is that they will quickly revise their policies in accordance with this new national standard.”
The guidelines, which are accompanied by a three-page checklist and a free digital app, were published online in Neurology.
Four years in the making
Work on the 85 recommendations in the new report began more than 4 years ago as a collaborative effort by the American Academy of Neurology, the American Academy of Pediatrics, the Child Neurology Society, and the Society of Critical Care Medicine.
A lack of high-quality evidence on brain death determination led panelists to devise an evidence-informed formal consensus process to develop the guidelines, which involved three rounds of anonymous voting on each recommendation and the rationales behind them.
The strength of each recommendation was based on the level of consensus reached through voting, with Level A denoting a recommendation that “must” be followed, Level B one that “should” be followed, and Level C one that “may” be followed.
The majority of recommendations received an A or B rating. Only one recommendation, about whether a second clinical exam is needed in adults, garnered a C rating.
In children, the guidelines recommend that clinicians must perform two clinical examinations and two apnea tests 12 hours apart. In adults, only one exam is required. Both of those recommendations were rated Level A. A recommendation for a second exam in adults received the single Level C rating.
A uniform set of guidelines?
The new guidelines replace adult practice guidance published by AAN in 2010 and guideline for infants and children released in 2011 by AAP, CNS, and SCCM, and for the first time combine brain death guidelines for adult and pediatric patients into one document.
“It is important for clinicians to review the new guideline carefully and ensure their hospital brain death guidelines are updated to be consistent with the new guideline in order to prevent inaccurate determinations of death,” guidelines coauthor Ariane Lewis, MD, NYU Langone Health, New York, said in an interview.
The 1981 Uniform Determination of Death Act (UDDA) is the legal foundation for the declaration of BD/DNC in the United States, but it only stipulates that brain death determination must be made in accordance with accepted medical standards.
There is no single national standard, and states and hospitals are free to adopt their own, which many have done. One goal of the new guidelines was to create a uniform set of guidelines that all institutions follow.
“This is a step toward having a set of guidelines that are accepted by most of the societies and clinical specialties involved in this sort of diagnosis,” that could lead to a national-level policy, Fernando Goldenberg, MD, professor of neurology and director of neuroscience critical care, University of Chicago Medicine, said in an interview.
Dr. Goldenberg was not part of the panel that developed the updated guidelines, but was a coauthor of a consensus statement from the World Brain Death Project in 2020.
Developing a singular global guideline for brain death determination is unlikely, Dr. Goldenberg said. Policies vary widely across the world, and some countries don’t even recognize brain death.
“But this attempts to unify things at the U.S. level, which is very important,” he said.
Permanent vs. irreversible
Dr. Goldenberg said that combining adult and pediatric guidelines into one document will be very helpful for clinicians like him who treat patients from age 16 years and up.
The expanded guidance on apnea testing, recommendations on specific ancillary tests to use or avoid, and inclusion of language stipulating that prior consent is not needed to perform a brain death evaluation are also useful.
He also noted that the section on credentialing and training of clinicians who perform BD/DNC evaluations recognizes advanced practice providers, the first time he recalls seeing these professionals included in brain death guidelines.
However, the panel’s decision to use the term “permanent” to describe loss of brain function instead of “irreversible” gave Dr. Goldenberg pause.
The UDDA provides that an individual is declared legally dead when “circulatory and respiratory functions irreversibly stop; or all functions of the entire brain, including the brain stem, irreversibly stop.”
Earlier in October, the American College of Physicians released a position paper on cardiorespiratory death determination that called for a revision of the UDDA language.
The ACP suggested that “irreversibly” be replaced with “permanently” with regard to the cessation of circulatory and respiratory functions, but that “irreversible” be kept in the description of brain death.
“Permanent means that there is damage that is potentially reversible and irreversible means that the damage is so profound, it cannot be reversed even if an attempt to do so is performed,” Dr. Goldenberg said.
Even though the World Brain Death Project, on which he worked, also used “permanent” to describe brain function loss, Dr. Goldenberg said he aligns with ACP’s position.
“The understanding of brain death is that the damage is so profound, it is irreversible, even if you were to try,” he said. “Therefore, I think that the most appropriate term for brain death should be irreversible as opposed to permanent.”
The report was funded by the American Academy of Neurology. Dr. Greer has received travel funding from Boston University; serves as editor-in-chief for Seminars in Neurology; receives publishing royalties for 50 Studies Every Neurologist Should Know and Successful Leadership in Academic Medicine; has received honoraria from AAN; has received research funding from Becton, Dickinson, and Company; and has served as expert witness in legal proceedings. Dr. Lewis has received honoraria from AAN and Neurodiem, serves as Neurology deputy editor of disputes and debates, and serves as deputy editor of seminars in Neurology. Dr. Goldenberg reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The consensus practice guideline on brain death, also known as death by neurologic criteria (BD/DNC), was developed by a panel of 20 experts from different specialties, institutions, and medical societies.
As with previous guidelines, the updated version stipulates that brain death should be declared when a patient with a known cause of catastrophic brain injury has permanent loss of function of the brain, including the brain stem, which results in coma, brain stem areflexia, and apnea in the setting of an adequate stimulus.
But the updated version also clarifies questions on neurological examinations and apnea testing and offers new guidance on pre-evaluation targets for blood pressure and body temperature and evaluating brain death in patients who are pregnant, are on extracorporeal membrane oxygenation, or have an injury to the base of the brain.
Also, for the first time, the guidance clarifies that clinicians don’t need to obtain consent before performing a brain death evaluation, unless institutional policy, state laws, or regulations stipulate otherwise.
“The 2023 guidelines will be considered the standard of care in the U.S.,” lead author David M. Greer, MD, chair and chief of neurology, Boston University, and chief of neurology, Boston Medical Center, said in an interview. “Each hospital in the U.S. is responsible for its own policy for BD/DNC determination, and our hope is that they will quickly revise their policies in accordance with this new national standard.”
The guidelines, which are accompanied by a three-page checklist and a free digital app, were published online in Neurology.
Four years in the making
Work on the 85 recommendations in the new report began more than 4 years ago as a collaborative effort by the American Academy of Neurology, the American Academy of Pediatrics, the Child Neurology Society, and the Society of Critical Care Medicine.
A lack of high-quality evidence on brain death determination led panelists to devise an evidence-informed formal consensus process to develop the guidelines, which involved three rounds of anonymous voting on each recommendation and the rationales behind them.
