User login
Antibody could reduce bone fractures in MM

Preclinical research suggests a targeted therapy may be able to rebuild and strengthen bone in patients with multiple myeloma (MM).
Researchers tested an antibody targeting the protein sclerostin, an important regulator of bone formation, in mouse models of MM.
The antibody prevented bone loss and even increased bone volume in the mice.
The treatment also made bones more resistant to fracture and worked synergistically with zoledronic acid.
Michelle McDonald, PhD, of the Garvan Institute of Medical Research in Sydney, New South Wales, Australia, and her colleagues reported these findings in Blood.
“Multiple myeloma is a cancer that grows in bone, and, in most patients, it is associated with widespread bone loss and recurrent bone fractures, which can be extremely painful and debilitating,” Dr McDonald said.
“The current treatment for myeloma-associated bone disease with bisphosphonate drugs prevents further bone loss, but it doesn’t fix damaged bones, so patients continue to fracture. We wanted to re-stimulate bone formation and increase bone strength and resistance to fracture.”
So the researchers tested an antibody that targets and neutralizes sclerostin. In clinical studies of osteoporosis, such antibodies have been shown to increase bone mass and reduce fracture incidence.
In the mouse models of MM, the antibody prevented further bone loss and even doubled bone volume in some of the mice.
“When we looked at the bones before and after treatment, the difference was remarkable,” Dr McDonald said. “We saw less lesions or ‘holes’ in the bones after anti-sclerostin treatment. These lesions are the primary cause of bone pain, so this is an extremely important result.”
The treatment also made the bones substantially stronger, with more than double the resistance to fracture observed in many of the tests.
The researchers then combined the antibody with the bisphosphonate zoledronic acid and observed positive results.
“Bisphosphonates work by preventing bone breakdown, so we combined zoledronic acid with the new anti-sclerostin antibody that rebuilds bone,” Dr McDonald said. “Together, the impact on bone thickness, strength, and resistance to fracture was greater than either treatment alone.”
The researchers said these findings suggest a potential new strategy for reducing fractures in patients with MM.
“[M]yelomas, like other cancers, vary from individual to individual and can therefore be difficult to target,” said study author Peter I. Croucher, PhD, of the Garvan Institute of Medical Research.
“By targeting sclerostin, we are blocking a protein that is active in every person’s bones and not something unique to a person’s cancer. Therefore, in the future, when we test this antibody in humans, we are hopeful to see a response in most, if not all, patients.”
“We are now looking towards clinical trials for this antibody and, in the future, development of this type of therapy for the clinical treatment of multiple myeloma. This therapeutic approach has the potential to transform the prognosis for myeloma patients, enhancing quality of life, and ultimately reducing mortality. It also has clinical implications for the treatment of other cancers that develop in the skeleton.” ![]()
Preclinical research suggests a targeted therapy may be able to rebuild and strengthen bone in patients with multiple myeloma (MM).
Researchers tested an antibody targeting the protein sclerostin, an important regulator of bone formation, in mouse models of MM.
The antibody prevented bone loss and even increased bone volume in the mice.
The treatment also made bones more resistant to fracture and worked synergistically with zoledronic acid.
Michelle McDonald, PhD, of the Garvan Institute of Medical Research in Sydney, New South Wales, Australia, and her colleagues reported these findings in Blood.
“Multiple myeloma is a cancer that grows in bone, and, in most patients, it is associated with widespread bone loss and recurrent bone fractures, which can be extremely painful and debilitating,” Dr McDonald said.
“The current treatment for myeloma-associated bone disease with bisphosphonate drugs prevents further bone loss, but it doesn’t fix damaged bones, so patients continue to fracture. We wanted to re-stimulate bone formation and increase bone strength and resistance to fracture.”
So the researchers tested an antibody that targets and neutralizes sclerostin. In clinical studies of osteoporosis, such antibodies have been shown to increase bone mass and reduce fracture incidence.
In the mouse models of MM, the antibody prevented further bone loss and even doubled bone volume in some of the mice.
