User login
Laparoscopic specimen retrieval bags in gyn surgery: Expert guidance on selection
The use of minimally invasive gynecologic surgery (MIGS) has grown rapidly over the past 20 years. MIGS, which includes vaginal hysterectomy and laparoscopic hysterectomy, is safe and has fewer complications and a more rapid recovery period than open abdominal surgery.1,2 In 2005, the role of MIGS was expanded further when the US Food and Drug Administration (FDA) approved robot-assisted surgery for the performance of gynecologic procedures.3 As knowledge and experience in the safe performance of MIGS progresses, the rates for MIGS procedures have skyrocketed and continue to grow. Between 2007 and 2010, laparoscopic hysterectomy rates rose from 23.5% to 30.5%, while robot-assisted laparoscopic hysterectomy rates increased from 0.5% to 9.5%, representing 40% of all hysterectomies.4 Due to the benefits of minimally invasive surgery over open abdominal surgery, patient and physician preference for minimally invasive procedures has grown significantly in popularity.1,5
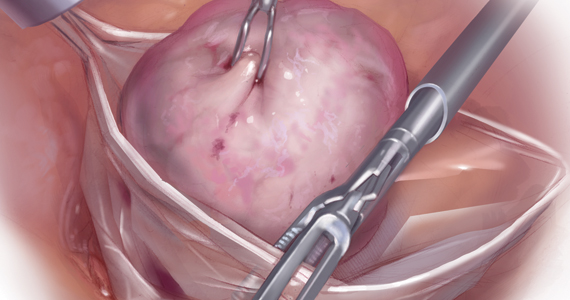
Because incisions are small in minimally invasive surgery, surgeons have been challenged with removing large specimens through incisions that are much smaller than the presenting pathology. One approach is to use a specimen retrieval bag for specimen extraction. Once the dissection is completed, the specimen is placed within the retrieval bag for removal, thus minimizing exposure of the specimen and its contents to the abdominopelvic cavity and incision.
The use of specimen retrieval devices has been advocated to prevent infection, avoid spillage into the peritoneal cavity, and minimize the risk of port-site metastases in cases of potentially cancerous specimens. Devices include affordable and readily available products, such as nonpowdered gloves, and commercially produced bags.6
While the use of specimen containment systems for tissue extraction has been well described in gynecology, the available systems vary widely in construction, size, durability, and shape, potentially leading to confusion and suboptimal bag selection during surgery.7 In this article, we review the most common laparoscopic bags available in the United States, provide an overview of bag characteristics, offer practice guidelines for bag selection, and review bag terminology to highlight important concepts for bag selection.
Controversy spurs change
In April 2014, the FDA warned against the use of power morcellation for specimen removal during minimally invasive surgery, citing a prevalence of 1 in 352 unsuspected uterine sarcomas and 1 in 498 unsuspected uterine leiomyosarcomas among women undergoing hysterectomy or myomectomy for presumed benign leiomyoma.8 Since then, the risk of occult uterine sarcomas, including leiomyosarcoma, in women undergoing surgery for benign gynecologic indications has been determined to be much lower.
Nonetheless, the clinical importance of contained specimen removal was clearly highlighted and the role of specimen retrieval bags soared to the forefront. Open power morcellation is no longer commonly practiced, and national societies such as the American Association of Gynecologic Laparoscopists (AAGL), the Society of Gynecologic Oncology (SGO), and the American College of Obstetricians and Gynecologists (ACOG) recommend that containment systems be used for safer specimen retrieval during gynecologic surgery.9-11 After the specimen is placed inside the containment system (typically a specimen bag), the surgeon may deliver the bag through a vaginal colpotomy or through a slightly extended laparoscopic incision to remove bulky specimens using cold-cutting extraction techniques.12-15
Continue to: Know the pathology’s characteristics...
Know the pathology’s characteristics
In most cases, based on imaging studies and physical examination, surgeons have a good idea of what to expect before proceeding with surgery. The 2 most common characteristics used for surgical planning are the specimen size (dimensions) and the tissue type (solid, cystic, soft tissue, or mixed). The mass size can range from less than 1 cm to larger than a 20-week sized fibroid uterus. Assessing the specimen in 3 dimensions is important. Tissue type also is a consideration, as soft and squishy masses, such as ovarian cysts, are easier to deflate and manipulate within the bag compared with solid or calcified tumors, such as a large fibroid uterus or a large dermoid with solid components.
Specimen shape also is a critical determinant for bag selection. Most specimen retrieval bags are tapered to varying degrees, and some have an irregular shape. Long tubular structures, such as fallopian tubes that are composed of soft tissue, fit easily into most bags regardless of bag shape or extent of bag taper, whereas the round shape of a bulky myoma may render certain bags ineffective even if the bag’s entrance accommodates the greatest diameter of the myoma. Often, a round mass will not fully fit into a bag because there is a poor fit between the mass’s shape and the bag’s shape and taper. (We discuss the concept of a poor “fit” below.) Knowing the pathology before starting a procedure can help optimize bag selection, streamline operative flow, and reduce waste.
Overview of laparoscopic bag characteristics and clinical applications
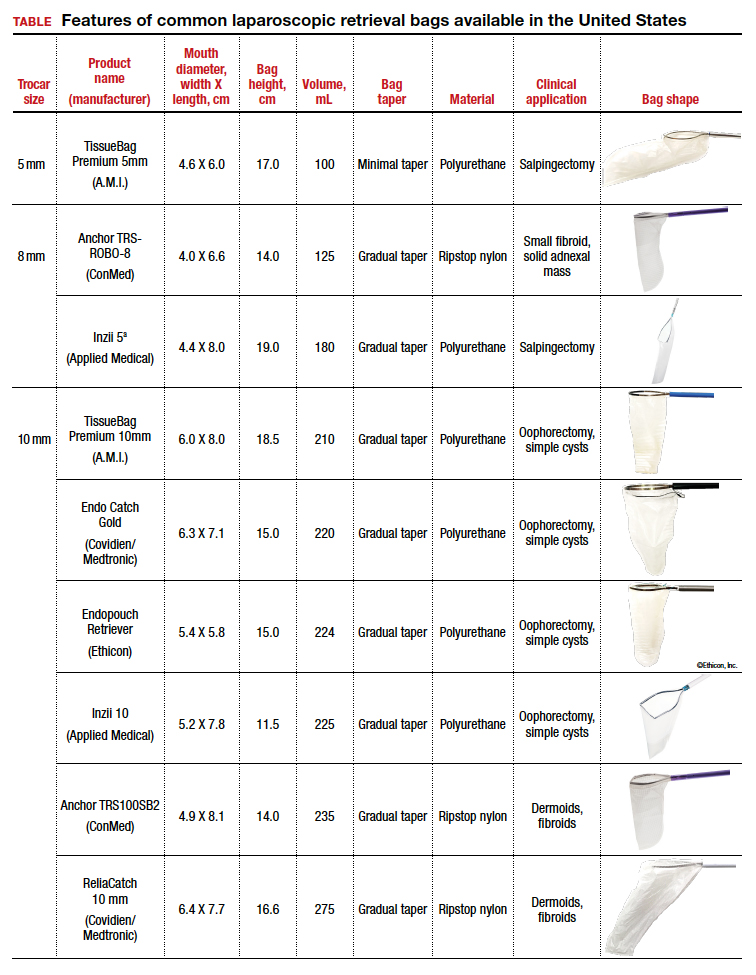
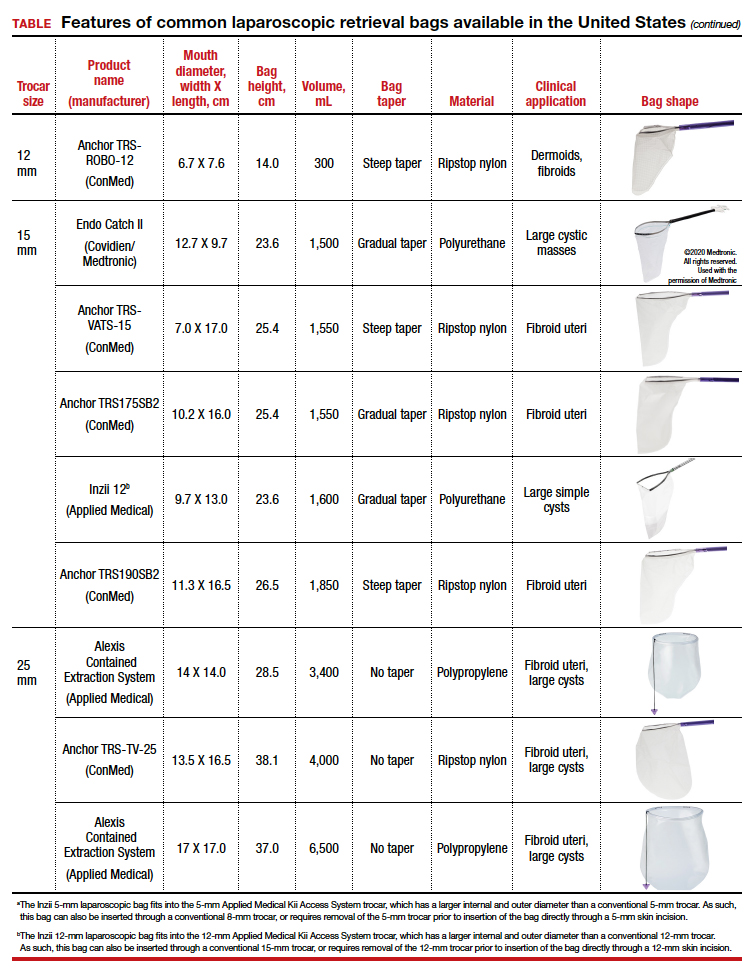
The TABLE lists the most common laparoscopic bags available for purchase in the United States. Details include the trocar size, manufacturer, product name, mouth diameter, volume, bag shape, construction material, and best clinical application.
The following are terms used to refer to the components of a laparoscopic retrieval bag:
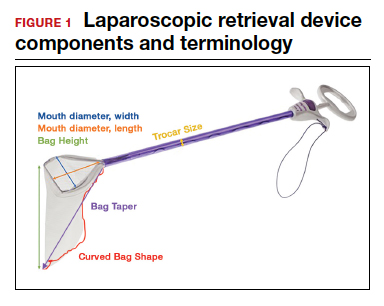
- Mouth diameter: diameter at the entrance of a fully opened bag (FIGURE 1)
- Bag volume: the total volume a bag can accommodate when completely full
- Bag rim: characteristics of the rim of the bag when opened (that is, rigid vs soft rim, complete vs partial rim mechanism to hold the bag open) (FIGURE 2)
- Bag shape: the shape of the bag when it is fully opened (square shaped vs cone shaped vs curved bag shape) (FIGURE 2)
- Bag taper (severity and type): extent the bag is tapered from the rim of the bag’s entrance to the base of the bag; categorized by taper severity (minimal, gradual, or steep taper) and type (continuous taper or curved taper) (FIGURE 3)
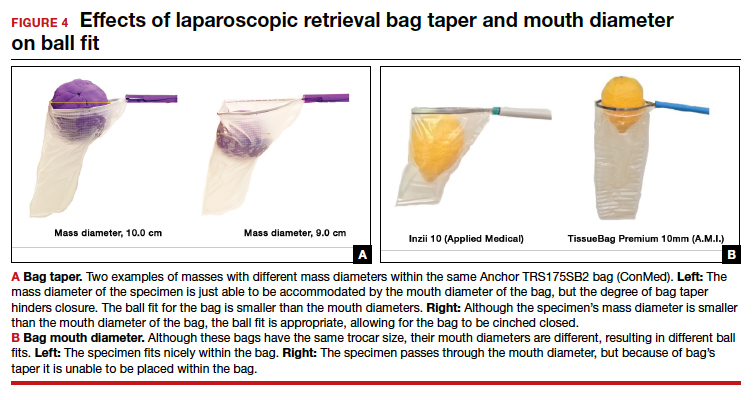
- Ball fit: the maximum spherical specimen size that completely fits into a bag and allows it to cinch closed (FIGURE 4)
- Bag strength: durability of a bag when placed on tension during specimen extraction (weak, moderate, or extremely durable).


Continue to: Mouth diameter...
Mouth diameter
Bag manufacturers often differentiate bag sizes by indicating “volume” in milliliters. Bag volume, however, offers little clinical value to surgeons, as pelvic mass dimensions are usually measured in centimeters on imaging. Rather, an important characteristic for bag selection is the diameter of the rim of the bag when it is fully opened—the so-called bag mouth diameter. For a specimen to fit, the 2 dimensions of the specimen must be smaller than the dimensions of the bag entrance.
Notably, the number often linked to the specimen bag—as, for example, in the 10-mm Endo Catch bag (Covidien/Medtronic)— describes the width of the shaft of the bag before it is opened rather than the mouth diameter of the opened bag. The number actually correlates with the trocar size necessary for bag insertion rather than with the specimen size that can fit into the bag. Therefore, a 10-mm Endo Catch bag cannot fit a 10-cm mass, but rather requires a trocar size of 10 mm or greater for insertion of the bag. Fully opened, the mouth diameters of the 10-mm Endo Catch bag are roughly 6 cm x 7 cm, which allows for delivery of a 6-cm mass.
Because 2 bags that use the same trocar size for insertion may have vastly differing bag dimensions, the surgeon must know the bag mouth diameters when selecting a bag to remove the presenting pathology. For example, the Inzii 12 (Applied Medical) laparoscopic bag has mouth diameters of 9.7 cm × 13.0 cm, whereas the Anchor TRSROBO-12 (ConMed) has mouth diameters of 6.7 cm × 7.6 cm (TABLE). Although both bags can be inserted through a 12-mm trocar, both bags cannot fit the same size mass for removal.
Shape and taper
Laparoscopic bags come in various shapes (curved, cone, or square shaped), with varying levels of bag taper (steep, gradual, or no taper) (FIGURES 2 and 3). While taper has little impact on long and skinny specimens, taper may hinder successful bagging of bulky or spherical specimens.
Each bag has different grades of taper regardless of mouth diameter or trocar size. For round masses, the steeper the taper, the smaller the mass that can comfortably fit within the bag. This concept is connected to the idea of “ball fit,” explained below.
In addition, bag shape may affect what mass size can fit into the bag. An irregularly shaped curved bag or a bag with a steep taper may be well suited for removal of multiple specimens of varying sizes or soft masses that are malleable enough to conform to the bag’s shape (such as a ruptured ovarian cyst). Alternatively, a square-shaped bag or a bag with minimal taper would better accommodate a round mass.
Ball fit
When thinking about large circular masses, such as myomas or ovarian cysts, one must consider the ball fit. This refers to the maximum spherical size of the specimen that fits completely within a bag while allowing the bag to cinch closed. Generally, this is an estimation that factors in the bag shape, extent of the bag taper, bag mouth diameter, and specimen shape and tissue type. At times, although a mass can fit through the bag’s mouth diameter, a steep taper may prevent the mass from being fully bagged and limit closure of the bag (FIGURE 4).
Curved bags like the Anchor TRSVATS-15 (ConMed), which have a very narrow bottom, are prone to a limited ball fit, and thus the bag mouth diameter will not correlate with the largest mass size that can be fitted within the bag. Therefore, if using a steeply tapered bag for removal of large round masses, do not rely on the bag’s mouth diameter for bag selection. The surgeon must visualize the ball fit within the bag, taking into account the specimen size and shape, bag shape, and bag taper. In these scenarios, using the diameter of the midportion of the opened bag may better reflect the mass size that can fit into that bag.
Bag strength
Bag strength depends on the material used for bag construction. Most laparoscopic bags in the United States are made of 3 different materials: polyurethane, polypropylene, and ripstop nylon.
Polyurethane and polypropylene are synthetic plastic polymers; in bag form they are stretchy and, under extreme force, may tear. They are best used for bagging fluid-filled cysts or soft pliable masses that will not require extensive bag or tissue handling, such as extraction of large leiomyomas. Polyurethane and polypropylene bags are more susceptible to puncture with sharp laparoscopic instruments or scalpels, and care must be taken to avoid accidentally cutting the bag during tissue extraction.
Alternatively, bags made of ripstop nylon are favored for their bag strength. Ripstop nylon is a synthetic fabric that is woven together in a crosshatch pattern that makes it resistant to tearing and ripping. It was developed originally during World War II as a replacement for silk parachutes. Modern applications include its use in sails, kites, and high-quality camping equipment. This material has a favorable strength-to-weight ratio, and, in case of a tear, it is less prone to extension of the tear. For surgical applications, these bags are best used for bagging specimens that will require a lot of bag manipulation and tissue extraction. However, the ripstop fabric takes up more space in the incision than polyurethane or polypropylene, leaving the surgeon with less space for tissue extraction. Thus, as a tradeoff for bag strength, the surgeon may need to extend the incision a little, and a small self-retracting wound retractor may be necessary to allow visibility for safe tissue extraction when using a ripstop nylon bag compared with others.
Continue to: Trocar selection is important...
Trocar selection is important
While considering bag selection, the surgeon also must consider trocar selection to allow for laparoscopic insertion of the bag. Trocar size for bag selection refers to the minimum trocar diameter needed to insert the laparoscopic bag. Most bags are designed to fit into a laparoscopic trocar or into the skin incision that previously housed the trocar. Trocar size does not directly correlate with bag mouth diameter; for example, a 10-mm laparoscopic bag that can be inserted through a 10- or 12-mm trocar size cannot fit a 10-cm mass (see the mouth diameter section above).
A tip to maximize operating room (OR) efficiency is to start off with a larger trocar, such as a 12-mm trocar, if it is known that a laparoscopic bag with a 12-mm trocar size will be used, rather than starting with a 5-mm trocar and upsizing the port site incision. This saves time and offers intraoperative flexibility, allowing for the use of larger instruments and quicker insufflation.
Furthermore, if the specimen has a solid component and tissue extraction is anticipated, consider starting off with a large trocar, one that is larger than the bag’s trocar size since the incision likely will be extended. For example, even if a myoma will fit within a 10-mm laparoscopic bag made of ripstop nylon, using a 15-mm trocar rather than a 10-mm trocar may be considered since the skin and fascial incisions will need to be extended to allow for cold-cut tissue extraction. Starting with the larger 15-mm trocar may offer surgical advantages, such as direct needle delivery of larger needles for myometrial closure after myomectomy or direct removal of smaller myomas through the trocar to avoid bagging multiple specimens.
Putting it all together
To optimize efficiency in the OR for specimen removal, we recommend streamlining OR flow and reducing waste by first considering the specimen size, tissue type, bag shape, and trocar selection. Choose a bag by taking into account the bag mouth diameter and the amount of taper you will need to obtain an appropriate ball fit. If the tissue type is soft and pliable, consider a polyurethane or polypropylene bag and the smallest bag size possible, even if it has a narrow bag shape and taper.
However, if the tissue type is solid, the shape is round, and the mass is large (requiring extensive tissue extraction for removal), consider a bag made of ripstop nylon and factor in the bag shape as well as the bag taper. Using a bag without a steep taper may allow a better fit.
After choosing a laparoscopic bag, select the appropriate trocars necessary for completion of the surgery. Consider starting off with a larger trocar rather than spending the time to upsize a trocar if you plan to use a large bag or intend to extend the trocar incision for a contained tissue extraction. These tips will help optimize efficiency, reduce equipment wastage, and prevent intra-abdominal spillage.