The strength of each recommendation was based on the level of consensus reached through voting, with Level A denoting a recommendation that “must” be followed, Level B one that “should” be followed, and Level C one that “may” be followed.
The majority of recommendations received an A or B rating. Only one recommendation, about whether a second clinical exam is needed in adults, garnered a C rating.
In children, the guidelines recommend that clinicians must perform two clinical examinations and two apnea tests 12 hours apart. In adults, only one exam is required. Both of those recommendations were rated Level A. A recommendation for a second exam in adults received the single Level C rating.
A uniform set of guidelines?
The new guidelines replace adult practice guidance published by AAN in 2010 and guideline for infants and children released in 2011 by AAP, CNS, and SCCM, and for the first time combine brain death guidelines for adult and pediatric patients into one document.
“It is important for clinicians to review the new guideline carefully and ensure their hospital brain death guidelines are updated to be consistent with the new guideline in order to prevent inaccurate determinations of death,” guidelines coauthor Ariane Lewis, MD, NYU Langone Health, New York, said in an interview.
The 1981 Uniform Determination of Death Act (UDDA) is the legal foundation for the declaration of BD/DNC in the United States, but it only stipulates that brain death determination must be made in accordance with accepted medical standards.
There is no single national standard, and states and hospitals are free to adopt their own, which many have done. One goal of the new guidelines was to create a uniform set of guidelines that all institutions follow.
“This is a step toward having a set of guidelines that are accepted by most of the societies and clinical specialties involved in this sort of diagnosis,” that could lead to a national-level policy, Fernando Goldenberg, MD, professor of neurology and director of neuroscience critical care, University of Chicago Medicine, said in an interview.
Dr. Goldenberg was not part of the panel that developed the updated guidelines, but was a coauthor of a consensus statement from the World Brain Death Project in 2020.
Developing a singular global guideline for brain death determination is unlikely, Dr. Goldenberg said. Policies vary widely across the world, and some countries don’t even recognize brain death.
“But this attempts to unify things at the U.S. level, which is very important,” he said.
Permanent vs. irreversible
Dr. Goldenberg said that combining adult and pediatric guidelines into one document will be very helpful for clinicians like him who treat patients from age 16 years and up.
The expanded guidance on apnea testing, recommendations on specific ancillary tests to use or avoid, and inclusion of language stipulating that prior consent is not needed to perform a brain death evaluation are also useful.
He also noted that the section on credentialing and training of clinicians who perform BD/DNC evaluations recognizes advanced practice providers, the first time he recalls seeing these professionals included in brain death guidelines.
However, the panel’s decision to use the term “permanent” to describe loss of brain function instead of “irreversible” gave Dr. Goldenberg pause.
The UDDA provides that an individual is declared legally dead when “circulatory and respiratory functions irreversibly stop; or all functions of the entire brain, including the brain stem, irreversibly stop.”
Earlier in October, the American College of Physicians released a position paper on cardiorespiratory death determination that called for a revision of the UDDA language.
The ACP suggested that “irreversibly” be replaced with “permanently” with regard to the cessation of circulatory and respiratory functions, but that “irreversible” be kept in the description of brain death.
“Permanent means that there is damage that is potentially reversible and irreversible means that the damage is so profound, it cannot be reversed even if an attempt to do so is performed,” Dr. Goldenberg said.
Even though the World Brain Death Project, on which he worked, also used “permanent” to describe brain function loss, Dr. Goldenberg said he aligns with ACP’s position.
“The understanding of brain death is that the damage is so profound, it is irreversible, even if you were to try,” he said. “Therefore, I think that the most appropriate term for brain death should be irreversible as opposed to permanent.”
The report was funded by the American Academy of Neurology. Dr. Greer has received travel funding from Boston University; serves as editor-in-chief for Seminars in Neurology; receives publishing royalties for 50 Studies Every Neurologist Should Know and Successful Leadership in Academic Medicine; has received honoraria from AAN; has received research funding from Becton, Dickinson, and Company; and has served as expert witness in legal proceedings. Dr. Lewis has received honoraria from AAN and Neurodiem, serves as Neurology deputy editor of disputes and debates, and serves as deputy editor of seminars in Neurology. Dr. Goldenberg reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The consensus practice guideline on brain death, also known as death by neurologic criteria (BD/DNC), was developed by a panel of 20 experts from different specialties, institutions, and medical societies.
As with previous guidelines, the updated version stipulates that brain death should be declared when a patient with a known cause of catastrophic brain injury has permanent loss of function of the brain, including the brain stem, which results in coma, brain stem areflexia, and apnea in the setting of an adequate stimulus.
But the updated version also clarifies questions on neurological examinations and apnea testing and offers new guidance on pre-evaluation targets for blood pressure and body temperature and evaluating brain death in patients who are pregnant, are on extracorporeal membrane oxygenation, or have an injury to the base of the brain.
Also, for the first time, the guidance clarifies that clinicians don’t need to obtain consent before performing a brain death evaluation, unless institutional policy, state laws, or regulations stipulate otherwise.
“The 2023 guidelines will be considered the standard of care in the U.S.,” lead author David M. Greer, MD, chair and chief of neurology, Boston University, and chief of neurology, Boston Medical Center, said in an interview. “Each hospital in the U.S. is responsible for its own policy for BD/DNC determination, and our hope is that they will quickly revise their policies in accordance with this new national standard.”
The guidelines, which are accompanied by a three-page checklist and a free digital app, were published online in Neurology.
Four years in the making
Work on the 85 recommendations in the new report began more than 4 years ago as a collaborative effort by the American Academy of Neurology, the American Academy of Pediatrics, the Child Neurology Society, and the Society of Critical Care Medicine.
A lack of high-quality evidence on brain death determination led panelists to devise an evidence-informed formal consensus process to develop the guidelines, which involved three rounds of anonymous voting on each recommendation and the rationales behind them.
The strength of each recommendation was based on the level of consensus reached through voting, with Level A denoting a recommendation that “must” be followed, Level B one that “should” be followed, and Level C one that “may” be followed.
The majority of recommendations received an A or B rating. Only one recommendation, about whether a second clinical exam is needed in adults, garnered a C rating.
In children, the guidelines recommend that clinicians must perform two clinical examinations and two apnea tests 12 hours apart. In adults, only one exam is required. Both of those recommendations were rated Level A. A recommendation for a second exam in adults received the single Level C rating.
A uniform set of guidelines?
The new guidelines replace adult practice guidance published by AAN in 2010 and guideline for infants and children released in 2011 by AAP, CNS, and SCCM, and for the first time combine brain death guidelines for adult and pediatric patients into one document.
“It is important for clinicians to review the new guideline carefully and ensure their hospital brain death guidelines are updated to be consistent with the new guideline in order to prevent inaccurate determinations of death,” guidelines coauthor Ariane Lewis, MD, NYU Langone Health, New York, said in an interview.
The 1981 Uniform Determination of Death Act (UDDA) is the legal foundation for the declaration of BD/DNC in the United States, but it only stipulates that brain death determination must be made in accordance with accepted medical standards.
There is no single national standard, and states and hospitals are free to adopt their own, which many have done. One goal of the new guidelines was to create a uniform set of guidelines that all institutions follow.
“This is a step toward having a set of guidelines that are accepted by most of the societies and clinical specialties involved in this sort of diagnosis,” that could lead to a national-level policy, Fernando Goldenberg, MD, professor of neurology and director of neuroscience critical care, University of Chicago Medicine, said in an interview.
Dr. Goldenberg was not part of the panel that developed the updated guidelines, but was a coauthor of a consensus statement from the World Brain Death Project in 2020.
Developing a singular global guideline for brain death determination is unlikely, Dr. Goldenberg said. Policies vary widely across the world, and some countries don’t even recognize brain death.
“But this attempts to unify things at the U.S. level, which is very important,” he said.
Permanent vs. irreversible
Dr. Goldenberg said that combining adult and pediatric guidelines into one document will be very helpful for clinicians like him who treat patients from age 16 years and up.
The expanded guidance on apnea testing, recommendations on specific ancillary tests to use or avoid, and inclusion of language stipulating that prior consent is not needed to perform a brain death evaluation are also useful.
He also noted that the section on credentialing and training of clinicians who perform BD/DNC evaluations recognizes advanced practice providers, the first time he recalls seeing these professionals included in brain death guidelines.
However, the panel’s decision to use the term “permanent” to describe loss of brain function instead of “irreversible” gave Dr. Goldenberg pause.
The UDDA provides that an individual is declared legally dead when “circulatory and respiratory functions irreversibly stop; or all functions of the entire brain, including the brain stem, irreversibly stop.”
Earlier in October, the American College of Physicians released a position paper on cardiorespiratory death determination that called for a revision of the UDDA language.
The ACP suggested that “irreversibly” be replaced with “permanently” with regard to the cessation of circulatory and respiratory functions, but that “irreversible” be kept in the description of brain death.
“Permanent means that there is damage that is potentially reversible and irreversible means that the damage is so profound, it cannot be reversed even if an attempt to do so is performed,” Dr. Goldenberg said.
Even though the World Brain Death Project, on which he worked, also used “permanent” to describe brain function loss, Dr. Goldenberg said he aligns with ACP’s position.
“The understanding of brain death is that the damage is so profound, it is irreversible, even if you were to try,” he said. “Therefore, I think that the most appropriate term for brain death should be irreversible as opposed to permanent.”
The report was funded by the American Academy of Neurology. Dr. Greer has received travel funding from Boston University; serves as editor-in-chief for Seminars in Neurology; receives publishing royalties for 50 Studies Every Neurologist Should Know and Successful Leadership in Academic Medicine; has received honoraria from AAN; has received research funding from Becton, Dickinson, and Company; and has served as expert witness in legal proceedings. Dr. Lewis has received honoraria from AAN and Neurodiem, serves as Neurology deputy editor of disputes and debates, and serves as deputy editor of seminars in Neurology. Dr. Goldenberg reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM NEUROLOGY
Proton pump inhibitors linked to increased dementia risk
TOPLINE:
and was highest among those diagnosed before age 70 years regardless of when PPI treatment was initiated.
METHODOLOGY:
- Researchers used four Danish registries to collect data on dementia diagnoses and prescription PPI use among 1,983,785 individuals aged 60-75 years between 2000 and 2018.
- The median follow-up time was 10.3 years.
TAKEAWAY:
- There were 99,384 (5.0%) cases of all-cause dementia during follow-up, with a median age of diagnosis of 79 years.
- Twenty-one-point-two percent of dementia cases and 18.9% of controls reported a history of PPI use.
- Risk for all-cause dementia before age 90 years was 36% higher with PPI use in people aged 60-69 years at baseline (adjusted incidence rate ratio, 1.36; 95% confidence interval, 1.29-1.43) and 6% higher in those who were age 80-89 years at baseline (aIRR, 1.06; 95% CI, 1.03-1.09).
- Investigators found significant increased dementia risk before age 90 years with PPI use regardless of when PPI treatment began and found no link between PPI use and dementia diagnoses after age 90 years.
IN PRACTICE:
“The association between PPI use and dementia was unambiguously largest among the youngest cases of dementia, potentially suggestive of a critical window of exposure where midlife PPI use affects dementia risk to a larger degree compared to late-life use,” the authors wrote. “Further, the finding could signify a declining impact of individual risk factors with advancing age owing to lengthy ongoing neuropathological processes.”
SOURCE:
Lead author of the study was Nelsan Pourhadi, MD, Danish Dementia Research Centre, department of neurology, Copenhagen University Hospital–Rigshospitalet. It was published online in Alzheimer’s and Dementia.
LIMITATIONS:
The study did not include data on PPI prescriptions before 1995, over-the-counter PPI use, and in-hospital intravenous use of PPI during the study period.
DISCLOSURES:
The study was funded by the Danish Ministry of Health. The authors reported no relevant conflicts.
A version of this article first appeared on Medscape.com.
TOPLINE:
and was highest among those diagnosed before age 70 years regardless of when PPI treatment was initiated.
METHODOLOGY:
- Researchers used four Danish registries to collect data on dementia diagnoses and prescription PPI use among 1,983,785 individuals aged 60-75 years between 2000 and 2018.
- The median follow-up time was 10.3 years.
TAKEAWAY:
- There were 99,384 (5.0%) cases of all-cause dementia during follow-up, with a median age of diagnosis of 79 years.
- Twenty-one-point-two percent of dementia cases and 18.9% of controls reported a history of PPI use.