Keep in mind that all procedures, including specimen removal using containment systems, have inherent risks. For example, visualization of the mass within the bag and visualization of vital structures may be hindered by bulkiness of the bag or specimen. There is also a risk of bag compromise and leakage, whether through manipulation of the bag or puncture during specimen extraction. Lastly, even though removing a specimen within a containment system minimizes spillage and reports of in-bag cold-knife tissue extraction in women with histologically proven endometrial cancer have suggested that it is safe, laparoscopic bags have not been proven to prevent the dissemination of malignant tissue fragments.16,17
Overall, the inherent risks of specimen extraction during minimally invasive surgery are far outweighed by the well-established advantages of laparoscopic surgery, which carries lower risks of surgical complications such as bleeding and infection, shorter hospital stay, and quicker recovery time compared to laparotomy. There is no doubt minimally invasive surgery offers many benefits.
In summary, for best bag selection, it is equally important to know the characteristics of the pathology as it is to know the features of the specimen retrieval systems available at your institution. Understanding both the pathology and the equipment available will allow the surgeon to make the best surgical decisions for the case. ●
- Desai VB, Wright JD, Lin H, et al. Laparoscopic hysterectomy route, resource use, and outcomes: change after power morcellation warning. Obstet Gynecol. 2019;134:227-238.
- American College of Obstetricians and Gynecologists. ACOG committee opinion No. 444: choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114:1156-1158.
- Liu H, Lu D, Wang L, et al. Robotic surgery for benign gynecological disease. Cochrane Database Syst Rev. 2012;2:CD008978.
- Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241.
- Turner LC, Shepherd JP, Wang L, et al. Hysterectomy surgery trends: a more accurate depiction of the last decade? Am J Obstet Gynecol. 2013;208:277.e1-7.
- Holme JB, Mortensen FV. A powder-free surgical glove bag for retraction of the gallbladder during laparoscopic cholecystectomy. Surg Laparosc Endosc Percutan Tech. 2005;15:209-211.
- Siedhoff MT, Cohen SL. Tissue extraction techniques for leiomyomas and uteri during minimally invasive surgery. Obstet Gynecol. 2017;130:1251-1260.
- US Food and Drug Administration. Laparoscopic uterine power morcellation in hysterectomy and myomectomy: FDA safety communication. April 17, 2014. https://wayback .archive-it.org/7993/20170722215731/https:/www.fda.gov /MedicalDevices/Safety/AlertsandNotices/ucm393576.htm. Accessed September 22, 2020.
- AAGL. AAGL practice report: morcellation during uterine tissue extraction. J Minim Invasive Gynecol. 2014;21:517-530.
- American College of Obstetricians and Gynecologists. ACOG committee opinion No. 770: uterine morcellation for presumed leiomyomas. Obstet Gynecol. 2019;133:e238-e248.
- Society of Gynecologic Oncology website. SGO position statement: morcellation. December 1, 2013. https://www .sgo.org/newsroom/position-statements-2/morcellation/. Accessed September 22, 2020.
- Advincula AP, Truong MD. ExCITE: minimally invasive tissue extraction made simple with simulation. OBG Manag. 2015;27(12):40-45.
- Solima E, Scagnelli G, Austoni V, et al. Vaginal uterine morcellation within a specimen containment system: a study of bag integrity. J Minim Invasive Gynecol. 2015;22:1244-1246.
- Ghezzi F, Casarin J, De Francesco G, et al. Transvaginal contained tissue extraction after laparoscopic myomectomy: a cohort study. BJOG. 2018;125:367-373.
- Dotson S, Landa A, Ehrisman J, et al. Safety and feasibility of contained uterine morcellation in women undergoing laparoscopic hysterectomy. Gynecol Oncol Res Pract. 2018;5:8.
- Favero G, Miglino G, Köhler C, et al. Vaginal morcellation inside protective pouch: a safe strategy for uterine extration in cases of bulky endometrial cancers: operative and oncological safety of the method. J Minim Invasive Gynecol. 2015;22:938-943.
- Montella F, Riboni F, Cosma S, et al. A safe method of vaginal longitudinal morcellation of bulky uterus with endometrial cancer in a bag at laparoscopy. Surg Endosc. 2014;28:1949-1953.

Dr. Sia is a Resident in Obstetrics and Gynecology, Columbia University College of Physicians and Surgeons, New York, New York.

Dr. Hur is an Assistant Professor of Obstetrics and Gynecology, Columbia University Irving Medical Center and New York Presbyterian Hospital.
The authors report no financial relationships relevant to this article.
Dr. Sia is a Resident in Obstetrics and Gynecology, Columbia University College of Physicians and Surgeons, New York, New York.
Dr. Hur is an Assistant Professor of Obstetrics and Gynecology, Columbia University Irving Medical Center and New York Presbyterian Hospital.
The authors report no financial relationships relevant to this article.
Dr. Sia is a Resident in Obstetrics and Gynecology, Columbia University College of Physicians and Surgeons, New York, New York.
Dr. Hur is an Assistant Professor of Obstetrics and Gynecology, Columbia University Irving Medical Center and New York Presbyterian Hospital.
The authors report no financial relationships relevant to this article.
The use of minimally invasive gynecologic surgery (MIGS) has grown rapidly over the past 20 years. MIGS, which includes vaginal hysterectomy and laparoscopic hysterectomy, is safe and has fewer complications and a more rapid recovery period than open abdominal surgery.1,2 In 2005, the role of MIGS was expanded further when the US Food and Drug Administration (FDA) approved robot-assisted surgery for the performance of gynecologic procedures.3 As knowledge and experience in the safe performance of MIGS progresses, the rates for MIGS procedures have skyrocketed and continue to grow. Between 2007 and 2010, laparoscopic hysterectomy rates rose from 23.5% to 30.5%, while robot-assisted laparoscopic hysterectomy rates increased from 0.5% to 9.5%, representing 40% of all hysterectomies.4 Due to the benefits of minimally invasive surgery over open abdominal surgery, patient and physician preference for minimally invasive procedures has grown significantly in popularity.1,5
Because incisions are small in minimally invasive surgery, surgeons have been challenged with removing large specimens through incisions that are much smaller than the presenting pathology. One approach is to use a specimen retrieval bag for specimen extraction. Once the dissection is completed, the specimen is placed within the retrieval bag for removal, thus minimizing exposure of the specimen and its contents to the abdominopelvic cavity and incision.
The use of specimen retrieval devices has been advocated to prevent infection, avoid spillage into the peritoneal cavity, and minimize the risk of port-site metastases in cases of potentially cancerous specimens. Devices include affordable and readily available products, such as nonpowdered gloves, and commercially produced bags.6
While the use of specimen containment systems for tissue extraction has been well described in gynecology, the available systems vary widely in construction, size, durability, and shape, potentially leading to confusion and suboptimal bag selection during surgery.7 In this article, we review the most common laparoscopic bags available in the United States, provide an overview of bag characteristics, offer practice guidelines for bag selection, and review bag terminology to highlight important concepts for bag selection.
Controversy spurs change
In April 2014, the FDA warned against the use of power morcellation for specimen removal during minimally invasive surgery, citing a prevalence of 1 in 352 unsuspected uterine sarcomas and 1 in 498 unsuspected uterine leiomyosarcomas among women undergoing hysterectomy or myomectomy for presumed benign leiomyoma.8 Since then, the risk of occult uterine sarcomas, including leiomyosarcoma, in women undergoing surgery for benign gynecologic indications has been determined to be much lower.
Nonetheless, the clinical importance of contained specimen removal was clearly highlighted and the role of specimen retrieval bags soared to the forefront. Open power morcellation is no longer commonly practiced, and national societies such as the American Association of Gynecologic Laparoscopists (AAGL), the Society of Gynecologic Oncology (SGO), and the American College of Obstetricians and Gynecologists (ACOG) recommend that containment systems be used for safer specimen retrieval during gynecologic surgery.9-11 After the specimen is placed inside the containment system (typically a specimen bag), the surgeon may deliver the bag through a vaginal colpotomy or through a slightly extended laparoscopic incision to remove bulky specimens using cold-cutting extraction techniques.12-15
Continue to: Know the pathology’s characteristics...
Know the pathology’s characteristics
In most cases, based on imaging studies and physical examination, surgeons have a good idea of what to expect before proceeding with surgery. The 2 most common characteristics used for surgical planning are the specimen size (dimensions) and the tissue type (solid, cystic, soft tissue, or mixed). The mass size can range from less than 1 cm to larger than a 20-week sized fibroid uterus. Assessing the specimen in 3 dimensions is important. Tissue type also is a consideration, as soft and squishy masses, such as ovarian cysts, are easier to deflate and manipulate within the bag compared with solid or calcified tumors, such as a large fibroid uterus or a large dermoid with solid components.
Specimen shape also is a critical determinant for bag selection. Most specimen retrieval bags are tapered to varying degrees, and some have an irregular shape. Long tubular structures, such as fallopian tubes that are composed of soft tissue, fit easily into most bags regardless of bag shape or extent of bag taper, whereas the round shape of a bulky myoma may render certain bags ineffective even if the bag’s entrance accommodates the greatest diameter of the myoma. Often, a round mass will not fully fit into a bag because there is a poor fit between the mass’s shape and the bag’s shape and taper. (We discuss the concept of a poor “fit” below.) Knowing the pathology before starting a procedure can help optimize bag selection, streamline operative flow, and reduce waste.
Overview of laparoscopic bag characteristics and clinical applications
The TABLE lists the most common laparoscopic bags available for purchase in the United States. Details include the trocar size, manufacturer, product name, mouth diameter, volume, bag shape, construction material, and best clinical application.
The following are terms used to refer to the components of a laparoscopic retrieval bag:
- Mouth diameter: diameter at the entrance of a fully opened bag (FIGURE 1)
- Bag volume: the total volume a bag can accommodate when completely full
- Bag rim: characteristics of the rim of the bag when opened (that is, rigid vs soft rim, complete vs partial rim mechanism to hold the bag open) (FIGURE 2)
- Bag shape: the shape of the bag when it is fully opened (square shaped vs cone shaped vs curved bag shape) (FIGURE 2)
- Bag taper (severity and type): extent the bag is tapered from the rim of the bag’s entrance to the base of the bag; categorized by taper severity (minimal, gradual, or steep taper) and type (continuous taper or curved taper) (FIGURE 3)
- Ball fit: the maximum spherical specimen size that completely fits into a bag and allows it to cinch closed (FIGURE 4)
- Bag strength: durability of a bag when placed on tension during specimen extraction (weak, moderate, or extremely durable).
Continue to: Mouth diameter...
Mouth diameter
Bag manufacturers often differentiate bag sizes by indicating “volume” in milliliters. Bag volume, however, offers little clinical value to surgeons, as pelvic mass dimensions are usually measured in centimeters on imaging. Rather, an important characteristic for bag selection is the diameter of the rim of the bag when it is fully opened—the so-called bag mouth diameter. For a specimen to fit, the 2 dimensions of the specimen must be smaller than the dimensions of the bag entrance.
Notably, the number often linked to the specimen bag—as, for example, in the 10-mm Endo Catch bag (Covidien/Medtronic)— describes the width of the shaft of the bag before it is opened rather than the mouth diameter of the opened bag. The number actually correlates with the trocar size necessary for bag insertion rather than with the specimen size that can fit into the bag. Therefore, a 10-mm Endo Catch bag cannot fit a 10-cm mass, but rather requires a trocar size of 10 mm or greater for insertion of the bag. Fully opened, the mouth diameters of the 10-mm Endo Catch bag are roughly 6 cm x 7 cm, which allows for delivery of a 6-cm mass.
Because 2 bags that use the same trocar size for insertion may have vastly differing bag dimensions, the surgeon must know the bag mouth diameters when selecting a bag to remove the presenting pathology. For example, the Inzii 12 (Applied Medical) laparoscopic bag has mouth diameters of 9.7 cm × 13.0 cm, whereas the Anchor TRSROBO-12 (ConMed) has mouth diameters of 6.7 cm × 7.6 cm (TABLE). Although both bags can be inserted through a 12-mm trocar, both bags cannot fit the same size mass for removal.
Shape and taper
Laparoscopic bags come in various shapes (curved, cone, or square shaped), with varying levels of bag taper (steep, gradual, or no taper) (FIGURES 2 and 3). While taper has little impact on long and skinny specimens, taper may hinder successful bagging of bulky or spherical specimens.
Each bag has different grades of taper regardless of mouth diameter or trocar size. For round masses, the steeper the taper, the smaller the mass that can comfortably fit within the bag. This concept is connected to the idea of “ball fit,” explained below.
In addition, bag shape may affect what mass size can fit into the bag. An irregularly shaped curved bag or a bag with a steep taper may be well suited for removal of multiple specimens of varying sizes or soft masses that are malleable enough to conform to the bag’s shape (such as a ruptured ovarian cyst). Alternatively, a square-shaped bag or a bag with minimal taper would better accommodate a round mass.
Ball fit
When thinking about large circular masses, such as myomas or ovarian cysts, one must consider the ball fit. This refers to the maximum spherical size of the specimen that fits completely within a bag while allowing the bag to cinch closed. Generally, this is an estimation that factors in the bag shape, extent of the bag taper, bag mouth diameter, and specimen shape and tissue type. At times, although a mass can fit through the bag’s mouth diameter, a steep taper may prevent the mass from being fully bagged and limit closure of the bag (FIGURE 4).
Curved bags like the Anchor TRSVATS-15 (ConMed), which have a very narrow bottom, are prone to a limited ball fit, and thus the bag mouth diameter will not correlate with the largest mass size that can be fitted within the bag. Therefore, if using a steeply tapered bag for removal of large round masses, do not rely on the bag’s mouth diameter for bag selection. The surgeon must visualize the ball fit within the bag, taking into account the specimen size and shape, bag shape, and bag taper. In these scenarios, using the diameter of the midportion of the opened bag may better reflect the mass size that can fit into that bag.
Bag strength
Bag strength depends on the material used for bag construction. Most laparoscopic bags in the United States are made of 3 different materials: polyurethane, polypropylene, and ripstop nylon.
Polyurethane and polypropylene are synthetic plastic polymers; in bag form they are stretchy and, under extreme force, may tear. They are best used for bagging fluid-filled cysts or soft pliable masses that will not require extensive bag or tissue handling, such as extraction of large leiomyomas. Polyurethane and polypropylene bags are more susceptible to puncture with sharp laparoscopic instruments or scalpels, and care must be taken to avoid accidentally cutting the bag during tissue extraction.
Alternatively, bags made of ripstop nylon are favored for their bag strength. Ripstop nylon is a synthetic fabric that is woven together in a crosshatch pattern that makes it resistant to tearing and ripping. It was developed originally during World War II as a replacement for silk parachutes. Modern applications include its use in sails, kites, and high-quality camping equipment. This material has a favorable strength-to-weight ratio, and, in case of a tear, it is less prone to extension of the tear. For surgical applications, these bags are best used for bagging specimens that will require a lot of bag manipulation and tissue extraction. However, the ripstop fabric takes up more space in the incision than polyurethane or polypropylene, leaving the surgeon with less space for tissue extraction. Thus, as a tradeoff for bag strength, the surgeon may need to extend the incision a little, and a small self-retracting wound retractor may be necessary to allow visibility for safe tissue extraction when using a ripstop nylon bag compared with others.
Continue to: Trocar selection is important...
Trocar selection is important
While considering bag selection, the surgeon also must consider trocar selection to allow for laparoscopic insertion of the bag. Trocar size for bag selection refers to the minimum trocar diameter needed to insert the laparoscopic bag. Most bags are designed to fit into a laparoscopic trocar or into the skin incision that previously housed the trocar. Trocar size does not directly correlate with bag mouth diameter; for example, a 10-mm laparoscopic bag that can be inserted through a 10- or 12-mm trocar size cannot fit a 10-cm mass (see the mouth diameter section above).
A tip to maximize operating room (OR) efficiency is to start off with a larger trocar, such as a 12-mm trocar, if it is known that a laparoscopic bag with a 12-mm trocar size will be used, rather than starting with a 5-mm trocar and upsizing the port site incision. This saves time and offers intraoperative flexibility, allowing for the use of larger instruments and quicker insufflation.
Furthermore, if the specimen has a solid component and tissue extraction is anticipated, consider starting off with a large trocar, one that is larger than the bag’s trocar size since the incision likely will be extended. For example, even if a myoma will fit within a 10-mm laparoscopic bag made of ripstop nylon, using a 15-mm trocar rather than a 10-mm trocar may be considered since the skin and fascial incisions will need to be extended to allow for cold-cut tissue extraction. Starting with the larger 15-mm trocar may offer surgical advantages, such as direct needle delivery of larger needles for myometrial closure after myomectomy or direct removal of smaller myomas through the trocar to avoid bagging multiple specimens.
Putting it all together
To optimize efficiency in the OR for specimen removal, we recommend streamlining OR flow and reducing waste by first considering the specimen size, tissue type, bag shape, and trocar selection. Choose a bag by taking into account the bag mouth diameter and the amount of taper you will need to obtain an appropriate ball fit. If the tissue type is soft and pliable, consider a polyurethane or polypropylene bag and the smallest bag size possible, even if it has a narrow bag shape and taper.
However, if the tissue type is solid, the shape is round, and the mass is large (requiring extensive tissue extraction for removal), consider a bag made of ripstop nylon and factor in the bag shape as well as the bag taper. Using a bag without a steep taper may allow a better fit.
After choosing a laparoscopic bag, select the appropriate trocars necessary for completion of the surgery. Consider starting off with a larger trocar rather than spending the time to upsize a trocar if you plan to use a large bag or intend to extend the trocar incision for a contained tissue extraction. These tips will help optimize efficiency, reduce equipment wastage, and prevent intra-abdominal spillage.
Keep in mind that all procedures, including specimen removal using containment systems, have inherent risks. For example, visualization of the mass within the bag and visualization of vital structures may be hindered by bulkiness of the bag or specimen. There is also a risk of bag compromise and leakage, whether through manipulation of the bag or puncture during specimen extraction. Lastly, even though removing a specimen within a containment system minimizes spillage and reports of in-bag cold-knife tissue extraction in women with histologically proven endometrial cancer have suggested that it is safe, laparoscopic bags have not been proven to prevent the dissemination of malignant tissue fragments.16,17
Overall, the inherent risks of specimen extraction during minimally invasive surgery are far outweighed by the well-established advantages of laparoscopic surgery, which carries lower risks of surgical complications such as bleeding and infection, shorter hospital stay, and quicker recovery time compared to laparotomy. There is no doubt minimally invasive surgery offers many benefits.
In summary, for best bag selection, it is equally important to know the characteristics of the pathology as it is to know the features of the specimen retrieval systems available at your institution. Understanding both the pathology and the equipment available will allow the surgeon to make the best surgical decisions for the case. ●
The use of minimally invasive gynecologic surgery (MIGS) has grown rapidly over the past 20 years. MIGS, which includes vaginal hysterectomy and laparoscopic hysterectomy, is safe and has fewer complications and a more rapid recovery period than open abdominal surgery.1,2 In 2005, the role of MIGS was expanded further when the US Food and Drug Administration (FDA) approved robot-assisted surgery for the performance of gynecologic procedures.3 As knowledge and experience in the safe performance of MIGS progresses, the rates for MIGS procedures have skyrocketed and continue to grow. Between 2007 and 2010, laparoscopic hysterectomy rates rose from 23.5% to 30.5%, while robot-assisted laparoscopic hysterectomy rates increased from 0.5% to 9.5%, representing 40% of all hysterectomies.4 Due to the benefits of minimally invasive surgery over open abdominal surgery, patient and physician preference for minimally invasive procedures has grown significantly in popularity.1,5
Because incisions are small in minimally invasive surgery, surgeons have been challenged with removing large specimens through incisions that are much smaller than the presenting pathology. One approach is to use a specimen retrieval bag for specimen extraction. Once the dissection is completed, the specimen is placed within the retrieval bag for removal, thus minimizing exposure of the specimen and its contents to the abdominopelvic cavity and incision.