- Risk for all-cause dementia before age 90 years was 36% higher with PPI use in people aged 60-69 years at baseline (adjusted incidence rate ratio, 1.36; 95% confidence interval, 1.29-1.43) and 6% higher in those who were age 80-89 years at baseline (aIRR, 1.06; 95% CI, 1.03-1.09).
- Investigators found significant increased dementia risk before age 90 years with PPI use regardless of when PPI treatment began and found no link between PPI use and dementia diagnoses after age 90 years.
IN PRACTICE:
“The association between PPI use and dementia was unambiguously largest among the youngest cases of dementia, potentially suggestive of a critical window of exposure where midlife PPI use affects dementia risk to a larger degree compared to late-life use,” the authors wrote. “Further, the finding could signify a declining impact of individual risk factors with advancing age owing to lengthy ongoing neuropathological processes.”
SOURCE:
Lead author of the study was Nelsan Pourhadi, MD, Danish Dementia Research Centre, department of neurology, Copenhagen University Hospital–Rigshospitalet. It was published online in Alzheimer’s and Dementia.
LIMITATIONS:
The study did not include data on PPI prescriptions before 1995, over-the-counter PPI use, and in-hospital intravenous use of PPI during the study period.
DISCLOSURES:
The study was funded by the Danish Ministry of Health. The authors reported no relevant conflicts.
A version of this article first appeared on Medscape.com.
TOPLINE:
and was highest among those diagnosed before age 70 years regardless of when PPI treatment was initiated.
METHODOLOGY:
- Researchers used four Danish registries to collect data on dementia diagnoses and prescription PPI use among 1,983,785 individuals aged 60-75 years between 2000 and 2018.
- The median follow-up time was 10.3 years.
TAKEAWAY:
- There were 99,384 (5.0%) cases of all-cause dementia during follow-up, with a median age of diagnosis of 79 years.
- Twenty-one-point-two percent of dementia cases and 18.9% of controls reported a history of PPI use.
- Risk for all-cause dementia before age 90 years was 36% higher with PPI use in people aged 60-69 years at baseline (adjusted incidence rate ratio, 1.36; 95% confidence interval, 1.29-1.43) and 6% higher in those who were age 80-89 years at baseline (aIRR, 1.06; 95% CI, 1.03-1.09).
- Investigators found significant increased dementia risk before age 90 years with PPI use regardless of when PPI treatment began and found no link between PPI use and dementia diagnoses after age 90 years.
IN PRACTICE:
“The association between PPI use and dementia was unambiguously largest among the youngest cases of dementia, potentially suggestive of a critical window of exposure where midlife PPI use affects dementia risk to a larger degree compared to late-life use,” the authors wrote. “Further, the finding could signify a declining impact of individual risk factors with advancing age owing to lengthy ongoing neuropathological processes.”
SOURCE:
Lead author of the study was Nelsan Pourhadi, MD, Danish Dementia Research Centre, department of neurology, Copenhagen University Hospital–Rigshospitalet. It was published online in Alzheimer’s and Dementia.
LIMITATIONS:
The study did not include data on PPI prescriptions before 1995, over-the-counter PPI use, and in-hospital intravenous use of PPI during the study period.
DISCLOSURES:
The study was funded by the Danish Ministry of Health. The authors reported no relevant conflicts.
A version of this article first appeared on Medscape.com.
Every click you make, the EHR is watching you
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.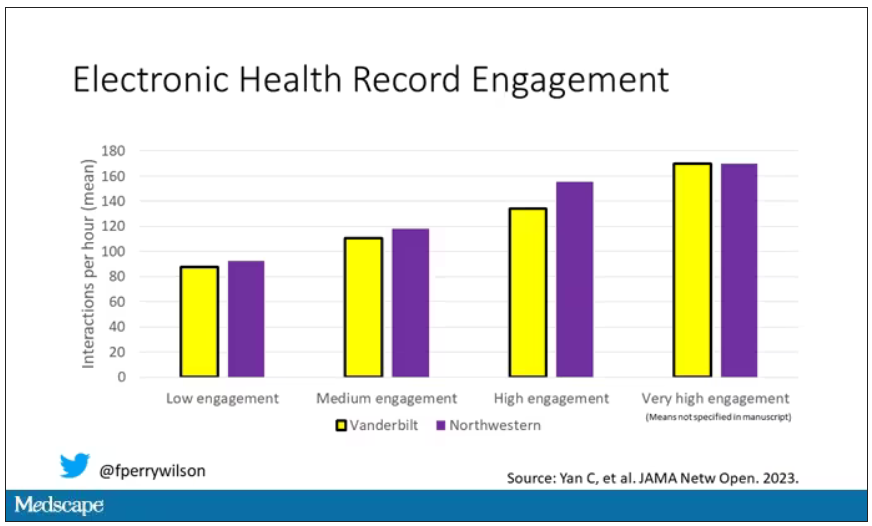
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.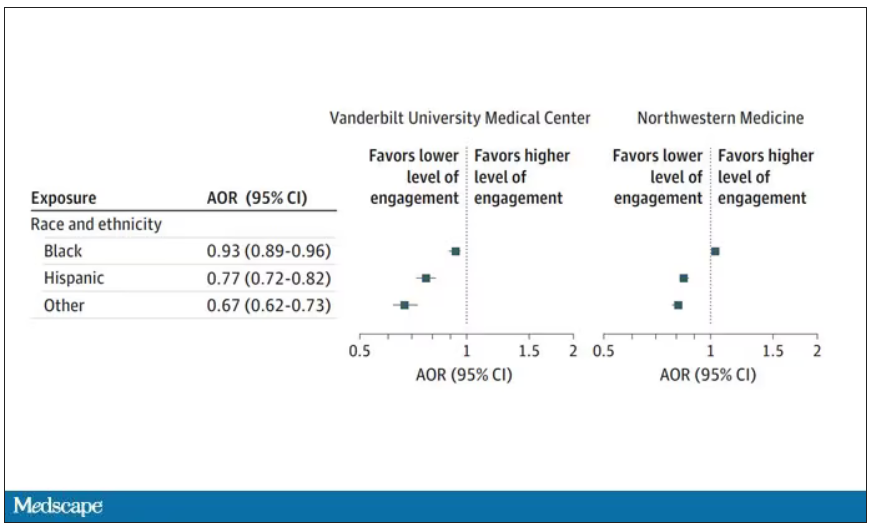
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.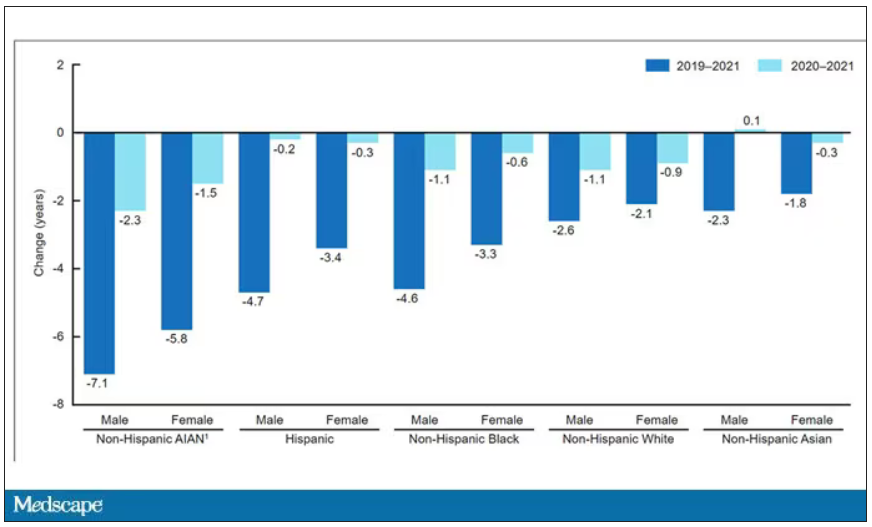
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
When I close my eyes and imagine what it is I do for a living, I see a computer screen.
I’m primarily a clinical researcher, so much of what I do is looking at statistical software, or, more recently, writing grant applications. But even when I think of my clinical duties, I see that computer screen.
The reason? The electronic health record (EHR) – the hot, beating heart of medical care in the modern era. Our most powerful tool and our greatest enemy.
The EHR records everything – not just the vital signs and lab values of our patients, not just our notes and billing codes. Everything. Every interaction we have is tracked and can be analyzed. The EHR is basically Sting in the song “Every Breath You Take.” Every click you make, it is watching you.
Researchers are leveraging that panopticon to give insight into something we don’t talk about frequently: the issue of racial bias in medicine. Is our true nature revealed by our interactions with the EHR?
We’re talking about this study in JAMA Network Open.
Researchers leveraged huge amounts of EHR data from two big academic medical centers, Vanderbilt University Medical Center and Northwestern University Medical Center. All told, there are data from nearly 250,000 hospitalizations here.
The researchers created a metric for EHR engagement. Basically, they summed the amount of clicks and other EHR interactions that occurred during the hospitalization, divided by the length of stay in days, to create a sort of average “engagement per day” metric. This number was categorized into four groups: low engagement, medium engagement, high engagement, and very high engagement.
What factors would predict higher engagement? Well, , except among Black patients who actually got a bit more engagement.
So, right away we need to be concerned about the obvious implications. Less engagement with the EHR may mean lower-quality care, right? Less attention to medical issues. And if that differs systematically by race, that’s a problem.
But we need to be careful here, because engagement in the health record is not random. Many factors would lead you to spend more time in one patient’s chart vs. another. Medical complexity is the most obvious one. The authors did their best to account for this, adjusting for patients’ age, sex, insurance status, comorbidity score, and social deprivation index based on their ZIP code. But notably, they did not account for the acuity of illness during the hospitalization. If individuals identifying as a minority were, all else being equal, less likely to be severely ill by the time they were hospitalized, you might see results like this.
The authors also restrict their analysis to individuals who were discharged alive. I’m not entirely clear why they made this choice. Most people don’t die in the hospital; the inpatient mortality rate at most centers is 1%-1.5%. But excluding those patients could potentially bias these results, especially if race is, all else being equal, a predictor of inpatient mortality, as some studies have shown.
But the truth is, these data aren’t coming out of nowhere; they don’t exist in a vacuum. Numerous studies demonstrate different intensity of care among minority vs. nonminority individuals. There is this study, which shows that minority populations are less likely to be placed on the liver transplant waitlist.
There is this study, which found that minority kids with type 1 diabetes were less likely to get insulin pumps than were their White counterparts. And this one, which showed that kids with acute appendicitis were less likely to get pain-control medications if they were Black.
This study shows that although life expectancy decreased across all races during the pandemic, it decreased the most among minority populations.
This list goes on. It’s why the CDC has called racism a “fundamental cause of ... disease.”
So, yes, it is clear that there are racial disparities in health care outcomes. It is clear that there are racial disparities in treatments. It is also clear that virtually every physician believes they deliver equitable care. Somewhere, this disconnect arises. Could the actions we take in the EHR reveal the unconscious biases we have? Does the all-seeing eye of the EHR see not only into our brains but into our hearts? And if it can, are we ready to confront what it sees?
F. Perry Wilson, MD, MSCE, is associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator in New Haven, Conn. He reported no conflicts of interest.
A version of this article first appeared on Medscape.com.
Take two pills and make a donation
I was a resident, on morning rounds. The attending neurologist was young and ambitious (weren’t we all once?), trying to get the hospital to help him fund a research program in his subspecialty of interest.
One of the patients we saw that morning was a locally known successful businessman who’d been admitted, fortunately not for anything too serious.
My attending took the history, verifying the one I’d presented, and examined the gentleman. He then made some teaching points and explained the care plan to the patient.
Pretty standard up to that point.
After answering questions, however, the attending suddenly went into a sales pitch on his new research program, asking the guy for a financial donation, and giving him the card for the person at his office handling the funding.
I don’t remember anymore if he repeated that with other patients, but even now it still leaves a bad taste in my mouth. As a resident I wasn’t in a position to criticize him, nor did I want to endanger my own standing in the program by talking to someone higher up.
He was, fortunately, the only attending I ever worked with who did that. It still stands out in my mind, perhaps as an example of what not to do, and sometimes I still think about it.
Perhaps I’m naive, but I assumed he was an aberration. Apparently not, as the American College of Physicians recently issued a position paper advising its members not to ask patients for donations to the doctor’s workplace. There’s actually an acronym, GPF (Grateful Patient Fundraising) for this.
I understand a lot of these doctors are in academics and need funding for research and other programs. I know that a lot of good comes from this research, and I fully support it.