The use of specimen retrieval devices has been advocated to prevent infection, avoid spillage into the peritoneal cavity, and minimize the risk of port-site metastases in cases of potentially cancerous specimens. Devices include affordable and readily available products, such as nonpowdered gloves, and commercially produced bags.6
While the use of specimen containment systems for tissue extraction has been well described in gynecology, the available systems vary widely in construction, size, durability, and shape, potentially leading to confusion and suboptimal bag selection during surgery.7 In this article, we review the most common laparoscopic bags available in the United States, provide an overview of bag characteristics, offer practice guidelines for bag selection, and review bag terminology to highlight important concepts for bag selection.
Controversy spurs change
In April 2014, the FDA warned against the use of power morcellation for specimen removal during minimally invasive surgery, citing a prevalence of 1 in 352 unsuspected uterine sarcomas and 1 in 498 unsuspected uterine leiomyosarcomas among women undergoing hysterectomy or myomectomy for presumed benign leiomyoma.8 Since then, the risk of occult uterine sarcomas, including leiomyosarcoma, in women undergoing surgery for benign gynecologic indications has been determined to be much lower.
Nonetheless, the clinical importance of contained specimen removal was clearly highlighted and the role of specimen retrieval bags soared to the forefront. Open power morcellation is no longer commonly practiced, and national societies such as the American Association of Gynecologic Laparoscopists (AAGL), the Society of Gynecologic Oncology (SGO), and the American College of Obstetricians and Gynecologists (ACOG) recommend that containment systems be used for safer specimen retrieval during gynecologic surgery.9-11 After the specimen is placed inside the containment system (typically a specimen bag), the surgeon may deliver the bag through a vaginal colpotomy or through a slightly extended laparoscopic incision to remove bulky specimens using cold-cutting extraction techniques.12-15
Continue to: Know the pathology’s characteristics...
Know the pathology’s characteristics
In most cases, based on imaging studies and physical examination, surgeons have a good idea of what to expect before proceeding with surgery. The 2 most common characteristics used for surgical planning are the specimen size (dimensions) and the tissue type (solid, cystic, soft tissue, or mixed). The mass size can range from less than 1 cm to larger than a 20-week sized fibroid uterus. Assessing the specimen in 3 dimensions is important. Tissue type also is a consideration, as soft and squishy masses, such as ovarian cysts, are easier to deflate and manipulate within the bag compared with solid or calcified tumors, such as a large fibroid uterus or a large dermoid with solid components.
Specimen shape also is a critical determinant for bag selection. Most specimen retrieval bags are tapered to varying degrees, and some have an irregular shape. Long tubular structures, such as fallopian tubes that are composed of soft tissue, fit easily into most bags regardless of bag shape or extent of bag taper, whereas the round shape of a bulky myoma may render certain bags ineffective even if the bag’s entrance accommodates the greatest diameter of the myoma. Often, a round mass will not fully fit into a bag because there is a poor fit between the mass’s shape and the bag’s shape and taper. (We discuss the concept of a poor “fit” below.) Knowing the pathology before starting a procedure can help optimize bag selection, streamline operative flow, and reduce waste.
Overview of laparoscopic bag characteristics and clinical applications
The TABLE lists the most common laparoscopic bags available for purchase in the United States. Details include the trocar size, manufacturer, product name, mouth diameter, volume, bag shape, construction material, and best clinical application.
The following are terms used to refer to the components of a laparoscopic retrieval bag:
- Mouth diameter: diameter at the entrance of a fully opened bag (FIGURE 1)
- Bag volume: the total volume a bag can accommodate when completely full
- Bag rim: characteristics of the rim of the bag when opened (that is, rigid vs soft rim, complete vs partial rim mechanism to hold the bag open) (FIGURE 2)
- Bag shape: the shape of the bag when it is fully opened (square shaped vs cone shaped vs curved bag shape) (FIGURE 2)
- Bag taper (severity and type): extent the bag is tapered from the rim of the bag’s entrance to the base of the bag; categorized by taper severity (minimal, gradual, or steep taper) and type (continuous taper or curved taper) (FIGURE 3)
- Ball fit: the maximum spherical specimen size that completely fits into a bag and allows it to cinch closed (FIGURE 4)
- Bag strength: durability of a bag when placed on tension during specimen extraction (weak, moderate, or extremely durable).
Continue to: Mouth diameter...
Mouth diameter
Bag manufacturers often differentiate bag sizes by indicating “volume” in milliliters. Bag volume, however, offers little clinical value to surgeons, as pelvic mass dimensions are usually measured in centimeters on imaging. Rather, an important characteristic for bag selection is the diameter of the rim of the bag when it is fully opened—the so-called bag mouth diameter. For a specimen to fit, the 2 dimensions of the specimen must be smaller than the dimensions of the bag entrance.
Notably, the number often linked to the specimen bag—as, for example, in the 10-mm Endo Catch bag (Covidien/Medtronic)— describes the width of the shaft of the bag before it is opened rather than the mouth diameter of the opened bag. The number actually correlates with the trocar size necessary for bag insertion rather than with the specimen size that can fit into the bag. Therefore, a 10-mm Endo Catch bag cannot fit a 10-cm mass, but rather requires a trocar size of 10 mm or greater for insertion of the bag. Fully opened, the mouth diameters of the 10-mm Endo Catch bag are roughly 6 cm x 7 cm, which allows for delivery of a 6-cm mass.
Because 2 bags that use the same trocar size for insertion may have vastly differing bag dimensions, the surgeon must know the bag mouth diameters when selecting a bag to remove the presenting pathology. For example, the Inzii 12 (Applied Medical) laparoscopic bag has mouth diameters of 9.7 cm × 13.0 cm, whereas the Anchor TRSROBO-12 (ConMed) has mouth diameters of 6.7 cm × 7.6 cm (TABLE). Although both bags can be inserted through a 12-mm trocar, both bags cannot fit the same size mass for removal.
Shape and taper
Laparoscopic bags come in various shapes (curved, cone, or square shaped), with varying levels of bag taper (steep, gradual, or no taper) (FIGURES 2 and 3). While taper has little impact on long and skinny specimens, taper may hinder successful bagging of bulky or spherical specimens.
Each bag has different grades of taper regardless of mouth diameter or trocar size. For round masses, the steeper the taper, the smaller the mass that can comfortably fit within the bag. This concept is connected to the idea of “ball fit,” explained below.
In addition, bag shape may affect what mass size can fit into the bag. An irregularly shaped curved bag or a bag with a steep taper may be well suited for removal of multiple specimens of varying sizes or soft masses that are malleable enough to conform to the bag’s shape (such as a ruptured ovarian cyst). Alternatively, a square-shaped bag or a bag with minimal taper would better accommodate a round mass.
Ball fit
When thinking about large circular masses, such as myomas or ovarian cysts, one must consider the ball fit. This refers to the maximum spherical size of the specimen that fits completely within a bag while allowing the bag to cinch closed. Generally, this is an estimation that factors in the bag shape, extent of the bag taper, bag mouth diameter, and specimen shape and tissue type. At times, although a mass can fit through the bag’s mouth diameter, a steep taper may prevent the mass from being fully bagged and limit closure of the bag (FIGURE 4).
Curved bags like the Anchor TRSVATS-15 (ConMed), which have a very narrow bottom, are prone to a limited ball fit, and thus the bag mouth diameter will not correlate with the largest mass size that can be fitted within the bag. Therefore, if using a steeply tapered bag for removal of large round masses, do not rely on the bag’s mouth diameter for bag selection. The surgeon must visualize the ball fit within the bag, taking into account the specimen size and shape, bag shape, and bag taper. In these scenarios, using the diameter of the midportion of the opened bag may better reflect the mass size that can fit into that bag.
Bag strength
Bag strength depends on the material used for bag construction. Most laparoscopic bags in the United States are made of 3 different materials: polyurethane, polypropylene, and ripstop nylon.
Polyurethane and polypropylene are synthetic plastic polymers; in bag form they are stretchy and, under extreme force, may tear. They are best used for bagging fluid-filled cysts or soft pliable masses that will not require extensive bag or tissue handling, such as extraction of large leiomyomas. Polyurethane and polypropylene bags are more susceptible to puncture with sharp laparoscopic instruments or scalpels, and care must be taken to avoid accidentally cutting the bag during tissue extraction.
Alternatively, bags made of ripstop nylon are favored for their bag strength. Ripstop nylon is a synthetic fabric that is woven together in a crosshatch pattern that makes it resistant to tearing and ripping. It was developed originally during World War II as a replacement for silk parachutes. Modern applications include its use in sails, kites, and high-quality camping equipment. This material has a favorable strength-to-weight ratio, and, in case of a tear, it is less prone to extension of the tear. For surgical applications, these bags are best used for bagging specimens that will require a lot of bag manipulation and tissue extraction. However, the ripstop fabric takes up more space in the incision than polyurethane or polypropylene, leaving the surgeon with less space for tissue extraction. Thus, as a tradeoff for bag strength, the surgeon may need to extend the incision a little, and a small self-retracting wound retractor may be necessary to allow visibility for safe tissue extraction when using a ripstop nylon bag compared with others.
Continue to: Trocar selection is important...
Trocar selection is important
While considering bag selection, the surgeon also must consider trocar selection to allow for laparoscopic insertion of the bag. Trocar size for bag selection refers to the minimum trocar diameter needed to insert the laparoscopic bag. Most bags are designed to fit into a laparoscopic trocar or into the skin incision that previously housed the trocar. Trocar size does not directly correlate with bag mouth diameter; for example, a 10-mm laparoscopic bag that can be inserted through a 10- or 12-mm trocar size cannot fit a 10-cm mass (see the mouth diameter section above).
A tip to maximize operating room (OR) efficiency is to start off with a larger trocar, such as a 12-mm trocar, if it is known that a laparoscopic bag with a 12-mm trocar size will be used, rather than starting with a 5-mm trocar and upsizing the port site incision. This saves time and offers intraoperative flexibility, allowing for the use of larger instruments and quicker insufflation.
Furthermore, if the specimen has a solid component and tissue extraction is anticipated, consider starting off with a large trocar, one that is larger than the bag’s trocar size since the incision likely will be extended. For example, even if a myoma will fit within a 10-mm laparoscopic bag made of ripstop nylon, using a 15-mm trocar rather than a 10-mm trocar may be considered since the skin and fascial incisions will need to be extended to allow for cold-cut tissue extraction. Starting with the larger 15-mm trocar may offer surgical advantages, such as direct needle delivery of larger needles for myometrial closure after myomectomy or direct removal of smaller myomas through the trocar to avoid bagging multiple specimens.
Putting it all together
To optimize efficiency in the OR for specimen removal, we recommend streamlining OR flow and reducing waste by first considering the specimen size, tissue type, bag shape, and trocar selection. Choose a bag by taking into account the bag mouth diameter and the amount of taper you will need to obtain an appropriate ball fit. If the tissue type is soft and pliable, consider a polyurethane or polypropylene bag and the smallest bag size possible, even if it has a narrow bag shape and taper.
However, if the tissue type is solid, the shape is round, and the mass is large (requiring extensive tissue extraction for removal), consider a bag made of ripstop nylon and factor in the bag shape as well as the bag taper. Using a bag without a steep taper may allow a better fit.
After choosing a laparoscopic bag, select the appropriate trocars necessary for completion of the surgery. Consider starting off with a larger trocar rather than spending the time to upsize a trocar if you plan to use a large bag or intend to extend the trocar incision for a contained tissue extraction. These tips will help optimize efficiency, reduce equipment wastage, and prevent intra-abdominal spillage.
Keep in mind that all procedures, including specimen removal using containment systems, have inherent risks. For example, visualization of the mass within the bag and visualization of vital structures may be hindered by bulkiness of the bag or specimen. There is also a risk of bag compromise and leakage, whether through manipulation of the bag or puncture during specimen extraction. Lastly, even though removing a specimen within a containment system minimizes spillage and reports of in-bag cold-knife tissue extraction in women with histologically proven endometrial cancer have suggested that it is safe, laparoscopic bags have not been proven to prevent the dissemination of malignant tissue fragments.16,17
Overall, the inherent risks of specimen extraction during minimally invasive surgery are far outweighed by the well-established advantages of laparoscopic surgery, which carries lower risks of surgical complications such as bleeding and infection, shorter hospital stay, and quicker recovery time compared to laparotomy. There is no doubt minimally invasive surgery offers many benefits.
In summary, for best bag selection, it is equally important to know the characteristics of the pathology as it is to know the features of the specimen retrieval systems available at your institution. Understanding both the pathology and the equipment available will allow the surgeon to make the best surgical decisions for the case. ●
- Desai VB, Wright JD, Lin H, et al. Laparoscopic hysterectomy route, resource use, and outcomes: change after power morcellation warning. Obstet Gynecol. 2019;134:227-238.
- American College of Obstetricians and Gynecologists. ACOG committee opinion No. 444: choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114:1156-1158.
- Liu H, Lu D, Wang L, et al. Robotic surgery for benign gynecological disease. Cochrane Database Syst Rev. 2012;2:CD008978.
- Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241.
- Turner LC, Shepherd JP, Wang L, et al. Hysterectomy surgery trends: a more accurate depiction of the last decade? Am J Obstet Gynecol. 2013;208:277.e1-7.
- Holme JB, Mortensen FV. A powder-free surgical glove bag for retraction of the gallbladder during laparoscopic cholecystectomy. Surg Laparosc Endosc Percutan Tech. 2005;15:209-211.
- Siedhoff MT, Cohen SL. Tissue extraction techniques for leiomyomas and uteri during minimally invasive surgery. Obstet Gynecol. 2017;130:1251-1260.
- US Food and Drug Administration. Laparoscopic uterine power morcellation in hysterectomy and myomectomy: FDA safety communication. April 17, 2014. https://wayback .archive-it.org/7993/20170722215731/https:/www.fda.gov /MedicalDevices/Safety/AlertsandNotices/ucm393576.htm. Accessed September 22, 2020.
- AAGL. AAGL practice report: morcellation during uterine tissue extraction. J Minim Invasive Gynecol. 2014;21:517-530.
- American College of Obstetricians and Gynecologists. ACOG committee opinion No. 770: uterine morcellation for presumed leiomyomas. Obstet Gynecol. 2019;133:e238-e248.
- Society of Gynecologic Oncology website. SGO position statement: morcellation. December 1, 2013. https://www .sgo.org/newsroom/position-statements-2/morcellation/. Accessed September 22, 2020.
- Advincula AP, Truong MD. ExCITE: minimally invasive tissue extraction made simple with simulation. OBG Manag. 2015;27(12):40-45.
- Solima E, Scagnelli G, Austoni V, et al. Vaginal uterine morcellation within a specimen containment system: a study of bag integrity. J Minim Invasive Gynecol. 2015;22:1244-1246.
- Ghezzi F, Casarin J, De Francesco G, et al. Transvaginal contained tissue extraction after laparoscopic myomectomy: a cohort study. BJOG. 2018;125:367-373.
- Dotson S, Landa A, Ehrisman J, et al. Safety and feasibility of contained uterine morcellation in women undergoing laparoscopic hysterectomy. Gynecol Oncol Res Pract. 2018;5:8.
- Favero G, Miglino G, Köhler C, et al. Vaginal morcellation inside protective pouch: a safe strategy for uterine extration in cases of bulky endometrial cancers: operative and oncological safety of the method. J Minim Invasive Gynecol. 2015;22:938-943.
- Montella F, Riboni F, Cosma S, et al. A safe method of vaginal longitudinal morcellation of bulky uterus with endometrial cancer in a bag at laparoscopy. Surg Endosc. 2014;28:1949-1953.
- Desai VB, Wright JD, Lin H, et al. Laparoscopic hysterectomy route, resource use, and outcomes: change after power morcellation warning. Obstet Gynecol. 2019;134:227-238.
- American College of Obstetricians and Gynecologists. ACOG committee opinion No. 444: choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2009;114:1156-1158.
- Liu H, Lu D, Wang L, et al. Robotic surgery for benign gynecological disease. Cochrane Database Syst Rev. 2012;2:CD008978.
- Wright JD, Herzog TJ, Tsui J, et al. Nationwide trends in the performance of inpatient hysterectomy in the United States. Obstet Gynecol. 2013;122(2 pt 1):233-241.
- Turner LC, Shepherd JP, Wang L, et al. Hysterectomy surgery trends: a more accurate depiction of the last decade? Am J Obstet Gynecol. 2013;208:277.e1-7.
- Holme JB, Mortensen FV. A powder-free surgical glove bag for retraction of the gallbladder during laparoscopic cholecystectomy. Surg Laparosc Endosc Percutan Tech. 2005;15:209-211.
- Siedhoff MT, Cohen SL. Tissue extraction techniques for leiomyomas and uteri during minimally invasive surgery. Obstet Gynecol. 2017;130:1251-1260.
- US Food and Drug Administration. Laparoscopic uterine power morcellation in hysterectomy and myomectomy: FDA safety communication. April 17, 2014. https://wayback .archive-it.org/7993/20170722215731/https:/www.fda.gov /MedicalDevices/Safety/AlertsandNotices/ucm393576.htm. Accessed September 22, 2020.
- AAGL. AAGL practice report: morcellation during uterine tissue extraction. J Minim Invasive Gynecol. 2014;21:517-530.
- American College of Obstetricians and Gynecologists. ACOG committee opinion No. 770: uterine morcellation for presumed leiomyomas. Obstet Gynecol. 2019;133:e238-e248.
- Society of Gynecologic Oncology website. SGO position statement: morcellation. December 1, 2013. https://www .sgo.org/newsroom/position-statements-2/morcellation/. Accessed September 22, 2020.
- Advincula AP, Truong MD. ExCITE: minimally invasive tissue extraction made simple with simulation. OBG Manag. 2015;27(12):40-45.
- Solima E, Scagnelli G, Austoni V, et al. Vaginal uterine morcellation within a specimen containment system: a study of bag integrity. J Minim Invasive Gynecol. 2015;22:1244-1246.
- Ghezzi F, Casarin J, De Francesco G, et al. Transvaginal contained tissue extraction after laparoscopic myomectomy: a cohort study. BJOG. 2018;125:367-373.
- Dotson S, Landa A, Ehrisman J, et al. Safety and feasibility of contained uterine morcellation in women undergoing laparoscopic hysterectomy. Gynecol Oncol Res Pract. 2018;5:8.
- Favero G, Miglino G, Köhler C, et al. Vaginal morcellation inside protective pouch: a safe strategy for uterine extration in cases of bulky endometrial cancers: operative and oncological safety of the method. J Minim Invasive Gynecol. 2015;22:938-943.
- Montella F, Riboni F, Cosma S, et al. A safe method of vaginal longitudinal morcellation of bulky uterus with endometrial cancer in a bag at laparoscopy. Surg Endosc. 2014;28:1949-1953.
HIT-6 may help track meaningful change in chronic migraine
, recent research suggests.
Using data from the phase 3 PROMISE-2 study, which evaluated intravenous eptinezumab in doses of 100 mg or 300 mg, or placebo every 12 weeks in 1,072 participants for the prevention of chronic migraine, Carrie R. Houts, PhD, director of psychometrics at the Vector Psychometric Group, in Chapel Hill, N.C., and colleagues determined that their finding of 6-point improvement of HIT-6 total score was consistent with other studies. However, they pointed out that little research has been done in evaluating how item-specific scores of HIT-6 impact individuals with chronic migraine. HIT-6 item scores examine whether individuals with headaches experience severe pain, limit their daily activities, have a desire to lie down, feel too tired to do daily activities, felt “fed up or irritated” because of headaches, and feel their headaches limit concentration on work or daily activities.
“The item-specific responder definitions give clinicians and researchers the ability to evaluate and track the impact of headache on specific item-level areas of patients’ lives. These responder definitions provide practical and easily interpreted results that can be used to evaluate treatment benefits over time and to improve clinician-patients communication focus on improvements in key aspects of functioning in individuals with chronic migraine,” Dr. Houts and colleagues wrote in their study, published in the October issue of Headache.
The 6-point value and the 1-2 category improvement values in item-specific scores, they suggested, could be used as a benchmark to help other clinicians and researchers detect meaningful change in individual patients with chronic migraine. Although the user guide for HIT-6 highlights a 5-point change in the total score as clinically meaningful, the authors of the guide do not provide evidence for why the 5-point value signifies clinically meaningful change, they said.
Determining thresholds of clinically meaningful change
In their study, Dr. Houts and colleagues used distribution-based methods to gauge responder values for the HIT-6 total score, while item-specific HIT-6 analyses were measured with Patients’ Global Impression of Change (PGIC), reduction in migraine frequency through monthly migraine days (MMDs), and EuroQol 5 dimensions 5 levels visual analog scale (EQ-5D-5L VAS). The researchers also used HIT-6 values from a literature review and from analyses in PROMISE-2 to calculate “a final chronic migraine-specific responder definition value” between baseline and 12 weeks. Participants in the PROMISE-2 study were mostly women (88.2%) and white (91.0%) with a mean age of 40.5 years.
The literature search revealed responder thresholds for the HIT-6 total score in a range between a decrease of 3 points and 8 points. Within PROMISE-2, the HIT-6 total score responder threshold was found to be between –2.6 and –2.2, which the researchers rounded down to a decrease of 3 points. When taking both sets of responder thresholds into account, the researchers calculated the median responder value as –5.5, which was rounded down to a decrease in 6 points in the HIT-6 total score. “[The estimate] appears most appropriate for discriminating between individuals with chronic migraine who have experienced meaningful change over time and those who have not,” Dr. Houts and colleagues said.
For item-specific HIT-6 scores, the mean score changes were –1 points for categories involving severe pain, limiting activities, lying down, and –2 points for categories involving feeling tired, being fed up or irritated, and limiting concentration.
“Taken together, the current chronic migraine-specific results are consistent with values derived from general headache/migraine samples and suggest that a decrease of 6 points or more on the HIT-6 total score would be considered meaningful to chronic migraine patients,” Dr. Houts and colleagues said. “This would translate to approximately a 4-category change on a single item, change on 2 items of approximately 2 and 3 categories, or a 1-category change on 3 or 4 of the 6 items, depending on the initial category.”
The researchers cautioned that the values outlined in the study “should not be used to determine clinically meaningful difference between treatment groups” and that “future work, similar to that reported here, will identify a chronic migraine-specific clinically meaningful difference between treatment groups value.”
A better measure of chronic migraine?
In an interview, J. D. Bartleson Jr., MD, a retired neurologist with the Mayo Clinic in Rochester, Minn., questioned why HIT-6 criteria was used in the initial PROMISE-2 study. “There is not a lot of difference between the significant and insignificant categories. Chronic migraine may be better measured with pain severity and number of headache days per month,” he said.
,“It may be appropriate to use just 1 or 2 symptoms for evaluating a given patient’s headache burden,” in terms of clinical application of the study for neurologists, Dr. Bartleson said. He emphasized that more research is needed.
This study was funded by H. Lundbeck A/S, which also provided funding of medical writing and editorial support for the manuscript. Three authors report being employees of Vector Psychometric Group at the time of the study, and the company received funding from H. Lundbeck A/S for their time conducting study-related research. Three other authors report relationships with pharmaceutical companies, medical societies, government agencies, and industry related to the study in the form of consultancies, advisory board memberships, honoraria, research support, stock or stock options, and employment. Dr. Bartleson reports no relevant conflicts of interest.
, recent research suggests.
Using data from the phase 3 PROMISE-2 study, which evaluated intravenous eptinezumab in doses of 100 mg or 300 mg, or placebo every 12 weeks in 1,072 participants for the prevention of chronic migraine, Carrie R. Houts, PhD, director of psychometrics at the Vector Psychometric Group, in Chapel Hill, N.C., and colleagues determined that their finding of 6-point improvement of HIT-6 total score was consistent with other studies. However, they pointed out that little research has been done in evaluating how item-specific scores of HIT-6 impact individuals with chronic migraine. HIT-6 item scores examine whether individuals with headaches experience severe pain, limit their daily activities, have a desire to lie down, feel too tired to do daily activities, felt “fed up or irritated” because of headaches, and feel their headaches limit concentration on work or daily activities.
“The item-specific responder definitions give clinicians and researchers the ability to evaluate and track the impact of headache on specific item-level areas of patients’ lives. These responder definitions provide practical and easily interpreted results that can be used to evaluate treatment benefits over time and to improve clinician-patients communication focus on improvements in key aspects of functioning in individuals with chronic migraine,” Dr. Houts and colleagues wrote in their study, published in the October issue of Headache.
The 6-point value and the 1-2 category improvement values in item-specific scores, they suggested, could be used as a benchmark to help other clinicians and researchers detect meaningful change in individual patients with chronic migraine. Although the user guide for HIT-6 highlights a 5-point change in the total score as clinically meaningful, the authors of the guide do not provide evidence for why the 5-point value signifies clinically meaningful change, they said.
Determining thresholds of clinically meaningful change
In their study, Dr. Houts and colleagues used distribution-based methods to gauge responder values for the HIT-6 total score, while item-specific HIT-6 analyses were measured with Patients’ Global Impression of Change (PGIC), reduction in migraine frequency through monthly migraine days (MMDs), and EuroQol 5 dimensions 5 levels visual analog scale (EQ-5D-5L VAS). The researchers also used HIT-6 values from a literature review and from analyses in PROMISE-2 to calculate “a final chronic migraine-specific responder definition value” between baseline and 12 weeks. Participants in the PROMISE-2 study were mostly women (88.2%) and white (91.0%) with a mean age of 40.5 years.
The literature search revealed responder thresholds for the HIT-6 total score in a range between a decrease of 3 points and 8 points. Within PROMISE-2, the HIT-6 total score responder threshold was found to be between –2.6 and –2.2, which the researchers rounded down to a decrease of 3 points. When taking both sets of responder thresholds into account, the researchers calculated the median responder value as –5.5, which was rounded down to a decrease in 6 points in the HIT-6 total score. “[The estimate] appears most appropriate for discriminating between individuals with chronic migraine who have experienced meaningful change over time and those who have not,” Dr. Houts and colleagues said.
For item-specific HIT-6 scores, the mean score changes were –1 points for categories involving severe pain, limiting activities, lying down, and –2 points for categories involving feeling tired, being fed up or irritated, and limiting concentration.
“Taken together, the current chronic migraine-specific results are consistent with values derived from general headache/migraine samples and suggest that a decrease of 6 points or more on the HIT-6 total score would be considered meaningful to chronic migraine patients,” Dr. Houts and colleagues said. “This would translate to approximately a 4-category change on a single item, change on 2 items of approximately 2 and 3 categories, or a 1-category change on 3 or 4 of the 6 items, depending on the initial category.”
The researchers cautioned that the values outlined in the study “should not be used to determine clinically meaningful difference between treatment groups” and that “future work, similar to that reported here, will identify a chronic migraine-specific clinically meaningful difference between treatment groups value.”
A better measure of chronic migraine?
In an interview, J. D. Bartleson Jr., MD, a retired neurologist with the Mayo Clinic in Rochester, Minn., questioned why HIT-6 criteria was used in the initial PROMISE-2 study. “There is not a lot of difference between the significant and insignificant categories. Chronic migraine may be better measured with pain severity and number of headache days per month,” he said.
,“It may be appropriate to use just 1 or 2 symptoms for evaluating a given patient’s headache burden,” in terms of clinical application of the study for neurologists, Dr. Bartleson said. He emphasized that more research is needed.
This study was funded by H. Lundbeck A/S, which also provided funding of medical writing and editorial support for the manuscript. Three authors report being employees of Vector Psychometric Group at the time of the study, and the company received funding from H. Lundbeck A/S for their time conducting study-related research. Three other authors report relationships with pharmaceutical companies, medical societies, government agencies, and industry related to the study in the form of consultancies, advisory board memberships, honoraria, research support, stock or stock options, and employment. Dr. Bartleson reports no relevant conflicts of interest.
, recent research suggests.
Using data from the phase 3 PROMISE-2 study, which evaluated intravenous eptinezumab in doses of 100 mg or 300 mg, or placebo every 12 weeks in 1,072 participants for the prevention of chronic migraine, Carrie R. Houts, PhD, director of psychometrics at the Vector Psychometric Group, in Chapel Hill, N.C., and colleagues determined that their finding of 6-point improvement of HIT-6 total score was consistent with other studies. However, they pointed out that little research has been done in evaluating how item-specific scores of HIT-6 impact individuals with chronic migraine. HIT-6 item scores examine whether individuals with headaches experience severe pain, limit their daily activities, have a desire to lie down, feel too tired to do daily activities, felt “fed up or irritated” because of headaches, and feel their headaches limit concentration on work or daily activities.
“The item-specific responder definitions give clinicians and researchers the ability to evaluate and track the impact of headache on specific item-level areas of patients’ lives. These responder definitions provide practical and easily interpreted results that can be used to evaluate treatment benefits over time and to improve clinician-patients communication focus on improvements in key aspects of functioning in individuals with chronic migraine,” Dr. Houts and colleagues wrote in their study, published in the October issue of Headache.
The 6-point value and the 1-2 category improvement values in item-specific scores, they suggested, could be used as a benchmark to help other clinicians and researchers detect meaningful change in individual patients with chronic migraine. Although the user guide for HIT-6 highlights a 5-point change in the total score as clinically meaningful, the authors of the guide do not provide evidence for why the 5-point value signifies clinically meaningful change, they said.
Determining thresholds of clinically meaningful change
In their study, Dr. Houts and colleagues used distribution-based methods to gauge responder values for the HIT-6 total score, while item-specific HIT-6 analyses were measured with Patients’ Global Impression of Change (PGIC), reduction in migraine frequency through monthly migraine days (MMDs), and EuroQol 5 dimensions 5 levels visual analog scale (EQ-5D-5L VAS). The researchers also used HIT-6 values from a literature review and from analyses in PROMISE-2 to calculate “a final chronic migraine-specific responder definition value” between baseline and 12 weeks. Participants in the PROMISE-2 study were mostly women (88.2%) and white (91.0%) with a mean age of 40.5 years.
The literature search revealed responder thresholds for the HIT-6 total score in a range between a decrease of 3 points and 8 points. Within PROMISE-2, the HIT-6 total score responder threshold was found to be between –2.6 and –2.2, which the researchers rounded down to a decrease of 3 points. When taking both sets of responder thresholds into account, the researchers calculated the median responder value as –5.5, which was rounded down to a decrease in 6 points in the HIT-6 total score. “[The estimate] appears most appropriate for discriminating between individuals with chronic migraine who have experienced meaningful change over time and those who have not,” Dr. Houts and colleagues said.
For item-specific HIT-6 scores, the mean score changes were –1 points for categories involving severe pain, limiting activities, lying down, and –2 points for categories involving feeling tired, being fed up or irritated, and limiting concentration.
“Taken together, the current chronic migraine-specific results are consistent with values derived from general headache/migraine samples and suggest that a decrease of 6 points or more on the HIT-6 total score would be considered meaningful to chronic migraine patients,” Dr. Houts and colleagues said. “This would translate to approximately a 4-category change on a single item, change on 2 items of approximately 2 and 3 categories, or a 1-category change on 3 or 4 of the 6 items, depending on the initial category.”
The researchers cautioned that the values outlined in the study “should not be used to determine clinically meaningful difference between treatment groups” and that “future work, similar to that reported here, will identify a chronic migraine-specific clinically meaningful difference between treatment groups value.”
A better measure of chronic migraine?
In an interview, J. D. Bartleson Jr., MD, a retired neurologist with the Mayo Clinic in Rochester, Minn., questioned why HIT-6 criteria was used in the initial PROMISE-2 study. “There is not a lot of difference between the significant and insignificant categories. Chronic migraine may be better measured with pain severity and number of headache days per month,” he said.
,“It may be appropriate to use just 1 or 2 symptoms for evaluating a given patient’s headache burden,” in terms of clinical application of the study for neurologists, Dr. Bartleson said. He emphasized that more research is needed.
This study was funded by H. Lundbeck A/S, which also provided funding of medical writing and editorial support for the manuscript. Three authors report being employees of Vector Psychometric Group at the time of the study, and the company received funding from H. Lundbeck A/S for their time conducting study-related research. Three other authors report relationships with pharmaceutical companies, medical societies, government agencies, and industry related to the study in the form of consultancies, advisory board memberships, honoraria, research support, stock or stock options, and employment. Dr. Bartleson reports no relevant conflicts of interest.
FROM HEADACHE
COVID-19: A second wave of mental illness 'imminent'
The mental health consequences of COVID-19 deaths are likely to overwhelm an already tattered U.S. mental health system, leading to a lack of access, particularly for the most vulnerable, experts warn.

“A second wave of devastation is imminent, attributable to mental health consequences of COVID-19,” write Naomi Simon, MD, and coauthors with the department of psychiatry, New York University.
In a Viewpoint article published in JAMA on Oct. 12, physicians offer some sobering statistics.
Since February 2020, COVID-19 has taken the lives of more than 214,000 Americans. The number of deaths currently attributed to the virus is nearly four times the number of Americans killed during the Vietnam War. The magnitude of death over a short period is a tragedy on a “historic scale,” wrote Dr. Simon and colleagues.
The surge in mental health problems related to COVID-19 deaths will bring further challenges to individuals, families, and communities, including a spike in deaths from suicide and drug overdoses, they warned.
It’s important to consider, they noted, that each COVID-19 death leaves an estimated nine family members bereaved, which is projected to lead to an estimated 2 million bereaved individuals in the United States.
The necessary social distancing and quarantine measures implemented to fight the virus have amplified emotional turmoil and have disrupted the ability of personal support networks and communities to come together and grieve.
“Of central concern is the transformation of normal grief and distress into prolonged grief and major depressive disorder and symptoms of posttraumatic stress disorder,” Simon and colleagues said.
“Once established, these conditions can become chronic with additional comorbidities such as substance use disorders. Prolonged grief affects approximately 10% of bereaved individuals, but this is likely an underestimate for grief related to deaths from COVID-19,” they wrote.
As with the first COVID-19 wave, the mental health wave will disproportionately affect Black persons, Hispanic persons, older adults, persons in lower socioeconomic groups of all races and ethnicities, and healthcare workers, they note.
The psychological risks for health care and other essential workers are of particular concern, they say. “Supporting the mental health of these and other essential workforce is critical to readiness for managing recurrent waves of the pandemic,” they stated.
How will the United States manage this impending wave of mental health problems?
“The solution will require increased funding for mental health; widespread screening to identify individuals at highest risk including suicide risk; availability of primary care clinicians and mental health professionals trained to treat those with prolonged grief, depression, traumatic stress, and substance abuse; and a diligent focus on families and communities to creatively restore the approaches by which they have managed tragedy and loss over generations,” the authors wrote.
“History has shown that societies recover from such devastation when leaders and members are joined by a shared purpose, acting in a unified way to facilitate recovery. In such societies, there is a shared understanding that its members must care for one another because the loss of one is a loss for all. Above all, this shared understanding must be restored,” they concluded.
Dr. Simon has received personal fees from Vanda Pharmaceuticals Inc, MGH Psychiatry Academy, Axovant Sciences, Springworks, Praxis Therapeutics, Aptinyx, Genomind, and Wiley (deputy editor, Depression and Anxiety). Saxe has received royalties from Guilford Press for the book Trauma Systems Therapy for Children and Teens (2016). Marmar serves on the scientific advisory board and owns equity in Receptor Life Sciences and serves on the PTSD advisory board for Otsuka Pharmaceutical.
A version of this article originally appeared on Medscape.com.
The mental health consequences of COVID-19 deaths are likely to overwhelm an already tattered U.S. mental health system, leading to a lack of access, particularly for the most vulnerable, experts warn.
“A second wave of devastation is imminent, attributable to mental health consequences of COVID-19,” write Naomi Simon, MD, and coauthors with the department of psychiatry, New York University.
In a Viewpoint article published in JAMA on Oct. 12, physicians offer some sobering statistics.
Since February 2020, COVID-19 has taken the lives of more than 214,000 Americans. The number of deaths currently attributed to the virus is nearly four times the number of Americans killed during the Vietnam War. The magnitude of death over a short period is a tragedy on a “historic scale,” wrote Dr. Simon and colleagues.
The surge in mental health problems related to COVID-19 deaths will bring further challenges to individuals, families, and communities, including a spike in deaths from suicide and drug overdoses, they warned.
It’s important to consider, they noted, that each COVID-19 death leaves an estimated nine family members bereaved, which is projected to lead to an estimated 2 million bereaved individuals in the United States.
The necessary social distancing and quarantine measures implemented to fight the virus have amplified emotional turmoil and have disrupted the ability of personal support networks and communities to come together and grieve.
“Of central concern is the transformation of normal grief and distress into prolonged grief and major depressive disorder and symptoms of posttraumatic stress disorder,” Simon and colleagues said.
“Once established, these conditions can become chronic with additional comorbidities such as substance use disorders. Prolonged grief affects approximately 10% of bereaved individuals, but this is likely an underestimate for grief related to deaths from COVID-19,” they wrote.
As with the first COVID-19 wave, the mental health wave will disproportionately affect Black persons, Hispanic persons, older adults, persons in lower socioeconomic groups of all races and ethnicities, and healthcare workers, they note.
The psychological risks for health care and other essential workers are of particular concern, they say. “Supporting the mental health of these and other essential workforce is critical to readiness for managing recurrent waves of the pandemic,” they stated.
How will the United States manage this impending wave of mental health problems?
“The solution will require increased funding for mental health; widespread screening to identify individuals at highest risk including suicide risk; availability of primary care clinicians and mental health professionals trained to treat those with prolonged grief, depression, traumatic stress, and substance abuse; and a diligent focus on families and communities to creatively restore the approaches by which they have managed tragedy and loss over generations,” the authors wrote.
“History has shown that societies recover from such devastation when leaders and members are joined by a shared purpose, acting in a unified way to facilitate recovery. In such societies, there is a shared understanding that its members must care for one another because the loss of one is a loss for all. Above all, this shared understanding must be restored,” they concluded.
Dr. Simon has received personal fees from Vanda Pharmaceuticals Inc, MGH Psychiatry Academy, Axovant Sciences, Springworks, Praxis Therapeutics, Aptinyx, Genomind, and Wiley (deputy editor, Depression and Anxiety). Saxe has received royalties from Guilford Press for the book Trauma Systems Therapy for Children and Teens (2016). Marmar serves on the scientific advisory board and owns equity in Receptor Life Sciences and serves on the PTSD advisory board for Otsuka Pharmaceutical.
A version of this article originally appeared on Medscape.com.
The mental health consequences of COVID-19 deaths are likely to overwhelm an already tattered U.S. mental health system, leading to a lack of access, particularly for the most vulnerable, experts warn.
“A second wave of devastation is imminent, attributable to mental health consequences of COVID-19,” write Naomi Simon, MD, and coauthors with the department of psychiatry, New York University.
In a Viewpoint article published in JAMA on Oct. 12, physicians offer some sobering statistics.