But this seems to be a bad way of doing it. Standing at the bedside on that long-ago morning, I remember thinking the patient (who looked kind of surprised) was going to wonder if this was a vague sort of hint: You’ll get better care if you pay up. Or a veiled threat that you may not get decent care if you don’t. I have no idea if he donated.
There must be a better way to get funding than hitting up a patient as part of the care plan. Perhaps discharge materials might include a brochure about how to make a donation, if interested. Or the ubiquitous portal might have a “donate” box in the task bar.
If the patient were to initiate this on his own, I wouldn’t have an issue with it. He gets out of the hospital, is grateful for his care, and calls the physician’s office to say he’d like to make a donation to whatever his program is (or just goes online to do it). That’s fine. I’ve even had the occasional patient call my office to say they’d like to make a donation to my favorite charity, and I give them a list of various neurology research foundations (none of which I’m affiliated with, for the record).
But to actively solicit donations from someone under your care is tasteless and inappropriate. It creates a conflict of interest for both parties.
The patient may believe he’ll get better care, and is obligated to keep giving – or else. The physician may feel like he’s stuck going beyond what’s really needed, ordering unnecessary tests and such to keep the financial VIP happy. And what happens if the big donor patient calls in because he hurt his ankle and needs a Percocet refill that another doctor won’t give him?
The statement by the ACP is appropriate. The only thing that bothers me about it is that it had to be made at all.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I was a resident, on morning rounds. The attending neurologist was young and ambitious (weren’t we all once?), trying to get the hospital to help him fund a research program in his subspecialty of interest.
One of the patients we saw that morning was a locally known successful businessman who’d been admitted, fortunately not for anything too serious.
My attending took the history, verifying the one I’d presented, and examined the gentleman. He then made some teaching points and explained the care plan to the patient.
Pretty standard up to that point.
After answering questions, however, the attending suddenly went into a sales pitch on his new research program, asking the guy for a financial donation, and giving him the card for the person at his office handling the funding.
I don’t remember anymore if he repeated that with other patients, but even now it still leaves a bad taste in my mouth. As a resident I wasn’t in a position to criticize him, nor did I want to endanger my own standing in the program by talking to someone higher up.
He was, fortunately, the only attending I ever worked with who did that. It still stands out in my mind, perhaps as an example of what not to do, and sometimes I still think about it.
Perhaps I’m naive, but I assumed he was an aberration. Apparently not, as the American College of Physicians recently issued a position paper advising its members not to ask patients for donations to the doctor’s workplace. There’s actually an acronym, GPF (Grateful Patient Fundraising) for this.
I understand a lot of these doctors are in academics and need funding for research and other programs. I know that a lot of good comes from this research, and I fully support it.
But this seems to be a bad way of doing it. Standing at the bedside on that long-ago morning, I remember thinking the patient (who looked kind of surprised) was going to wonder if this was a vague sort of hint: You’ll get better care if you pay up. Or a veiled threat that you may not get decent care if you don’t. I have no idea if he donated.
There must be a better way to get funding than hitting up a patient as part of the care plan. Perhaps discharge materials might include a brochure about how to make a donation, if interested. Or the ubiquitous portal might have a “donate” box in the task bar.
If the patient were to initiate this on his own, I wouldn’t have an issue with it. He gets out of the hospital, is grateful for his care, and calls the physician’s office to say he’d like to make a donation to whatever his program is (or just goes online to do it). That’s fine. I’ve even had the occasional patient call my office to say they’d like to make a donation to my favorite charity, and I give them a list of various neurology research foundations (none of which I’m affiliated with, for the record).
But to actively solicit donations from someone under your care is tasteless and inappropriate. It creates a conflict of interest for both parties.
The patient may believe he’ll get better care, and is obligated to keep giving – or else. The physician may feel like he’s stuck going beyond what’s really needed, ordering unnecessary tests and such to keep the financial VIP happy. And what happens if the big donor patient calls in because he hurt his ankle and needs a Percocet refill that another doctor won’t give him?
The statement by the ACP is appropriate. The only thing that bothers me about it is that it had to be made at all.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
I was a resident, on morning rounds. The attending neurologist was young and ambitious (weren’t we all once?), trying to get the hospital to help him fund a research program in his subspecialty of interest.
One of the patients we saw that morning was a locally known successful businessman who’d been admitted, fortunately not for anything too serious.
My attending took the history, verifying the one I’d presented, and examined the gentleman. He then made some teaching points and explained the care plan to the patient.
Pretty standard up to that point.
After answering questions, however, the attending suddenly went into a sales pitch on his new research program, asking the guy for a financial donation, and giving him the card for the person at his office handling the funding.
I don’t remember anymore if he repeated that with other patients, but even now it still leaves a bad taste in my mouth. As a resident I wasn’t in a position to criticize him, nor did I want to endanger my own standing in the program by talking to someone higher up.
He was, fortunately, the only attending I ever worked with who did that. It still stands out in my mind, perhaps as an example of what not to do, and sometimes I still think about it.
Perhaps I’m naive, but I assumed he was an aberration. Apparently not, as the American College of Physicians recently issued a position paper advising its members not to ask patients for donations to the doctor’s workplace. There’s actually an acronym, GPF (Grateful Patient Fundraising) for this.
I understand a lot of these doctors are in academics and need funding for research and other programs. I know that a lot of good comes from this research, and I fully support it.
But this seems to be a bad way of doing it. Standing at the bedside on that long-ago morning, I remember thinking the patient (who looked kind of surprised) was going to wonder if this was a vague sort of hint: You’ll get better care if you pay up. Or a veiled threat that you may not get decent care if you don’t. I have no idea if he donated.
There must be a better way to get funding than hitting up a patient as part of the care plan. Perhaps discharge materials might include a brochure about how to make a donation, if interested. Or the ubiquitous portal might have a “donate” box in the task bar.
If the patient were to initiate this on his own, I wouldn’t have an issue with it. He gets out of the hospital, is grateful for his care, and calls the physician’s office to say he’d like to make a donation to whatever his program is (or just goes online to do it). That’s fine. I’ve even had the occasional patient call my office to say they’d like to make a donation to my favorite charity, and I give them a list of various neurology research foundations (none of which I’m affiliated with, for the record).
But to actively solicit donations from someone under your care is tasteless and inappropriate. It creates a conflict of interest for both parties.