Since February 2020, COVID-19 has taken the lives of more than 214,000 Americans. The number of deaths currently attributed to the virus is nearly four times the number of Americans killed during the Vietnam War. The magnitude of death over a short period is a tragedy on a “historic scale,” wrote Dr. Simon and colleagues.
The surge in mental health problems related to COVID-19 deaths will bring further challenges to individuals, families, and communities, including a spike in deaths from suicide and drug overdoses, they warned.
It’s important to consider, they noted, that each COVID-19 death leaves an estimated nine family members bereaved, which is projected to lead to an estimated 2 million bereaved individuals in the United States.
The necessary social distancing and quarantine measures implemented to fight the virus have amplified emotional turmoil and have disrupted the ability of personal support networks and communities to come together and grieve.
“Of central concern is the transformation of normal grief and distress into prolonged grief and major depressive disorder and symptoms of posttraumatic stress disorder,” Simon and colleagues said.
“Once established, these conditions can become chronic with additional comorbidities such as substance use disorders. Prolonged grief affects approximately 10% of bereaved individuals, but this is likely an underestimate for grief related to deaths from COVID-19,” they wrote.
As with the first COVID-19 wave, the mental health wave will disproportionately affect Black persons, Hispanic persons, older adults, persons in lower socioeconomic groups of all races and ethnicities, and healthcare workers, they note.
The psychological risks for health care and other essential workers are of particular concern, they say. “Supporting the mental health of these and other essential workforce is critical to readiness for managing recurrent waves of the pandemic,” they stated.
How will the United States manage this impending wave of mental health problems?
“The solution will require increased funding for mental health; widespread screening to identify individuals at highest risk including suicide risk; availability of primary care clinicians and mental health professionals trained to treat those with prolonged grief, depression, traumatic stress, and substance abuse; and a diligent focus on families and communities to creatively restore the approaches by which they have managed tragedy and loss over generations,” the authors wrote.
“History has shown that societies recover from such devastation when leaders and members are joined by a shared purpose, acting in a unified way to facilitate recovery. In such societies, there is a shared understanding that its members must care for one another because the loss of one is a loss for all. Above all, this shared understanding must be restored,” they concluded.
Dr. Simon has received personal fees from Vanda Pharmaceuticals Inc, MGH Psychiatry Academy, Axovant Sciences, Springworks, Praxis Therapeutics, Aptinyx, Genomind, and Wiley (deputy editor, Depression and Anxiety). Saxe has received royalties from Guilford Press for the book Trauma Systems Therapy for Children and Teens (2016). Marmar serves on the scientific advisory board and owns equity in Receptor Life Sciences and serves on the PTSD advisory board for Otsuka Pharmaceutical.
A version of this article originally appeared on Medscape.com.
Heterosexual men likely to have unmet HIV treatment needs
according to findings presented at the HIV Glasgow 2020 Virtual Meeting. MSM had better overall health outcomes than the other two groups, the study found, suggesting that MSW and women have unmet needs that require providers’ attention.
Chinyere Okoli of ViiV Healthcare Global Medical Affairs in Brentford, England, and her associates administered a Web-based survey about HIV-related perceptions and behaviors to 2,389 adults with HIV in 25 countries. The respondents included 1,018 MSM, 479 MSW, and 696 women.
In high-income countries, MSM respondents had been diagnosed a median 9 years earlier, MSW respondents a median 4 years earlier, and women respondents a median 5 years earlier. In middle-income countries, diagnosis was a median 3 years ago for MSM respondents and a median 6 years for MSW and women respondents.
Rates of suboptimal adherence to antiretroviral therapy (ART) were lowest (15.5%) among MSM, compared with MSW (38.8%) and women (28%). Similarly, viral nonsuppression had occurred in only 10.9% of MSM, whereas it had occurred in 43.2% of MSW and 37.1% of women. A little more than one-third (36.5%) of MSM had suboptimal overall health, whereas 47.2% of MSW and 46.2% of women had suboptimal overall health (P < .05).
A similar percentage of MSM (38%) and women (38.2%) reported polypharmacy; both percentages were significantly lower than for MSW (45.1%; P = .020). Yet MSW were less likely than the other two groups to have comorbidities unrelated to HIV: 46.1%, compared with 64.6% of MSM and 56.7% of women (P < .001).
Although a higher proportion (63%) of MSW than MSM (44%) or women (55%) were receiving a multitablet ART regimen, MSW were least likely to consider the impact of side effects when they began ART and were most likely to experience side effects. Only 45% of MSW prioritized minimizing side effects when they began receiving ART, and more than half (52%) were experiencing side effects with their current regimen.
By contrast, a majority of MSM (60%) prioritized minimizing side effects at ART initiation, and only 35% currently had side effects. Women fell in the middle with 48% considering side effects when starting ART and 49% reporting current side effects.
The proportion of respondents who said ART side effects were affecting their lives was not significantly different: 69% of MSM, 73% of MSW, and 74% of women. However, 56% of MSW reported skipping at least one dose in the past month because of side effects, which was more than twice the percentage of MSM (24%; P < .001). One-third of women (33%) reported skipping at least one dose.
MSW were also least comfortable talking to their health care provider about ART side effects: 55% reported discomfort, compared with 34% of MSM and 43% of women. A high majority of MSW (87.9%) said they experienced barriers to talking to their providers about relevant health concerns. The proportion who reported barriers was lower for MSM (59%) and women (72.7%; P < .001).
The substantial differences between MSM and MSW, which were even greater than those between MSW and women, suggest this population has the greatest amount of unmet needs, the researchers concluded. “Acknowledging these differences when planning/administering care can help narrow disparities,” they wrote.
A version of this article originally appeared on Medscape.com.
according to findings presented at the HIV Glasgow 2020 Virtual Meeting. MSM had better overall health outcomes than the other two groups, the study found, suggesting that MSW and women have unmet needs that require providers’ attention.
Chinyere Okoli of ViiV Healthcare Global Medical Affairs in Brentford, England, and her associates administered a Web-based survey about HIV-related perceptions and behaviors to 2,389 adults with HIV in 25 countries. The respondents included 1,018 MSM, 479 MSW, and 696 women.
In high-income countries, MSM respondents had been diagnosed a median 9 years earlier, MSW respondents a median 4 years earlier, and women respondents a median 5 years earlier. In middle-income countries, diagnosis was a median 3 years ago for MSM respondents and a median 6 years for MSW and women respondents.
Rates of suboptimal adherence to antiretroviral therapy (ART) were lowest (15.5%) among MSM, compared with MSW (38.8%) and women (28%). Similarly, viral nonsuppression had occurred in only 10.9% of MSM, whereas it had occurred in 43.2% of MSW and 37.1% of women. A little more than one-third (36.5%) of MSM had suboptimal overall health, whereas 47.2% of MSW and 46.2% of women had suboptimal overall health (P < .05).
A similar percentage of MSM (38%) and women (38.2%) reported polypharmacy; both percentages were significantly lower than for MSW (45.1%; P = .020). Yet MSW were less likely than the other two groups to have comorbidities unrelated to HIV: 46.1%, compared with 64.6% of MSM and 56.7% of women (P < .001).
Although a higher proportion (63%) of MSW than MSM (44%) or women (55%) were receiving a multitablet ART regimen, MSW were least likely to consider the impact of side effects when they began ART and were most likely to experience side effects. Only 45% of MSW prioritized minimizing side effects when they began receiving ART, and more than half (52%) were experiencing side effects with their current regimen.
By contrast, a majority of MSM (60%) prioritized minimizing side effects at ART initiation, and only 35% currently had side effects. Women fell in the middle with 48% considering side effects when starting ART and 49% reporting current side effects.
The proportion of respondents who said ART side effects were affecting their lives was not significantly different: 69% of MSM, 73% of MSW, and 74% of women. However, 56% of MSW reported skipping at least one dose in the past month because of side effects, which was more than twice the percentage of MSM (24%; P < .001). One-third of women (33%) reported skipping at least one dose.
MSW were also least comfortable talking to their health care provider about ART side effects: 55% reported discomfort, compared with 34% of MSM and 43% of women. A high majority of MSW (87.9%) said they experienced barriers to talking to their providers about relevant health concerns. The proportion who reported barriers was lower for MSM (59%) and women (72.7%; P < .001).
The substantial differences between MSM and MSW, which were even greater than those between MSW and women, suggest this population has the greatest amount of unmet needs, the researchers concluded. “Acknowledging these differences when planning/administering care can help narrow disparities,” they wrote.
A version of this article originally appeared on Medscape.com.
according to findings presented at the HIV Glasgow 2020 Virtual Meeting. MSM had better overall health outcomes than the other two groups, the study found, suggesting that MSW and women have unmet needs that require providers’ attention.
Chinyere Okoli of ViiV Healthcare Global Medical Affairs in Brentford, England, and her associates administered a Web-based survey about HIV-related perceptions and behaviors to 2,389 adults with HIV in 25 countries. The respondents included 1,018 MSM, 479 MSW, and 696 women.
In high-income countries, MSM respondents had been diagnosed a median 9 years earlier, MSW respondents a median 4 years earlier, and women respondents a median 5 years earlier. In middle-income countries, diagnosis was a median 3 years ago for MSM respondents and a median 6 years for MSW and women respondents.
Rates of suboptimal adherence to antiretroviral therapy (ART) were lowest (15.5%) among MSM, compared with MSW (38.8%) and women (28%). Similarly, viral nonsuppression had occurred in only 10.9% of MSM, whereas it had occurred in 43.2% of MSW and 37.1% of women. A little more than one-third (36.5%) of MSM had suboptimal overall health, whereas 47.2% of MSW and 46.2% of women had suboptimal overall health (P < .05).
A similar percentage of MSM (38%) and women (38.2%) reported polypharmacy; both percentages were significantly lower than for MSW (45.1%; P = .020). Yet MSW were less likely than the other two groups to have comorbidities unrelated to HIV: 46.1%, compared with 64.6% of MSM and 56.7% of women (P < .001).
Although a higher proportion (63%) of MSW than MSM (44%) or women (55%) were receiving a multitablet ART regimen, MSW were least likely to consider the impact of side effects when they began ART and were most likely to experience side effects. Only 45% of MSW prioritized minimizing side effects when they began receiving ART, and more than half (52%) were experiencing side effects with their current regimen.
By contrast, a majority of MSM (60%) prioritized minimizing side effects at ART initiation, and only 35% currently had side effects. Women fell in the middle with 48% considering side effects when starting ART and 49% reporting current side effects.
The proportion of respondents who said ART side effects were affecting their lives was not significantly different: 69% of MSM, 73% of MSW, and 74% of women. However, 56% of MSW reported skipping at least one dose in the past month because of side effects, which was more than twice the percentage of MSM (24%; P < .001). One-third of women (33%) reported skipping at least one dose.
MSW were also least comfortable talking to their health care provider about ART side effects: 55% reported discomfort, compared with 34% of MSM and 43% of women. A high majority of MSW (87.9%) said they experienced barriers to talking to their providers about relevant health concerns. The proportion who reported barriers was lower for MSM (59%) and women (72.7%; P < .001).
The substantial differences between MSM and MSW, which were even greater than those between MSW and women, suggest this population has the greatest amount of unmet needs, the researchers concluded. “Acknowledging these differences when planning/administering care can help narrow disparities,” they wrote.
A version of this article originally appeared on Medscape.com.
Mastering mask communicating
. For those specialties not accustomed to wearing a mask all day, it’s frustrating: How many times have you had to repeat yourself today? Or ask your patient to say something again? (Ain’t no one got time to repeat a third time how to do that prednisone taper). Worse, we’re losing important nonverbal cues that help us connect with our patients. How can we be understood when our faces are covered and 6 feet away?

Masks muffle both verbal and nonverbal communication. For soft-spoken or high-pitched speakers, the verbal effect is significant. In particular, masks make hearing consonants more difficult. They can make the “sh,” “th,” “f,” and “s” sounds difficult to distinguish. Typically, we’d use context and lip reading to boost the signal, but this fix is blocked (and the clear mouth-window masks are kinda creepy).
Masks also prevent us from seeing facial microexpressions, critical information when you are trying to connect with someone or to build trust. A randomized controlled trial published in 2013 indeed showed that doctors wearing a mask were perceived as less empathetic and had diminished relational continuity with patients as compared to doctors not wearing a mask. There are a few things we can do to help.
Speak more loudly is obvious advice. Loud talking has limitations though, as it can feel rude, and it blunts inflections, which add richness and emotion. (Shouting “THIS WILL ONLY HURT A LITTLE” seems a mixed message). More important than the volume is your choice of words. Try to use simple terms and short sentences. Pause between points. Hit your consonants harder.
It’s also important that you have their full attention and are giving yours. As much as possible, try to align squared up with patients. Facing your computer exacerbates the problem. Look them in their eyes and be sure they are connected with you before any complex or difficult conversations. Hearing-impaired patients are now sometimes leaving out their aids because it’s too uncomfortable to wear them with their mask. You might ask them to put them back in. Check in with patients and repeat back what you heard them say. This can help with clarity and with connecting. Use your face more: if you’ve ever acted on stage, this would be your on-stage face. Exaggerate your expressions so it’s a little easier for them to read you.
Lastly, there are apps such as Ava or Google Live Translator, which can transcribe your speech real time. You could then share your screen with the patient so they can read exactly what you’ve said.
Some of us are natural communicators. Even if you are not, you can mitigate some of our current challenges. I’ll admit, it’s been a bit easier for me than for others. Between my prominent eyebrows and Italian-American upbringing, I can express my way through pretty much any face covering. If you’d like to learn how to use your hands better, then just watch this little girl: https://youtu.be/Z5wAWyqDrnc.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@mdedge.com.
. For those specialties not accustomed to wearing a mask all day, it’s frustrating: How many times have you had to repeat yourself today? Or ask your patient to say something again? (Ain’t no one got time to repeat a third time how to do that prednisone taper). Worse, we’re losing important nonverbal cues that help us connect with our patients. How can we be understood when our faces are covered and 6 feet away?
Masks muffle both verbal and nonverbal communication. For soft-spoken or high-pitched speakers, the verbal effect is significant. In particular, masks make hearing consonants more difficult. They can make the “sh,” “th,” “f,” and “s” sounds difficult to distinguish. Typically, we’d use context and lip reading to boost the signal, but this fix is blocked (and the clear mouth-window masks are kinda creepy).
Masks also prevent us from seeing facial microexpressions, critical information when you are trying to connect with someone or to build trust. A randomized controlled trial published in 2013 indeed showed that doctors wearing a mask were perceived as less empathetic and had diminished relational continuity with patients as compared to doctors not wearing a mask. There are a few things we can do to help.
Speak more loudly is obvious advice. Loud talking has limitations though, as it can feel rude, and it blunts inflections, which add richness and emotion. (Shouting “THIS WILL ONLY HURT A LITTLE” seems a mixed message). More important than the volume is your choice of words. Try to use simple terms and short sentences. Pause between points. Hit your consonants harder.
It’s also important that you have their full attention and are giving yours. As much as possible, try to align squared up with patients. Facing your computer exacerbates the problem. Look them in their eyes and be sure they are connected with you before any complex or difficult conversations. Hearing-impaired patients are now sometimes leaving out their aids because it’s too uncomfortable to wear them with their mask. You might ask them to put them back in. Check in with patients and repeat back what you heard them say. This can help with clarity and with connecting. Use your face more: if you’ve ever acted on stage, this would be your on-stage face. Exaggerate your expressions so it’s a little easier for them to read you.
Lastly, there are apps such as Ava or Google Live Translator, which can transcribe your speech real time. You could then share your screen with the patient so they can read exactly what you’ve said.
Some of us are natural communicators. Even if you are not, you can mitigate some of our current challenges. I’ll admit, it’s been a bit easier for me than for others. Between my prominent eyebrows and Italian-American upbringing, I can express my way through pretty much any face covering. If you’d like to learn how to use your hands better, then just watch this little girl: https://youtu.be/Z5wAWyqDrnc.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@mdedge.com.
. For those specialties not accustomed to wearing a mask all day, it’s frustrating: How many times have you had to repeat yourself today? Or ask your patient to say something again? (Ain’t no one got time to repeat a third time how to do that prednisone taper). Worse, we’re losing important nonverbal cues that help us connect with our patients. How can we be understood when our faces are covered and 6 feet away?
Masks muffle both verbal and nonverbal communication. For soft-spoken or high-pitched speakers, the verbal effect is significant. In particular, masks make hearing consonants more difficult. They can make the “sh,” “th,” “f,” and “s” sounds difficult to distinguish. Typically, we’d use context and lip reading to boost the signal, but this fix is blocked (and the clear mouth-window masks are kinda creepy).
Masks also prevent us from seeing facial microexpressions, critical information when you are trying to connect with someone or to build trust. A randomized controlled trial published in 2013 indeed showed that doctors wearing a mask were perceived as less empathetic and had diminished relational continuity with patients as compared to doctors not wearing a mask. There are a few things we can do to help.
Speak more loudly is obvious advice. Loud talking has limitations though, as it can feel rude, and it blunts inflections, which add richness and emotion. (Shouting “THIS WILL ONLY HURT A LITTLE” seems a mixed message). More important than the volume is your choice of words. Try to use simple terms and short sentences. Pause between points. Hit your consonants harder.
It’s also important that you have their full attention and are giving yours. As much as possible, try to align squared up with patients. Facing your computer exacerbates the problem. Look them in their eyes and be sure they are connected with you before any complex or difficult conversations. Hearing-impaired patients are now sometimes leaving out their aids because it’s too uncomfortable to wear them with their mask. You might ask them to put them back in. Check in with patients and repeat back what you heard them say. This can help with clarity and with connecting. Use your face more: if you’ve ever acted on stage, this would be your on-stage face. Exaggerate your expressions so it’s a little easier for them to read you.
Lastly, there are apps such as Ava or Google Live Translator, which can transcribe your speech real time. You could then share your screen with the patient so they can read exactly what you’ve said.
Some of us are natural communicators. Even if you are not, you can mitigate some of our current challenges. I’ll admit, it’s been a bit easier for me than for others. Between my prominent eyebrows and Italian-American upbringing, I can express my way through pretty much any face covering. If you’d like to learn how to use your hands better, then just watch this little girl: https://youtu.be/Z5wAWyqDrnc.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@mdedge.com.
Entresto halves renal events in preserved EF heart failure patients
Patients with heart failure with preserved ejection fraction (HFpEF) who received sacubitril/valsartan in the PARAGON-HF trial had significant protection against progression of renal dysfunction in a prespecified secondary analysis.
The 2,419 patients with HFpEF who received sacubitril/valsartan (Entresto) had half the rate of the primary adverse renal outcome, compared with the 2,403 patients randomized to valsartan alone in the comparator group, a significant difference, according to the results published online Sept. 29 in Circulation by Finnian R. McCausland, MBBCh, and colleagues.
In absolute terms, sacubitril/valsartan treatment, an angiotensin-receptor/neprilysin inhibitor (ARNI), cut the incidence of the combined renal endpoint – renal death, end-stage renal disease, or at least a 50% drop in estimated glomerular filtration rate (eGFR) – from 2.7% in the control group to 1.4% in the sacubitril/valsartan group during a median follow-up of 35 months.
The absolute difference of 1.3% equated to a number needed to treat of 51 to prevent one of these events.
Also notable was that renal protection from sacubitril/valsartan was equally robust across the range of baseline kidney function.
‘An important therapeutic option’
The efficacy “across the spectrum of baseline renal function” indicates treatment with sacubitril/valsartan is “an important therapeutic option to slow renal-function decline in patients with heart failure,” wrote Dr. McCausland, a nephrologist at Brigham and Women’s Hospital in Boston, and colleagues.