The patient may believe he’ll get better care, and is obligated to keep giving – or else. The physician may feel like he’s stuck going beyond what’s really needed, ordering unnecessary tests and such to keep the financial VIP happy. And what happens if the big donor patient calls in because he hurt his ankle and needs a Percocet refill that another doctor won’t give him?
The statement by the ACP is appropriate. The only thing that bothers me about it is that it had to be made at all.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
A new clue into the cause, spread of Parkinson’s disease?
.
While defects in mitochondrial functions and in mitochondrial DNA have been implicated in PD in the past, the current study demonstrates “for the first time how damaged mitochondrial DNA can underlie the mechanisms of PD initiation and spread in brain,” lead investigator Shohreh Issazadeh-Navikas, PhD, with the University of Copenhagen, told this news organization.
“This has direct implication for clinical diagnosis” – if damaged mtDNA can be detected in blood, it could serve as an early biomarker for disease, she explained.
The study was published online in Molecular Psychiatry.
“Infectious-like” spread of PD pathology
In earlier work, the researchers identified dysregulated interferon-beta (IFN-beta) signaling as a “top candidate pathway” associated with sporadic PD and its progression to PD with dementia (PDD).
In mice PD models that were deficient in IFN-beta signaling, the investigators showed that neuronal IFN-beta is required to maintain mitochondrial homeostasis and metabolism.
Lack of neuronal IFN-beta or disruption of its downstream signaling causes the accumulation of damaged mitochondria with excessive oxidative stress and insufficient adenosine triphosphate production.
In the current study, using postmortem brain tissue samples from patients with sporadic PD, they confirmed that there were deletions of mtDNA in the medial frontal gyrus, a region implicated in cognitive impairments in PD, suggesting a potential role of damaged mtDNA in disease pathophysiology.
They also identified mtDNA deletions in a “hotspot” in complex I respiratory chain subunits that were associated with dysregulation of oxidative stress and DNA damage response pathways in cohorts with sporadic PD and PDD.
They confirmed the contribution of mtDNA damage to PD pathology in the PD mouse models. They showed that lack of neuronal IFN-beta signaling leads to oxidative damage and mutations in mtDNA in neurons, which are subsequently released outside the neurons.
Injecting damaged mtDNA into mouse brain induced PDD-like behavioral symptoms, including neuropsychiatric, motor, and cognitive impairments. It also caused neurodegeneration in brain regions distant from the injection site, suggesting that damaged mtDNA triggers spread of PDD characteristics in an “infectious-like” manner, the researchers report.
Further study revealed that the mechanism through which damaged mtDNA causes pathology in healthy neurons involves dual activation of Toll-like receptor (TLR) 9 and 4 pathways, leading to increased oxidative stress and neuronal cell death, respectively.
“Our proteomic analysis of extracellular vesicles containing damaged mtDNA identified the TLR4 activator, ribosomal protein S3, as a key protein involved in recognizing and extruding damaged mtDNA,” the investigators write.
In the future they plan to investigate how mtDNA damage can serve as a predictive marker for different disease stages and progression and to explore potential therapeutic strategies aimed at restoring normal mitochondrial function to rectify the mitochondrial dysfunctions implicated in PD.
Making a comeback?
Commenting on the research for this news organization, James Beck, PhD, chief scientific officer at the Parkinson’s Foundation, noted that the role of mitochondria in PD is “like a starlet that burst onto the scene in the 80s, faded into obscurity, and through diligence and continued research has moved beyond being a solid character actor and is reemerging as a force to reckon with.
“This paper only adds to the allure that mitochondria may have in contributing to PD by providing evidence of a novel process by which mitochondria may be not only contributing to PD and loss of dopamine neurons but may play a larger role in the subsequent effects that many people with PD experience – dementia,” Dr. Beck said.
He noted that the authors identified several proteins as facilitating the neurodegeneration that is wrought by damaged mitochondrial DNA.
“These could be potential targets for future drug development. In addition, this work implicates alterations in immune signaling and drugs in development to target inflammatory responses may also bring ancillary benefit,” Dr. Beck said.
However, he said, “while very interesting findings, this is really the first effort that demonstrates how damaged mitochondrial DNA may contribute to neurodegeneration in the context of PD and PD dementia. Further work needs to validate these findings as well as to elucidate mechanisms underlying the propagation of the mitochondrial DNA from cell to cell.”
Funding for this research was provided by the European Union’s Horizon 2020 Research and Innovation Program, the Lundbeck Foundation, and the Danish Council for Independent Research–Medicine. Dr. Issazadeh-Navikas and Dr. Beck have disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
.
While defects in mitochondrial functions and in mitochondrial DNA have been implicated in PD in the past, the current study demonstrates “for the first time how damaged mitochondrial DNA can underlie the mechanisms of PD initiation and spread in brain,” lead investigator Shohreh Issazadeh-Navikas, PhD, with the University of Copenhagen, told this news organization.
“This has direct implication for clinical diagnosis” – if damaged mtDNA can be detected in blood, it could serve as an early biomarker for disease, she explained.
The study was published online in Molecular Psychiatry.
“Infectious-like” spread of PD pathology
In earlier work, the researchers identified dysregulated interferon-beta (IFN-beta) signaling as a “top candidate pathway” associated with sporadic PD and its progression to PD with dementia (PDD).
In mice PD models that were deficient in IFN-beta signaling, the investigators showed that neuronal IFN-beta is required to maintain mitochondrial homeostasis and metabolism.
Lack of neuronal IFN-beta or disruption of its downstream signaling causes the accumulation of damaged mitochondria with excessive oxidative stress and insufficient adenosine triphosphate production.
In the current study, using postmortem brain tissue samples from patients with sporadic PD, they confirmed that there were deletions of mtDNA in the medial frontal gyrus, a region implicated in cognitive impairments in PD, suggesting a potential role of damaged mtDNA in disease pathophysiology.
They also identified mtDNA deletions in a “hotspot” in complex I respiratory chain subunits that were associated with dysregulation of oxidative stress and DNA damage response pathways in cohorts with sporadic PD and PDD.
They confirmed the contribution of mtDNA damage to PD pathology in the PD mouse models. They showed that lack of neuronal IFN-beta signaling leads to oxidative damage and mutations in mtDNA in neurons, which are subsequently released outside the neurons.
Injecting damaged mtDNA into mouse brain induced PDD-like behavioral symptoms, including neuropsychiatric, motor, and cognitive impairments. It also caused neurodegeneration in brain regions distant from the injection site, suggesting that damaged mtDNA triggers spread of PDD characteristics in an “infectious-like” manner, the researchers report.
Further study revealed that the mechanism through which damaged mtDNA causes pathology in healthy neurons involves dual activation of Toll-like receptor (TLR) 9 and 4 pathways, leading to increased oxidative stress and neuronal cell death, respectively.
“Our proteomic analysis of extracellular vesicles containing damaged mtDNA identified the TLR4 activator, ribosomal protein S3, as a key protein involved in recognizing and extruding damaged mtDNA,” the investigators write.
In the future they plan to investigate how mtDNA damage can serve as a predictive marker for different disease stages and progression and to explore potential therapeutic strategies aimed at restoring normal mitochondrial function to rectify the mitochondrial dysfunctions implicated in PD.
Making a comeback?
Commenting on the research for this news organization, James Beck, PhD, chief scientific officer at the Parkinson’s Foundation, noted that the role of mitochondria in PD is “like a starlet that burst onto the scene in the 80s, faded into obscurity, and through diligence and continued research has moved beyond being a solid character actor and is reemerging as a force to reckon with.
“This paper only adds to the allure that mitochondria may have in contributing to PD by providing evidence of a novel process by which mitochondria may be not only contributing to PD and loss of dopamine neurons but may play a larger role in the subsequent effects that many people with PD experience – dementia,” Dr. Beck said.
He noted that the authors identified several proteins as facilitating the neurodegeneration that is wrought by damaged mitochondrial DNA.
“These could be potential targets for future drug development. In addition, this work implicates alterations in immune signaling and drugs in development to target inflammatory responses may also bring ancillary benefit,” Dr. Beck said.
However, he said, “while very interesting findings, this is really the first effort that demonstrates how damaged mitochondrial DNA may contribute to neurodegeneration in the context of PD and PD dementia. Further work needs to validate these findings as well as to elucidate mechanisms underlying the propagation of the mitochondrial DNA from cell to cell.”
Funding for this research was provided by the European Union’s Horizon 2020 Research and Innovation Program, the Lundbeck Foundation, and the Danish Council for Independent Research–Medicine. Dr. Issazadeh-Navikas and Dr. Beck have disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
.
While defects in mitochondrial functions and in mitochondrial DNA have been implicated in PD in the past, the current study demonstrates “for the first time how damaged mitochondrial DNA can underlie the mechanisms of PD initiation and spread in brain,” lead investigator Shohreh Issazadeh-Navikas, PhD, with the University of Copenhagen, told this news organization.
“This has direct implication for clinical diagnosis” – if damaged mtDNA can be detected in blood, it could serve as an early biomarker for disease, she explained.
The study was published online in Molecular Psychiatry.
“Infectious-like” spread of PD pathology
In earlier work, the researchers identified dysregulated interferon-beta (IFN-beta) signaling as a “top candidate pathway” associated with sporadic PD and its progression to PD with dementia (PDD).
In mice PD models that were deficient in IFN-beta signaling, the investigators showed that neuronal IFN-beta is required to maintain mitochondrial homeostasis and metabolism.
Lack of neuronal IFN-beta or disruption of its downstream signaling causes the accumulation of damaged mitochondria with excessive oxidative stress and insufficient adenosine triphosphate production.
In the current study, using postmortem brain tissue samples from patients with sporadic PD, they confirmed that there were deletions of mtDNA in the medial frontal gyrus, a region implicated in cognitive impairments in PD, suggesting a potential role of damaged mtDNA in disease pathophysiology.
They also identified mtDNA deletions in a “hotspot” in complex I respiratory chain subunits that were associated with dysregulation of oxidative stress and DNA damage response pathways in cohorts with sporadic PD and PDD.
They confirmed the contribution of mtDNA damage to PD pathology in the PD mouse models. They showed that lack of neuronal IFN-beta signaling leads to oxidative damage and mutations in mtDNA in neurons, which are subsequently released outside the neurons.
Injecting damaged mtDNA into mouse brain induced PDD-like behavioral symptoms, including neuropsychiatric, motor, and cognitive impairments. It also caused neurodegeneration in brain regions distant from the injection site, suggesting that damaged mtDNA triggers spread of PDD characteristics in an “infectious-like” manner, the researchers report.
Further study revealed that the mechanism through which damaged mtDNA causes pathology in healthy neurons involves dual activation of Toll-like receptor (TLR) 9 and 4 pathways, leading to increased oxidative stress and neuronal cell death, respectively.
“Our proteomic analysis of extracellular vesicles containing damaged mtDNA identified the TLR4 activator, ribosomal protein S3, as a key protein involved in recognizing and extruding damaged mtDNA,” the investigators write.
In the future they plan to investigate how mtDNA damage can serve as a predictive marker for different disease stages and progression and to explore potential therapeutic strategies aimed at restoring normal mitochondrial function to rectify the mitochondrial dysfunctions implicated in PD.
Making a comeback?
Commenting on the research for this news organization, James Beck, PhD, chief scientific officer at the Parkinson’s Foundation, noted that the role of mitochondria in PD is “like a starlet that burst onto the scene in the 80s, faded into obscurity, and through diligence and continued research has moved beyond being a solid character actor and is reemerging as a force to reckon with.
“This paper only adds to the allure that mitochondria may have in contributing to PD by providing evidence of a novel process by which mitochondria may be not only contributing to PD and loss of dopamine neurons but may play a larger role in the subsequent effects that many people with PD experience – dementia,” Dr. Beck said.
He noted that the authors identified several proteins as facilitating the neurodegeneration that is wrought by damaged mitochondrial DNA.
“These could be potential targets for future drug development. In addition, this work implicates alterations in immune signaling and drugs in development to target inflammatory responses may also bring ancillary benefit,” Dr. Beck said.
However, he said, “while very interesting findings, this is really the first effort that demonstrates how damaged mitochondrial DNA may contribute to neurodegeneration in the context of PD and PD dementia. Further work needs to validate these findings as well as to elucidate mechanisms underlying the propagation of the mitochondrial DNA from cell to cell.”
Funding for this research was provided by the European Union’s Horizon 2020 Research and Innovation Program, the Lundbeck Foundation, and the Danish Council for Independent Research–Medicine. Dr. Issazadeh-Navikas and Dr. Beck have disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
FROM MOLECULAR PSYCHIATRY