The authors’ conclusion is striking because currently no drug class has produced clear evidence for efficacy in HFpEF.
On the other hand, the PARAGON-HF trial that provided the data for this new analysis was statistically neutral for its primary endpoint – a reduction in the combined rate of cardiovascular death and hospitalizations for heart failure – with a P value of .06 and 95% confidence interval of 0.75-1.01.
“Because this difference [in the primary endpoint incidence between the two study group] did not meet the predetermined level of statistical significance, subsequent analyses were considered to be exploratory,” noted the authors of the primary analysis of PARAGON-HF, as reported by Medscape Medical News.
Despite this limitation in interpreting secondary outcomes from the trial, the new report of a significant renal benefit “opens the potential to provide evidence-based treatment for patients with HFpEF,” commented Sheldon W. Tobe, MD, and Stephanie Poon, MD, in an editorial accompanying the latest analysis.
“At the very least, these results are certainly intriguing and suggest that there may be important patient subgroups with HFpEF who might benefit from using sacubitril/valsartan,” they emphasized.
First large trial to show renal improvement in HFpEF
The editorialists’ enthusiasm for the implications of the new findings relate in part to the fact that “PARAGON-HF is the first large trial to demonstrate improvement in renal parameters in HFpEF,” they noted.
“The finding that the composite renal outcome did not differ according to baseline eGFR is significant and suggests that the beneficial effect on renal function was indirect, possibly linked to improved cardiac function,” say Dr. Tobe, a nephrologist, and Dr. Poon, a cardiologist, both at Sunnybrook Health Sciences Centre in Toronto.
PARAGON-HF enrolled 4,822 HFpEF patients at 848 centers in 43 countries, and the efficacy analysis included 4,796 patients.
The composite renal outcome was mainly driven by the incidence of a 50% or greater drop from baseline in eGFR, which occurred in 27 patients (1.1%) in the sacubitril/valsartan group and 60 patients (2.5%) who received valsartan alone.
The annual average drop in eGFR during the study was 2.0 mL/min per 1.73m2 in the sacubitril/valsartan group and 2.7 mL/min per 1.73m2 in the control group.
Although the heart failure community was disappointed that sacubitril/valsartan failed to show a significant benefit for the study’s primary outcome in HFpEF, the combination has become a mainstay of treatment for patients with HFpEF based on its performance in the PARADIGM-HF trial.
And despite the unqualified support sacubitril/valsartan now receives in guidelines and its label as a foundational treatment for HFpEF, the formulation has had a hard time gaining traction in U.S. practice, often because of barriers placed by third-party payers.
PARAGON-HF was sponsored by Novartis, which markets sacubitril/valsartan (Entresto). Dr. McCausland has reported no relevant financial relationships. Dr. Tobe has reported participating on a steering committee for Bayer Fidelio/Figaro studies and being a speaker on behalf of Pfizer and Servier. Dr. Poon has reported being an adviser to Novartis, Boehringer Ingelheim, and Servier.
A version of this article originally appeared on Medscape.com.
Patients with heart failure with preserved ejection fraction (HFpEF) who received sacubitril/valsartan in the PARAGON-HF trial had significant protection against progression of renal dysfunction in a prespecified secondary analysis.
The 2,419 patients with HFpEF who received sacubitril/valsartan (Entresto) had half the rate of the primary adverse renal outcome, compared with the 2,403 patients randomized to valsartan alone in the comparator group, a significant difference, according to the results published online Sept. 29 in Circulation by Finnian R. McCausland, MBBCh, and colleagues.
In absolute terms, sacubitril/valsartan treatment, an angiotensin-receptor/neprilysin inhibitor (ARNI), cut the incidence of the combined renal endpoint – renal death, end-stage renal disease, or at least a 50% drop in estimated glomerular filtration rate (eGFR) – from 2.7% in the control group to 1.4% in the sacubitril/valsartan group during a median follow-up of 35 months.
The absolute difference of 1.3% equated to a number needed to treat of 51 to prevent one of these events.
Also notable was that renal protection from sacubitril/valsartan was equally robust across the range of baseline kidney function.
‘An important therapeutic option’
The efficacy “across the spectrum of baseline renal function” indicates treatment with sacubitril/valsartan is “an important therapeutic option to slow renal-function decline in patients with heart failure,” wrote Dr. McCausland, a nephrologist at Brigham and Women’s Hospital in Boston, and colleagues.
The authors’ conclusion is striking because currently no drug class has produced clear evidence for efficacy in HFpEF.
On the other hand, the PARAGON-HF trial that provided the data for this new analysis was statistically neutral for its primary endpoint – a reduction in the combined rate of cardiovascular death and hospitalizations for heart failure – with a P value of .06 and 95% confidence interval of 0.75-1.01.
“Because this difference [in the primary endpoint incidence between the two study group] did not meet the predetermined level of statistical significance, subsequent analyses were considered to be exploratory,” noted the authors of the primary analysis of PARAGON-HF, as reported by Medscape Medical News.
Despite this limitation in interpreting secondary outcomes from the trial, the new report of a significant renal benefit “opens the potential to provide evidence-based treatment for patients with HFpEF,” commented Sheldon W. Tobe, MD, and Stephanie Poon, MD, in an editorial accompanying the latest analysis.
“At the very least, these results are certainly intriguing and suggest that there may be important patient subgroups with HFpEF who might benefit from using sacubitril/valsartan,” they emphasized.
First large trial to show renal improvement in HFpEF
The editorialists’ enthusiasm for the implications of the new findings relate in part to the fact that “PARAGON-HF is the first large trial to demonstrate improvement in renal parameters in HFpEF,” they noted.
“The finding that the composite renal outcome did not differ according to baseline eGFR is significant and suggests that the beneficial effect on renal function was indirect, possibly linked to improved cardiac function,” say Dr. Tobe, a nephrologist, and Dr. Poon, a cardiologist, both at Sunnybrook Health Sciences Centre in Toronto.
PARAGON-HF enrolled 4,822 HFpEF patients at 848 centers in 43 countries, and the efficacy analysis included 4,796 patients.
The composite renal outcome was mainly driven by the incidence of a 50% or greater drop from baseline in eGFR, which occurred in 27 patients (1.1%) in the sacubitril/valsartan group and 60 patients (2.5%) who received valsartan alone.
The annual average drop in eGFR during the study was 2.0 mL/min per 1.73m2 in the sacubitril/valsartan group and 2.7 mL/min per 1.73m2 in the control group.
Although the heart failure community was disappointed that sacubitril/valsartan failed to show a significant benefit for the study’s primary outcome in HFpEF, the combination has become a mainstay of treatment for patients with HFpEF based on its performance in the PARADIGM-HF trial.
And despite the unqualified support sacubitril/valsartan now receives in guidelines and its label as a foundational treatment for HFpEF, the formulation has had a hard time gaining traction in U.S. practice, often because of barriers placed by third-party payers.
PARAGON-HF was sponsored by Novartis, which markets sacubitril/valsartan (Entresto). Dr. McCausland has reported no relevant financial relationships. Dr. Tobe has reported participating on a steering committee for Bayer Fidelio/Figaro studies and being a speaker on behalf of Pfizer and Servier. Dr. Poon has reported being an adviser to Novartis, Boehringer Ingelheim, and Servier.
A version of this article originally appeared on Medscape.com.
Patients with heart failure with preserved ejection fraction (HFpEF) who received sacubitril/valsartan in the PARAGON-HF trial had significant protection against progression of renal dysfunction in a prespecified secondary analysis.
The 2,419 patients with HFpEF who received sacubitril/valsartan (Entresto) had half the rate of the primary adverse renal outcome, compared with the 2,403 patients randomized to valsartan alone in the comparator group, a significant difference, according to the results published online Sept. 29 in Circulation by Finnian R. McCausland, MBBCh, and colleagues.
In absolute terms, sacubitril/valsartan treatment, an angiotensin-receptor/neprilysin inhibitor (ARNI), cut the incidence of the combined renal endpoint – renal death, end-stage renal disease, or at least a 50% drop in estimated glomerular filtration rate (eGFR) – from 2.7% in the control group to 1.4% in the sacubitril/valsartan group during a median follow-up of 35 months.
The absolute difference of 1.3% equated to a number needed to treat of 51 to prevent one of these events.
Also notable was that renal protection from sacubitril/valsartan was equally robust across the range of baseline kidney function.
‘An important therapeutic option’
The efficacy “across the spectrum of baseline renal function” indicates treatment with sacubitril/valsartan is “an important therapeutic option to slow renal-function decline in patients with heart failure,” wrote Dr. McCausland, a nephrologist at Brigham and Women’s Hospital in Boston, and colleagues.
The authors’ conclusion is striking because currently no drug class has produced clear evidence for efficacy in HFpEF.
On the other hand, the PARAGON-HF trial that provided the data for this new analysis was statistically neutral for its primary endpoint – a reduction in the combined rate of cardiovascular death and hospitalizations for heart failure – with a P value of .06 and 95% confidence interval of 0.75-1.01.
“Because this difference [in the primary endpoint incidence between the two study group] did not meet the predetermined level of statistical significance, subsequent analyses were considered to be exploratory,” noted the authors of the primary analysis of PARAGON-HF, as reported by Medscape Medical News.
Despite this limitation in interpreting secondary outcomes from the trial, the new report of a significant renal benefit “opens the potential to provide evidence-based treatment for patients with HFpEF,” commented Sheldon W. Tobe, MD, and Stephanie Poon, MD, in an editorial accompanying the latest analysis.
“At the very least, these results are certainly intriguing and suggest that there may be important patient subgroups with HFpEF who might benefit from using sacubitril/valsartan,” they emphasized.
First large trial to show renal improvement in HFpEF
The editorialists’ enthusiasm for the implications of the new findings relate in part to the fact that “PARAGON-HF is the first large trial to demonstrate improvement in renal parameters in HFpEF,” they noted.
“The finding that the composite renal outcome did not differ according to baseline eGFR is significant and suggests that the beneficial effect on renal function was indirect, possibly linked to improved cardiac function,” say Dr. Tobe, a nephrologist, and Dr. Poon, a cardiologist, both at Sunnybrook Health Sciences Centre in Toronto.
PARAGON-HF enrolled 4,822 HFpEF patients at 848 centers in 43 countries, and the efficacy analysis included 4,796 patients.
The composite renal outcome was mainly driven by the incidence of a 50% or greater drop from baseline in eGFR, which occurred in 27 patients (1.1%) in the sacubitril/valsartan group and 60 patients (2.5%) who received valsartan alone.
The annual average drop in eGFR during the study was 2.0 mL/min per 1.73m2 in the sacubitril/valsartan group and 2.7 mL/min per 1.73m2 in the control group.
Although the heart failure community was disappointed that sacubitril/valsartan failed to show a significant benefit for the study’s primary outcome in HFpEF, the combination has become a mainstay of treatment for patients with HFpEF based on its performance in the PARADIGM-HF trial.
And despite the unqualified support sacubitril/valsartan now receives in guidelines and its label as a foundational treatment for HFpEF, the formulation has had a hard time gaining traction in U.S. practice, often because of barriers placed by third-party payers.
PARAGON-HF was sponsored by Novartis, which markets sacubitril/valsartan (Entresto). Dr. McCausland has reported no relevant financial relationships. Dr. Tobe has reported participating on a steering committee for Bayer Fidelio/Figaro studies and being a speaker on behalf of Pfizer and Servier. Dr. Poon has reported being an adviser to Novartis, Boehringer Ingelheim, and Servier.
A version of this article originally appeared on Medscape.com.
Manners matter
Have you been surprised and impressed by a child who says after a visit, “Thank you, Doctor [Howard]”? While it may seem antiquated to teach such manners to children these days, there are several important benefits to this education.

Manners serve important functions in benefiting a person’s group with cohesiveness and the individuals themselves with acceptance in the group. Use of manners instantly suggests a more trustworthy person.
There are three main categories of manners: hygiene, courtesy, and cultural norm manners.
Hygiene manners, from using the toilet to refraining from picking one’s nose, have obvious health benefits of not spreading disease. Hygiene manners take time to teach, but parents are motivated and helped by natural reactions of disgust that even infants recognize.
Courtesy manners, on the other hand, are habits of self-control and good-faith behaviors that signal that one is putting the interests of others ahead of one’s own for the moment. Taking another’s comfort into account, basic to kindness and respect, does not require agreeing with or submitting to the other. Courtesy manners require a developing self-awareness (I can choose to act this way) and awareness of social status (I am not more important than everyone else) that begins in toddlerhood. Modeling manners around the child is the most important way to teach courtesy. Parents usually start actively teaching the child to say “please” and “thank you,” and show pride in this apparent “demonstration of appreciation” even when it is simply reinforced behavior at first. The delight of grandparents reinforces both the parents and children, and reflects manners as building tribe cohesiveness.
Good manners become a habit
Manners such as warm greetings, a firm handshake (before COVID-19), and prompt thanks are most believable when occurring promptly when appropriate – when they come from habit. This immediate reaction, a result of so-called “fast thinking,” develops when behaviors learned from “slow thinking” are instilled early and often until they are automatic. The other benefit of this overlearning is that the behavior then looks unambivalent; a lag of too many milliseconds makes the recipient doubt genuineness.
Parents often ask us how to handle their child‘s rude or disrespectful behavior. Praise for manners is a simple start. Toddlers and preschoolers are taught manners best by adult modeling, but also by reinforcement and praise for the basics: to say “Hello,” ask “Please,” and say “Thank you,” “Excuse me,” “You’re welcome,” or “Would you help me, please?” The behaviors also include avoiding raising one’s voice, suppressing interrupting, and apologizing when appropriate. Even shy children can learn eye contact by making a game of figuring out the other’s eye color. Shaming, yelling, and punishing for poor manners usually backfires because it shows disrespect of the child who will likely give this back.
Older children can be taught to offer other people the opportunity to go through a door first, to be first to select a seat, speak first and without interruption, or order first. There are daily opportunities for these manners of showing respect. Opening doors for others, or standing when a guest enters the room are more formal but still appreciated. Parents who use and expect courtesy manners with everyone – irrespective of gender, race, ethnicity, or role as a server versus professional – show that they value others and build antiracism.

School age is a time to learn to wait before speaking to consider whether what they say could be experienced as hurtful to the other person. This requires taking someone else’s point of view, an ability that emerges around age 6 years and can be promoted when parents review with their child “How would you feel if it were you?” Role playing common scenarios of how to behave and speak when seeing a person who looks or acts different is also effective. Avoiding interrupting may be more difficult for very talkative or impulsive children, especially those with ADHD. Practicing waiting for permission to speak by being handed a “talking stick” at the dinner table can be good practice for everyone.
Manners are a group asset
Beyond personal benefits, manners are the basis of a civil society. Cultural norm manners are particular to groups, helping members feel affiliated, as well as identifying those with different manners as “other.”
Teens are particularly likely to use a different code of behavior to fit in with a subgroup. This may be acceptable if restricted to within their group (such as swear words) or within certain agreed-upon limits with family members. But teens need to understand the value of learning, practicing, and using manners for their own, as well as their group’s and nation’s, well-being.
As a developmental-behavioral pediatrician, I have cared for many children with intellectual disabilities and autism spectrum disorder (ASD). Deficits in social interaction skills are a basic criterion for the diagnosis of ASD. Overtraining is especially needed for children with ASD whose mirror movements, social attention, and imitation are weak. For children with these conditions, making manners a strong habit takes more effort but is even more vital than for neurotypical children. Temple Grandin, a famous adult with ASD, has described how her mother taught her manners as a survival skill. She reports incorporating manners very consciously and methodically because they did not come naturally. Children with even rote social skills are liked better by peers and teachers, their atypical behaviors is better tolerated, and they get more positive feedback that encourages integration inside and outside the classroom. Manners may make the difference between being allowed in or expelled from classrooms, libraries, clubs, teams, or religious institutions. When it is time to get a job, social skills are the key factor for employment for these individuals and a significant help for neurotypical individuals as well. Failure to signal socially appropriate behavior can make a person appear threatening and has had the rare but tragic result of rough or fatal handling by police.
Has the teaching of manners waned? Perhaps, because, for some families, the child is being socialized mostly by nonfamily caregivers who have low use of manners. Some parents have made teaching manners a low priority or even resisted using manners themselves as inauthentic. This may reflect prioritizing a “laid-back” lifestyle and speaking crudely as a sign of independence, perhaps in reaction to lack of autonomy at work. Mastering the careful interactions developed over time to avoid invoking an aggressive response depend on direct feedback from reactions of the recipient. With so much of our communication done electronically, asynchronously, even anonymously, the usual feedback has been reduced. Practicing curses, insults, and put-downs online easily extends to in-person interactions without the perpetrator even noticing and are generally reinforced and repeated without parental supervision. Disrespectful behavior from community leaders also reduces the threshold for society.
When people are ignorant of or choose not to use manners they may be perceived as “other” and hostile. This may lead to distrust, dislike, and lowered ability to find the common ground needed for making decisions that benefit the greater society. Oliver Wendell Holmes said “Under bad manners ... lies very commonly an overestimate of our special individuality, as distinguished from our generic humanity (“The Professor at the Breakfast Table,” 1858). Working for major goals that benefit all of humanity is essential to survival in our highly interconnected world. Considering all of humanity is a difficult concept for children, and even for many adults, but it starts with using civil behavior at home, in school, and in one’s community.
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to MDedge News. E-mail her at pdnews@mdedge.com.
Have you been surprised and impressed by a child who says after a visit, “Thank you, Doctor [Howard]”? While it may seem antiquated to teach such manners to children these days, there are several important benefits to this education.
Manners serve important functions in benefiting a person’s group with cohesiveness and the individuals themselves with acceptance in the group. Use of manners instantly suggests a more trustworthy person.
There are three main categories of manners: hygiene, courtesy, and cultural norm manners.
Hygiene manners, from using the toilet to refraining from picking one’s nose, have obvious health benefits of not spreading disease. Hygiene manners take time to teach, but parents are motivated and helped by natural reactions of disgust that even infants recognize.
Courtesy manners, on the other hand, are habits of self-control and good-faith behaviors that signal that one is putting the interests of others ahead of one’s own for the moment. Taking another’s comfort into account, basic to kindness and respect, does not require agreeing with or submitting to the other. Courtesy manners require a developing self-awareness (I can choose to act this way) and awareness of social status (I am not more important than everyone else) that begins in toddlerhood. Modeling manners around the child is the most important way to teach courtesy. Parents usually start actively teaching the child to say “please” and “thank you,” and show pride in this apparent “demonstration of appreciation” even when it is simply reinforced behavior at first. The delight of grandparents reinforces both the parents and children, and reflects manners as building tribe cohesiveness.
Good manners become a habit
Manners such as warm greetings, a firm handshake (before COVID-19), and prompt thanks are most believable when occurring promptly when appropriate – when they come from habit. This immediate reaction, a result of so-called “fast thinking,” develops when behaviors learned from “slow thinking” are instilled early and often until they are automatic. The other benefit of this overlearning is that the behavior then looks unambivalent; a lag of too many milliseconds makes the recipient doubt genuineness.
Parents often ask us how to handle their child‘s rude or disrespectful behavior. Praise for manners is a simple start. Toddlers and preschoolers are taught manners best by adult modeling, but also by reinforcement and praise for the basics: to say “Hello,” ask “Please,” and say “Thank you,” “Excuse me,” “You’re welcome,” or “Would you help me, please?” The behaviors also include avoiding raising one’s voice, suppressing interrupting, and apologizing when appropriate. Even shy children can learn eye contact by making a game of figuring out the other’s eye color. Shaming, yelling, and punishing for poor manners usually backfires because it shows disrespect of the child who will likely give this back.
Older children can be taught to offer other people the opportunity to go through a door first, to be first to select a seat, speak first and without interruption, or order first. There are daily opportunities for these manners of showing respect. Opening doors for others, or standing when a guest enters the room are more formal but still appreciated. Parents who use and expect courtesy manners with everyone – irrespective of gender, race, ethnicity, or role as a server versus professional – show that they value others and build antiracism.
School age is a time to learn to wait before speaking to consider whether what they say could be experienced as hurtful to the other person. This requires taking someone else’s point of view, an ability that emerges around age 6 years and can be promoted when parents review with their child “How would you feel if it were you?” Role playing common scenarios of how to behave and speak when seeing a person who looks or acts different is also effective. Avoiding interrupting may be more difficult for very talkative or impulsive children, especially those with ADHD. Practicing waiting for permission to speak by being handed a “talking stick” at the dinner table can be good practice for everyone.
Manners are a group asset
Beyond personal benefits, manners are the basis of a civil society. Cultural norm manners are particular to groups, helping members feel affiliated, as well as identifying those with different manners as “other.”
Teens are particularly likely to use a different code of behavior to fit in with a subgroup. This may be acceptable if restricted to within their group (such as swear words) or within certain agreed-upon limits with family members. But teens need to understand the value of learning, practicing, and using manners for their own, as well as their group’s and nation’s, well-being.
As a developmental-behavioral pediatrician, I have cared for many children with intellectual disabilities and autism spectrum disorder (ASD). Deficits in social interaction skills are a basic criterion for the diagnosis of ASD. Overtraining is especially needed for children with ASD whose mirror movements, social attention, and imitation are weak. For children with these conditions, making manners a strong habit takes more effort but is even more vital than for neurotypical children. Temple Grandin, a famous adult with ASD, has described how her mother taught her manners as a survival skill. She reports incorporating manners very consciously and methodically because they did not come naturally. Children with even rote social skills are liked better by peers and teachers, their atypical behaviors is better tolerated, and they get more positive feedback that encourages integration inside and outside the classroom. Manners may make the difference between being allowed in or expelled from classrooms, libraries, clubs, teams, or religious institutions. When it is time to get a job, social skills are the key factor for employment for these individuals and a significant help for neurotypical individuals as well. Failure to signal socially appropriate behavior can make a person appear threatening and has had the rare but tragic result of rough or fatal handling by police.
Has the teaching of manners waned? Perhaps, because, for some families, the child is being socialized mostly by nonfamily caregivers who have low use of manners. Some parents have made teaching manners a low priority or even resisted using manners themselves as inauthentic. This may reflect prioritizing a “laid-back” lifestyle and speaking crudely as a sign of independence, perhaps in reaction to lack of autonomy at work. Mastering the careful interactions developed over time to avoid invoking an aggressive response depend on direct feedback from reactions of the recipient. With so much of our communication done electronically, asynchronously, even anonymously, the usual feedback has been reduced. Practicing curses, insults, and put-downs online easily extends to in-person interactions without the perpetrator even noticing and are generally reinforced and repeated without parental supervision. Disrespectful behavior from community leaders also reduces the threshold for society.
When people are ignorant of or choose not to use manners they may be perceived as “other” and hostile. This may lead to distrust, dislike, and lowered ability to find the common ground needed for making decisions that benefit the greater society. Oliver Wendell Holmes said “Under bad manners ... lies very commonly an overestimate of our special individuality, as distinguished from our generic humanity (“The Professor at the Breakfast Table,” 1858). Working for major goals that benefit all of humanity is essential to survival in our highly interconnected world. Considering all of humanity is a difficult concept for children, and even for many adults, but it starts with using civil behavior at home, in school, and in one’s community.
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to MDedge News. E-mail her at pdnews@mdedge.com.
Have you been surprised and impressed by a child who says after a visit, “Thank you, Doctor [Howard]”? While it may seem antiquated to teach such manners to children these days, there are several important benefits to this education.
Manners serve important functions in benefiting a person’s group with cohesiveness and the individuals themselves with acceptance in the group. Use of manners instantly suggests a more trustworthy person.
There are three main categories of manners: hygiene, courtesy, and cultural norm manners.
Hygiene manners, from using the toilet to refraining from picking one’s nose, have obvious health benefits of not spreading disease. Hygiene manners take time to teach, but parents are motivated and helped by natural reactions of disgust that even infants recognize.
Courtesy manners, on the other hand, are habits of self-control and good-faith behaviors that signal that one is putting the interests of others ahead of one’s own for the moment. Taking another’s comfort into account, basic to kindness and respect, does not require agreeing with or submitting to the other. Courtesy manners require a developing self-awareness (I can choose to act this way) and awareness of social status (I am not more important than everyone else) that begins in toddlerhood. Modeling manners around the child is the most important way to teach courtesy. Parents usually start actively teaching the child to say “please” and “thank you,” and show pride in this apparent “demonstration of appreciation” even when it is simply reinforced behavior at first. The delight of grandparents reinforces both the parents and children, and reflects manners as building tribe cohesiveness.
Good manners become a habit
Manners such as warm greetings, a firm handshake (before COVID-19), and prompt thanks are most believable when occurring promptly when appropriate – when they come from habit. This immediate reaction, a result of so-called “fast thinking,” develops when behaviors learned from “slow thinking” are instilled early and often until they are automatic. The other benefit of this overlearning is that the behavior then looks unambivalent; a lag of too many milliseconds makes the recipient doubt genuineness.
Parents often ask us how to handle their child‘s rude or disrespectful behavior. Praise for manners is a simple start. Toddlers and preschoolers are taught manners best by adult modeling, but also by reinforcement and praise for the basics: to say “Hello,” ask “Please,” and say “Thank you,” “Excuse me,” “You’re welcome,” or “Would you help me, please?” The behaviors also include avoiding raising one’s voice, suppressing interrupting, and apologizing when appropriate. Even shy children can learn eye contact by making a game of figuring out the other’s eye color. Shaming, yelling, and punishing for poor manners usually backfires because it shows disrespect of the child who will likely give this back.
Older children can be taught to offer other people the opportunity to go through a door first, to be first to select a seat, speak first and without interruption, or order first. There are daily opportunities for these manners of showing respect. Opening doors for others, or standing when a guest enters the room are more formal but still appreciated. Parents who use and expect courtesy manners with everyone – irrespective of gender, race, ethnicity, or role as a server versus professional – show that they value others and build antiracism.
School age is a time to learn to wait before speaking to consider whether what they say could be experienced as hurtful to the other person. This requires taking someone else’s point of view, an ability that emerges around age 6 years and can be promoted when parents review with their child “How would you feel if it were you?” Role playing common scenarios of how to behave and speak when seeing a person who looks or acts different is also effective. Avoiding interrupting may be more difficult for very talkative or impulsive children, especially those with ADHD. Practicing waiting for permission to speak by being handed a “talking stick” at the dinner table can be good practice for everyone.
Manners are a group asset
Beyond personal benefits, manners are the basis of a civil society. Cultural norm manners are particular to groups, helping members feel affiliated, as well as identifying those with different manners as “other.”
Teens are particularly likely to use a different code of behavior to fit in with a subgroup. This may be acceptable if restricted to within their group (such as swear words) or within certain agreed-upon limits with family members. But teens need to understand the value of learning, practicing, and using manners for their own, as well as their group’s and nation’s, well-being.
As a developmental-behavioral pediatrician, I have cared for many children with intellectual disabilities and autism spectrum disorder (ASD). Deficits in social interaction skills are a basic criterion for the diagnosis of ASD. Overtraining is especially needed for children with ASD whose mirror movements, social attention, and imitation are weak. For children with these conditions, making manners a strong habit takes more effort but is even more vital than for neurotypical children. Temple Grandin, a famous adult with ASD, has described how her mother taught her manners as a survival skill. She reports incorporating manners very consciously and methodically because they did not come naturally. Children with even rote social skills are liked better by peers and teachers, their atypical behaviors is better tolerated, and they get more positive feedback that encourages integration inside and outside the classroom. Manners may make the difference between being allowed in or expelled from classrooms, libraries, clubs, teams, or religious institutions. When it is time to get a job, social skills are the key factor for employment for these individuals and a significant help for neurotypical individuals as well. Failure to signal socially appropriate behavior can make a person appear threatening and has had the rare but tragic result of rough or fatal handling by police.
Has the teaching of manners waned? Perhaps, because, for some families, the child is being socialized mostly by nonfamily caregivers who have low use of manners. Some parents have made teaching manners a low priority or even resisted using manners themselves as inauthentic. This may reflect prioritizing a “laid-back” lifestyle and speaking crudely as a sign of independence, perhaps in reaction to lack of autonomy at work. Mastering the careful interactions developed over time to avoid invoking an aggressive response depend on direct feedback from reactions of the recipient. With so much of our communication done electronically, asynchronously, even anonymously, the usual feedback has been reduced. Practicing curses, insults, and put-downs online easily extends to in-person interactions without the perpetrator even noticing and are generally reinforced and repeated without parental supervision. Disrespectful behavior from community leaders also reduces the threshold for society.
When people are ignorant of or choose not to use manners they may be perceived as “other” and hostile. This may lead to distrust, dislike, and lowered ability to find the common ground needed for making decisions that benefit the greater society. Oliver Wendell Holmes said “Under bad manners ... lies very commonly an overestimate of our special individuality, as distinguished from our generic humanity (“The Professor at the Breakfast Table,” 1858). Working for major goals that benefit all of humanity is essential to survival in our highly interconnected world. Considering all of humanity is a difficult concept for children, and even for many adults, but it starts with using civil behavior at home, in school, and in one’s community.
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to MDedge News. E-mail her at pdnews@mdedge.com.
Gene signature found similarly prognostic in ILC and IDC

ILC is enriched with features indicating low proliferative activity, noted investigator Otto Metzger, MD, of the Dana Farber Cancer Institute in Boston.
“Data from retrospective series have indicated no benefit with adjuvant chemotherapy for patients diagnosed with early-stage ILC,” he said. “It’s fair to say that chemotherapy decisions for patients with ILC remain controversial.”
With this in mind, Dr. Metzger and colleagues analyzed data for 5,313 women who underwent surgery for early-stage breast cancer (node-negative or up to three positive lymph nodes) and were risk-stratified to receive or skip adjuvant chemotherapy based on both clinical risk and the MammaPrint score for genomic risk. Fully 44% of women with ILC had discordant clinical and genomic risks.
With a median follow-up of 8.7 years, the 5-year rate of distant metastasis–free survival among all patients classified as genomic high risk was 92.1% in women with IDC and 88.1% in women with ILC, with overlapping 95% confidence intervals. Rates of distant metastasis–free survival for patients with genomic low risk were 96.4% in women with IDC and 96.6% in women with ILC, again with confidence intervals that overlapped.
The pattern was essentially the same for overall survival, and results carried over into 8-year outcomes as well.
“We believe that MammaPrint is a clinically useful test for patients diagnosed with ILC,” Dr. Metzger said. “There are similar survival outcomes for ILC and IDC when matched by genomic risk. This is an important message.”
It should be standard to omit chemotherapy for patients who have ILC classified as high clinical risk but low genomic risk by MammaPrint, Dr. Metzger recommended. “By contrast, MammaPrint should facilitate chemotherapy treatment decisions for patients diagnosed with ILC and high-risk MammaPrint,” he said.
Prognostic, but predictive?
“This is a well-designed prospective multicenter trial and provides the best evidence to date that MammaPrint is an important prognostic tool for ILC,” Todd Tuttle, MD, of University of Minnesota in Minneapolis, said in an interview.

Dr. Tuttle said he mainly uses the MammaPrint test and the OncoType 21-gene recurrence score to estimate prognosis for his patients with ILC.
These new data establish that MammaPrint is prognostic in ILC, but the value of MammaPrint’s genomic high risk result for making the decision about chemotherapy is still unclear, according to Dr. Tuttle.
“I don’t think we know whether MammaPrint can predict the benefit of chemotherapy for patients with stage I or II ILC,” he elaborated. “We need further high-quality studies such as this one to determine the best treatment strategies for ILC, which is a difficult breast cancer.”
Study details
Of the 5,313 patients studied, 487 had ILC (255 classic and 232 variant) and 4,826 had IDC according to central pathology assessment, Dr. Metzger reported.
MammaPrint classified 39% of the IDC group and 16% of the ILC group (10% of those with classic disease and 23% of those with variant disease) as genomically high risk for recurrence. The Adjuvant! Online tool classified 48.3% of ILC and 51.5% of IDC patients as clinically high risk.
Among the 44% of women with ILC having discordant genomic and clinical risk, discordance was usually due to the combination of low genomic risk and high clinical risk, seen in 38%.
The curves for 5-year distant metastasis–free survival stratified by genomic risk essentially overlapped for the IDC and ILC groups. Furthermore, there was no significant interaction of histologic type and genomic risk on this outcome (P = .547).
The 5-year rate of overall survival among women with genomic high risk was 95.6% in the IDC group and 93.5% in the ILC group. Among women with genomic low risk, 5-year overall survival was 98.1% in the IDC group and 97.7% in the ILC group, again with overlapping confidence intervals within each risk category.
The study was funded with support from the Breast Cancer Research Foundation. Dr. Metzger disclosed consulting fees from AbbVie, Genentech, Roche, and Pfizer. Dr. Tuttle disclosed no conflicts of interest.
SOURCE: Metzger O et al. EBCC-12 Virtual Conference. Abstract 6.
ILC is enriched with features indicating low proliferative activity, noted investigator Otto Metzger, MD, of the Dana Farber Cancer Institute in Boston.
“Data from retrospective series have indicated no benefit with adjuvant chemotherapy for patients diagnosed with early-stage ILC,” he said. “It’s fair to say that chemotherapy decisions for patients with ILC remain controversial.”
With this in mind, Dr. Metzger and colleagues analyzed data for 5,313 women who underwent surgery for early-stage breast cancer (node-negative or up to three positive lymph nodes) and were risk-stratified to receive or skip adjuvant chemotherapy based on both clinical risk and the MammaPrint score for genomic risk. Fully 44% of women with ILC had discordant clinical and genomic risks.
With a median follow-up of 8.7 years, the 5-year rate of distant metastasis–free survival among all patients classified as genomic high risk was 92.1% in women with IDC and 88.1% in women with ILC, with overlapping 95% confidence intervals. Rates of distant metastasis–free survival for patients with genomic low risk were 96.4% in women with IDC and 96.6% in women with ILC, again with confidence intervals that overlapped.
The pattern was essentially the same for overall survival, and results carried over into 8-year outcomes as well.
“We believe that MammaPrint is a clinically useful test for patients diagnosed with ILC,” Dr. Metzger said. “There are similar survival outcomes for ILC and IDC when matched by genomic risk. This is an important message.”
It should be standard to omit chemotherapy for patients who have ILC classified as high clinical risk but low genomic risk by MammaPrint, Dr. Metzger recommended. “By contrast, MammaPrint should facilitate chemotherapy treatment decisions for patients diagnosed with ILC and high-risk MammaPrint,” he said.
Prognostic, but predictive?
“This is a well-designed prospective multicenter trial and provides the best evidence to date that MammaPrint is an important prognostic tool for ILC,” Todd Tuttle, MD, of University of Minnesota in Minneapolis, said in an interview.
Dr. Tuttle said he mainly uses the MammaPrint test and the OncoType 21-gene recurrence score to estimate prognosis for his patients with ILC.
These new data establish that MammaPrint is prognostic in ILC, but the value of MammaPrint’s genomic high risk result for making the decision about chemotherapy is still unclear, according to Dr. Tuttle.
“I don’t think we know whether MammaPrint can predict the benefit of chemotherapy for patients with stage I or II ILC,” he elaborated. “We need further high-quality studies such as this one to determine the best treatment strategies for ILC, which is a difficult breast cancer.”
Study details
Of the 5,313 patients studied, 487 had ILC (255 classic and 232 variant) and 4,826 had IDC according to central pathology assessment, Dr. Metzger reported.
MammaPrint classified 39% of the IDC group and 16% of the ILC group (10% of those with classic disease and 23% of those with variant disease) as genomically high risk for recurrence. The Adjuvant! Online tool classified 48.3% of ILC and 51.5% of IDC patients as clinically high risk.
Among the 44% of women with ILC having discordant genomic and clinical risk, discordance was usually due to the combination of low genomic risk and high clinical risk, seen in 38%.
The curves for 5-year distant metastasis–free survival stratified by genomic risk essentially overlapped for the IDC and ILC groups. Furthermore, there was no significant interaction of histologic type and genomic risk on this outcome (P = .547).
The 5-year rate of overall survival among women with genomic high risk was 95.6% in the IDC group and 93.5% in the ILC group. Among women with genomic low risk, 5-year overall survival was 98.1% in the IDC group and 97.7% in the ILC group, again with overlapping confidence intervals within each risk category.
The study was funded with support from the Breast Cancer Research Foundation. Dr. Metzger disclosed consulting fees from AbbVie, Genentech, Roche, and Pfizer. Dr. Tuttle disclosed no conflicts of interest.
SOURCE: Metzger O et al. EBCC-12 Virtual Conference. Abstract 6.
ILC is enriched with features indicating low proliferative activity, noted investigator Otto Metzger, MD, of the Dana Farber Cancer Institute in Boston.
“Data from retrospective series have indicated no benefit with adjuvant chemotherapy for patients diagnosed with early-stage ILC,” he said. “It’s fair to say that chemotherapy decisions for patients with ILC remain controversial.”
With this in mind, Dr. Metzger and colleagues analyzed data for 5,313 women who underwent surgery for early-stage breast cancer (node-negative or up to three positive lymph nodes) and were risk-stratified to receive or skip adjuvant chemotherapy based on both clinical risk and the MammaPrint score for genomic risk. Fully 44% of women with ILC had discordant clinical and genomic risks.
With a median follow-up of 8.7 years, the 5-year rate of distant metastasis–free survival among all patients classified as genomic high risk was 92.1% in women with IDC and 88.1% in women with ILC, with overlapping 95% confidence intervals. Rates of distant metastasis–free survival for patients with genomic low risk were 96.4% in women with IDC and 96.6% in women with ILC, again with confidence intervals that overlapped.
The pattern was essentially the same for overall survival, and results carried over into 8-year outcomes as well.
“We believe that MammaPrint is a clinically useful test for patients diagnosed with ILC,” Dr. Metzger said. “There are similar survival outcomes for ILC and IDC when matched by genomic risk. This is an important message.”
It should be standard to omit chemotherapy for patients who have ILC classified as high clinical risk but low genomic risk by MammaPrint, Dr. Metzger recommended. “By contrast, MammaPrint should facilitate chemotherapy treatment decisions for patients diagnosed with ILC and high-risk MammaPrint,” he said.
Prognostic, but predictive?
“This is a well-designed prospective multicenter trial and provides the best evidence to date that MammaPrint is an important prognostic tool for ILC,” Todd Tuttle, MD, of University of Minnesota in Minneapolis, said in an interview.
Dr. Tuttle said he mainly uses the MammaPrint test and the OncoType 21-gene recurrence score to estimate prognosis for his patients with ILC.
These new data establish that MammaPrint is prognostic in ILC, but the value of MammaPrint’s genomic high risk result for making the decision about chemotherapy is still unclear, according to Dr. Tuttle.
“I don’t think we know whether MammaPrint can predict the benefit of chemotherapy for patients with stage I or II ILC,” he elaborated. “We need further high-quality studies such as this one to determine the best treatment strategies for ILC, which is a difficult breast cancer.”
Study details
Of the 5,313 patients studied, 487 had ILC (255 classic and 232 variant) and 4,826 had IDC according to central pathology assessment, Dr. Metzger reported.
MammaPrint classified 39% of the IDC group and 16% of the ILC group (10% of those with classic disease and 23% of those with variant disease) as genomically high risk for recurrence. The Adjuvant! Online tool classified 48.3% of ILC and 51.5% of IDC patients as clinically high risk.
Among the 44% of women with ILC having discordant genomic and clinical risk, discordance was usually due to the combination of low genomic risk and high clinical risk, seen in 38%.
The curves for 5-year distant metastasis–free survival stratified by genomic risk essentially overlapped for the IDC and ILC groups. Furthermore, there was no significant interaction of histologic type and genomic risk on this outcome (P = .547).
The 5-year rate of overall survival among women with genomic high risk was 95.6% in the IDC group and 93.5% in the ILC group. Among women with genomic low risk, 5-year overall survival was 98.1% in the IDC group and 97.7% in the ILC group, again with overlapping confidence intervals within each risk category.
The study was funded with support from the Breast Cancer Research Foundation. Dr. Metzger disclosed consulting fees from AbbVie, Genentech, Roche, and Pfizer. Dr. Tuttle disclosed no conflicts of interest.
SOURCE: Metzger O et al. EBCC-12 Virtual Conference. Abstract 6.
FROM EBCC-12 VIRTUAL CONFERENCE
Guselkumab improvements for psoriatic arthritis persist through 1 year
Adults with active psoriatic arthritis (PsA) treated with guselkumab (Tremfya) showed significant improvement in American College of Rheumatology response criteria and disease activity after 1 year, based on data from the phase 3 DISCOVER-2 trial.

The findings, published in Arthritis & Rheumatology, extend the previously published 24-week, primary endpoint results of the trial, which tested guselkumab for adults with PsA who had not previously taken a biologic drug. Guselkumab was approved in July 2020 in the United States.
Iain B. McInnes, MD, PhD, of the University of Glasgow and his colleagues described guselkumab as “a fully-human monoclonal antibody specific to interleukin (IL)-23’s p19-subunit” that offers a potential alternative for PsA patients who discontinue their index tumor necrosis factor inhibitor because of insufficient efficacy.
The study enrolled 739 PsA patients at 118 sites worldwide. Participants were randomized to receive subcutaneous injections of 100 mg guselkumab every 4 weeks; 100 mg guselkumab at week 0 and 4, then every 8 weeks; or a placebo; 238 placebo-treated patients crossed over at 24 weeks to receive 100 mg guselkumab every 4 weeks. Patients on nonbiologic disease-modifying antirheumatic drugs at baseline were allowed to continue stable doses. Overall, about 93% of patients originally randomized to the three groups remained on guselkumab at 52 weeks.
Overall, 71% and 75% of 4-week and 8-week guselkumab patients, respectively, showed an improvement of at least 20% from baseline in ACR response criteria components at 52 weeks, which was up from 64% of patients seen at 24 weeks in both groups.
The study participants had an average disease duration of more than 5 years with no biologic treatment, and an average of 12-13 swollen joints and 20-22 tender joints at baseline. Approximately half were male, half had psoriasis or dactylitis, and two-thirds had enthesitis. Skin disease severity was assessed using the Investigator’s Global Assessment and Psoriasis Area Severity Index (PASI).
At 52 weeks, 75% and 58% of patients in the guselkumab groups had resolution of dactylitis and enthesitis, respectively. In addition, 86% of patients in both guselkumab groups achieved PASI 75 at 52 weeks, and 58% and 53% of the 4-week and 8-week groups, respectively, achieved PASI 100.
In addition, patients treated with guselkumab showed low levels of radiographic progression and significant improvements from baseline in measures of physical function and quality of life.
The most frequently reported adverse events in guselkumab patients were upper respiratory tract infections, nasopharyngitis, bronchitis, and investigator-reported laboratory values of increased alanine aminotransferase and aspartate aminotransferase; these rates were similar to those seen in the previously published 24-week data. Approximately 2% of guselkumab and placebo patients discontinued their treatments because of adverse events.
No patient developed an opportunistic infection or died during the study period.
The study findings were limited by several factors including the relatively short 1-year duration, the shorter duration of placebo, compared with guselkumab, and by potential confounding from missing data on patients who discontinued, the researchers noted. However, the results support the effectiveness of guselkumab for improving a range of manifestations of active PsA, and the overall treatment and safety profiles seen at 24 weeks were maintained, they said.
“Data obtained during the second year of DISCOVER-2 will augment current knowledge of the guselkumab benefit-risk profile and further our understanding of longer-term radiographic outcomes with both guselkumab dosing regimens,” they concluded.
The study was supported by Janssen. Many authors reported financial relationships with Janssen and other pharmaceutical companies. Nine of the 15 authors are employees of Janssen (a subsidiary of Johnson & Johnson) and own Johnson & Johnson stock or stock options.
SOURCE: McInnes IB et al. Arthritis Rheumatol. 2020 Oct 11. doi: 10.1002/art.41553.
Adults with active psoriatic arthritis (PsA) treated with guselkumab (Tremfya) showed significant improvement in American College of Rheumatology response criteria and disease activity after 1 year, based on data from the phase 3 DISCOVER-2 trial.
The findings, published in Arthritis & Rheumatology, extend the previously published 24-week, primary endpoint results of the trial, which tested guselkumab for adults with PsA who had not previously taken a biologic drug. Guselkumab was approved in July 2020 in the United States.
Iain B. McInnes, MD, PhD, of the University of Glasgow and his colleagues described guselkumab as “a fully-human monoclonal antibody specific to interleukin (IL)-23’s p19-subunit” that offers a potential alternative for PsA patients who discontinue their index tumor necrosis factor inhibitor because of insufficient efficacy.
The study enrolled 739 PsA patients at 118 sites worldwide. Participants were randomized to receive subcutaneous injections of 100 mg guselkumab every 4 weeks; 100 mg guselkumab at week 0 and 4, then every 8 weeks; or a placebo; 238 placebo-treated patients crossed over at 24 weeks to receive 100 mg guselkumab every 4 weeks. Patients on nonbiologic disease-modifying antirheumatic drugs at baseline were allowed to continue stable doses. Overall, about 93% of patients originally randomized to the three groups remained on guselkumab at 52 weeks.
Overall, 71% and 75% of 4-week and 8-week guselkumab patients, respectively, showed an improvement of at least 20% from baseline in ACR response criteria components at 52 weeks, which was up from 64% of patients seen at 24 weeks in both groups.
The study participants had an average disease duration of more than 5 years with no biologic treatment, and an average of 12-13 swollen joints and 20-22 tender joints at baseline. Approximately half were male, half had psoriasis or dactylitis, and two-thirds had enthesitis. Skin disease severity was assessed using the Investigator’s Global Assessment and Psoriasis Area Severity Index (PASI).
At 52 weeks, 75% and 58% of patients in the guselkumab groups had resolution of dactylitis and enthesitis, respectively. In addition, 86% of patients in both guselkumab groups achieved PASI 75 at 52 weeks, and 58% and 53% of the 4-week and 8-week groups, respectively, achieved PASI 100.
In addition, patients treated with guselkumab showed low levels of radiographic progression and significant improvements from baseline in measures of physical function and quality of life.
The most frequently reported adverse events in guselkumab patients were upper respiratory tract infections, nasopharyngitis, bronchitis, and investigator-reported laboratory values of increased alanine aminotransferase and aspartate aminotransferase; these rates were similar to those seen in the previously published 24-week data. Approximately 2% of guselkumab and placebo patients discontinued their treatments because of adverse events.
No patient developed an opportunistic infection or died during the study period.
The study findings were limited by several factors including the relatively short 1-year duration, the shorter duration of placebo, compared with guselkumab, and by potential confounding from missing data on patients who discontinued, the researchers noted. However, the results support the effectiveness of guselkumab for improving a range of manifestations of active PsA, and the overall treatment and safety profiles seen at 24 weeks were maintained, they said.
“Data obtained during the second year of DISCOVER-2 will augment current knowledge of the guselkumab benefit-risk profile and further our understanding of longer-term radiographic outcomes with both guselkumab dosing regimens,” they concluded.
The study was supported by Janssen. Many authors reported financial relationships with Janssen and other pharmaceutical companies. Nine of the 15 authors are employees of Janssen (a subsidiary of Johnson & Johnson) and own Johnson & Johnson stock or stock options.
SOURCE: McInnes IB et al. Arthritis Rheumatol. 2020 Oct 11. doi: 10.1002/art.41553.
Adults with active psoriatic arthritis (PsA) treated with guselkumab (Tremfya) showed significant improvement in American College of Rheumatology response criteria and disease activity after 1 year, based on data from the phase 3 DISCOVER-2 trial.
The findings, published in Arthritis & Rheumatology, extend the previously published 24-week, primary endpoint results of the trial, which tested guselkumab for adults with PsA who had not previously taken a biologic drug. Guselkumab was approved in July 2020 in the United States.
Iain B. McInnes, MD, PhD, of the University of Glasgow and his colleagues described guselkumab as “a fully-human monoclonal antibody specific to interleukin (IL)-23’s p19-subunit” that offers a potential alternative for PsA patients who discontinue their index tumor necrosis factor inhibitor because of insufficient efficacy.
The study enrolled 739 PsA patients at 118 sites worldwide. Participants were randomized to receive subcutaneous injections of 100 mg guselkumab every 4 weeks; 100 mg guselkumab at week 0 and 4, then every 8 weeks; or a placebo; 238 placebo-treated patients crossed over at 24 weeks to receive 100 mg guselkumab every 4 weeks. Patients on nonbiologic disease-modifying antirheumatic drugs at baseline were allowed to continue stable doses. Overall, about 93% of patients originally randomized to the three groups remained on guselkumab at 52 weeks.
Overall, 71% and 75% of 4-week and 8-week guselkumab patients, respectively, showed an improvement of at least 20% from baseline in ACR response criteria components at 52 weeks, which was up from 64% of patients seen at 24 weeks in both groups.
The study participants had an average disease duration of more than 5 years with no biologic treatment, and an average of 12-13 swollen joints and 20-22 tender joints at baseline. Approximately half were male, half had psoriasis or dactylitis, and two-thirds had enthesitis. Skin disease severity was assessed using the Investigator’s Global Assessment and Psoriasis Area Severity Index (PASI).
At 52 weeks, 75% and 58% of patients in the guselkumab groups had resolution of dactylitis and enthesitis, respectively. In addition, 86% of patients in both guselkumab groups achieved PASI 75 at 52 weeks, and 58% and 53% of the 4-week and 8-week groups, respectively, achieved PASI 100.
In addition, patients treated with guselkumab showed low levels of radiographic progression and significant improvements from baseline in measures of physical function and quality of life.
The most frequently reported adverse events in guselkumab patients were upper respiratory tract infections, nasopharyngitis, bronchitis, and investigator-reported laboratory values of increased alanine aminotransferase and aspartate aminotransferase; these rates were similar to those seen in the previously published 24-week data. Approximately 2% of guselkumab and placebo patients discontinued their treatments because of adverse events.
No patient developed an opportunistic infection or died during the study period.
The study findings were limited by several factors including the relatively short 1-year duration, the shorter duration of placebo, compared with guselkumab, and by potential confounding from missing data on patients who discontinued, the researchers noted. However, the results support the effectiveness of guselkumab for improving a range of manifestations of active PsA, and the overall treatment and safety profiles seen at 24 weeks were maintained, they said.
“Data obtained during the second year of DISCOVER-2 will augment current knowledge of the guselkumab benefit-risk profile and further our understanding of longer-term radiographic outcomes with both guselkumab dosing regimens,” they concluded.
The study was supported by Janssen. Many authors reported financial relationships with Janssen and other pharmaceutical companies. Nine of the 15 authors are employees of Janssen (a subsidiary of Johnson & Johnson) and own Johnson & Johnson stock or stock options.
SOURCE: McInnes IB et al. Arthritis Rheumatol. 2020 Oct 11. doi: 10.1002/art.41553.
FROM ARTHRITIS & RHEUMATOLOGY
Decline in febuxostat use trends with cardiovascular concerns
Use of febuxostat (Uloric) decreased among patients with gout in the United States following a Food and Drug Administration–mandated black-box warning that cited cardiovascular concerns, but overall use of urate-lowering therapy remained stable, according to data from a study of commercial insurance enrollees in the United States between 2009 and 2019.

Initiation of urate-lowering therapy (ULT) is recommended for gout patients, and allopurinol remains the first-line treatment, but it is not effective in all patients, and febuxostat was developed as an alternative, wrote Seoyoung C. Kim, MD, ScD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, and colleagues.
However, based on data from a postmarketing safety trial (the CARES trial) mandated by the FDA, the agency first issued a safety announcement about an increased risk of cardiovascular mortality with febuxostat in November 2017, followed in February 2019 with a black-box warning after full CARES trial results were published in March 2018 showing a greater risk of cardiovascular and all-cause mortality in febuxostat versus allopurinol.
In a study published in Arthritis & Rheumatology, the researchers examined trends in the use of ULT before and after the FDA warning. They analyzed claims data from a national commercial health database that included 838,432 adult ULT users and 633,229 gout patients.
Overall, allopurinol accounted for the majority of ULT use between 2009 (95% in the first quarter) and 2019 (92% in the fourth quarter).
Febuxostat use peaked at 10% of all ULT use in 2013 and 2014, after a gradual increase following its introduction into the market in 2009, the researchers noted, but decreased to 6% of all ULT use in the fourth quarter of 2019. Other medications, including probenecid, lesinurad (Zurampic), and pegloticase (Krystexxa), accounted for no more than 5% of ULT use.
When the researchers examined gout patients in particular, they found a slight increase in any ULT use from 567 per 1,000 patients in the first quarter of 2009 to 656 per 1,000 patients in the fourth quarter of 2019.
The study findings were limited by several factors, including potential lack of generalizability to other health plans and lack of adjustment for comorbid conditions, the researchers noted.
However, the results highlight the suboptimal use of ULT as a class and the need to address the treatment gap in gout patients “with appropriate ULT prescribing and monitoring,” they said. “While the decrease in febuxostat use was accompanied by a compensatory increase in allopurinol use, the proportion of patients with gout without any ULT remained high throughout the study period,” they concluded.
The study was supported by the division of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital. Dr. Kim disclosed receiving research grants to Brigham and Women’s Hospital from Pfizer, AbbVie, Roche, and Bristol-Myers Squibb for research unrelated to the current study.
SOURCE: Kim SC et al. Arthritis Rheumatol. 2020 Oct 7. doi: 10.1002/art.41550.
Use of febuxostat (Uloric) decreased among patients with gout in the United States following a Food and Drug Administration–mandated black-box warning that cited cardiovascular concerns, but overall use of urate-lowering therapy remained stable, according to data from a study of commercial insurance enrollees in the United States between 2009 and 2019.
Initiation of urate-lowering therapy (ULT) is recommended for gout patients, and allopurinol remains the first-line treatment, but it is not effective in all patients, and febuxostat was developed as an alternative, wrote Seoyoung C. Kim, MD, ScD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, and colleagues.
However, based on data from a postmarketing safety trial (the CARES trial) mandated by the FDA, the agency first issued a safety announcement about an increased risk of cardiovascular mortality with febuxostat in November 2017, followed in February 2019 with a black-box warning after full CARES trial results were published in March 2018 showing a greater risk of cardiovascular and all-cause mortality in febuxostat versus allopurinol.
In a study published in Arthritis & Rheumatology, the researchers examined trends in the use of ULT before and after the FDA warning. They analyzed claims data from a national commercial health database that included 838,432 adult ULT users and 633,229 gout patients.
Overall, allopurinol accounted for the majority of ULT use between 2009 (95% in the first quarter) and 2019 (92% in the fourth quarter).
Febuxostat use peaked at 10% of all ULT use in 2013 and 2014, after a gradual increase following its introduction into the market in 2009, the researchers noted, but decreased to 6% of all ULT use in the fourth quarter of 2019. Other medications, including probenecid, lesinurad (Zurampic), and pegloticase (Krystexxa), accounted for no more than 5% of ULT use.
When the researchers examined gout patients in particular, they found a slight increase in any ULT use from 567 per 1,000 patients in the first quarter of 2009 to 656 per 1,000 patients in the fourth quarter of 2019.
The study findings were limited by several factors, including potential lack of generalizability to other health plans and lack of adjustment for comorbid conditions, the researchers noted.
However, the results highlight the suboptimal use of ULT as a class and the need to address the treatment gap in gout patients “with appropriate ULT prescribing and monitoring,” they said. “While the decrease in febuxostat use was accompanied by a compensatory increase in allopurinol use, the proportion of patients with gout without any ULT remained high throughout the study period,” they concluded.
The study was supported by the division of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital. Dr. Kim disclosed receiving research grants to Brigham and Women’s Hospital from Pfizer, AbbVie, Roche, and Bristol-Myers Squibb for research unrelated to the current study.
SOURCE: Kim SC et al. Arthritis Rheumatol. 2020 Oct 7. doi: 10.1002/art.41550.
Use of febuxostat (Uloric) decreased among patients with gout in the United States following a Food and Drug Administration–mandated black-box warning that cited cardiovascular concerns, but overall use of urate-lowering therapy remained stable, according to data from a study of commercial insurance enrollees in the United States between 2009 and 2019.
Initiation of urate-lowering therapy (ULT) is recommended for gout patients, and allopurinol remains the first-line treatment, but it is not effective in all patients, and febuxostat was developed as an alternative, wrote Seoyoung C. Kim, MD, ScD, of Brigham and Women’s Hospital and Harvard Medical School, Boston, and colleagues.
However, based on data from a postmarketing safety trial (the CARES trial) mandated by the FDA, the agency first issued a safety announcement about an increased risk of cardiovascular mortality with febuxostat in November 2017, followed in February 2019 with a black-box warning after full CARES trial results were published in March 2018 showing a greater risk of cardiovascular and all-cause mortality in febuxostat versus allopurinol.
In a study published in Arthritis & Rheumatology, the researchers examined trends in the use of ULT before and after the FDA warning. They analyzed claims data from a national commercial health database that included 838,432 adult ULT users and 633,229 gout patients.
Overall, allopurinol accounted for the majority of ULT use between 2009 (95% in the first quarter) and 2019 (92% in the fourth quarter).
Febuxostat use peaked at 10% of all ULT use in 2013 and 2014, after a gradual increase following its introduction into the market in 2009, the researchers noted, but decreased to 6% of all ULT use in the fourth quarter of 2019. Other medications, including probenecid, lesinurad (Zurampic), and pegloticase (Krystexxa), accounted for no more than 5% of ULT use.
When the researchers examined gout patients in particular, they found a slight increase in any ULT use from 567 per 1,000 patients in the first quarter of 2009 to 656 per 1,000 patients in the fourth quarter of 2019.
The study findings were limited by several factors, including potential lack of generalizability to other health plans and lack of adjustment for comorbid conditions, the researchers noted.
However, the results highlight the suboptimal use of ULT as a class and the need to address the treatment gap in gout patients “with appropriate ULT prescribing and monitoring,” they said. “While the decrease in febuxostat use was accompanied by a compensatory increase in allopurinol use, the proportion of patients with gout without any ULT remained high throughout the study period,” they concluded.
The study was supported by the division of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital. Dr. Kim disclosed receiving research grants to Brigham and Women’s Hospital from Pfizer, AbbVie, Roche, and Bristol-Myers Squibb for research unrelated to the current study.
SOURCE: Kim SC et al. Arthritis Rheumatol. 2020 Oct 7. doi: 10.1002/art.41550.
FROM ARTHRITIS & RHEUMATOLOGY