“When we looked at the bones before and after treatment, the difference was remarkable,” Dr McDonald said. “We saw less lesions or ‘holes’ in the bones after anti-sclerostin treatment. These lesions are the primary cause of bone pain, so this is an extremely important result.”
The treatment also made the bones substantially stronger, with more than double the resistance to fracture observed in many of the tests.
The researchers then combined the antibody with the bisphosphonate zoledronic acid and observed positive results.
“Bisphosphonates work by preventing bone breakdown, so we combined zoledronic acid with the new anti-sclerostin antibody that rebuilds bone,” Dr McDonald said. “Together, the impact on bone thickness, strength, and resistance to fracture was greater than either treatment alone.”
The researchers said these findings suggest a potential new strategy for reducing fractures in patients with MM.
“[M]yelomas, like other cancers, vary from individual to individual and can therefore be difficult to target,” said study author Peter I. Croucher, PhD, of the Garvan Institute of Medical Research.
“By targeting sclerostin, we are blocking a protein that is active in every person’s bones and not something unique to a person’s cancer. Therefore, in the future, when we test this antibody in humans, we are hopeful to see a response in most, if not all, patients.”
“We are now looking towards clinical trials for this antibody and, in the future, development of this type of therapy for the clinical treatment of multiple myeloma. This therapeutic approach has the potential to transform the prognosis for myeloma patients, enhancing quality of life, and ultimately reducing mortality. It also has clinical implications for the treatment of other cancers that develop in the skeleton.” ![]()
Preclinical research suggests a targeted therapy may be able to rebuild and strengthen bone in patients with multiple myeloma (MM).
Researchers tested an antibody targeting the protein sclerostin, an important regulator of bone formation, in mouse models of MM.
The antibody prevented bone loss and even increased bone volume in the mice.
The treatment also made bones more resistant to fracture and worked synergistically with zoledronic acid.
Michelle McDonald, PhD, of the Garvan Institute of Medical Research in Sydney, New South Wales, Australia, and her colleagues reported these findings in Blood.
“Multiple myeloma is a cancer that grows in bone, and, in most patients, it is associated with widespread bone loss and recurrent bone fractures, which can be extremely painful and debilitating,” Dr McDonald said.
“The current treatment for myeloma-associated bone disease with bisphosphonate drugs prevents further bone loss, but it doesn’t fix damaged bones, so patients continue to fracture. We wanted to re-stimulate bone formation and increase bone strength and resistance to fracture.”
So the researchers tested an antibody that targets and neutralizes sclerostin. In clinical studies of osteoporosis, such antibodies have been shown to increase bone mass and reduce fracture incidence.
In the mouse models of MM, the antibody prevented further bone loss and even doubled bone volume in some of the mice.
“When we looked at the bones before and after treatment, the difference was remarkable,” Dr McDonald said. “We saw less lesions or ‘holes’ in the bones after anti-sclerostin treatment. These lesions are the primary cause of bone pain, so this is an extremely important result.”
The treatment also made the bones substantially stronger, with more than double the resistance to fracture observed in many of the tests.
The researchers then combined the antibody with the bisphosphonate zoledronic acid and observed positive results.
“Bisphosphonates work by preventing bone breakdown, so we combined zoledronic acid with the new anti-sclerostin antibody that rebuilds bone,” Dr McDonald said. “Together, the impact on bone thickness, strength, and resistance to fracture was greater than either treatment alone.”
The researchers said these findings suggest a potential new strategy for reducing fractures in patients with MM.
“[M]yelomas, like other cancers, vary from individual to individual and can therefore be difficult to target,” said study author Peter I. Croucher, PhD, of the Garvan Institute of Medical Research.
“By targeting sclerostin, we are blocking a protein that is active in every person’s bones and not something unique to a person’s cancer. Therefore, in the future, when we test this antibody in humans, we are hopeful to see a response in most, if not all, patients.”
“We are now looking towards clinical trials for this antibody and, in the future, development of this type of therapy for the clinical treatment of multiple myeloma. This therapeutic approach has the potential to transform the prognosis for myeloma patients, enhancing quality of life, and ultimately reducing mortality. It also has clinical implications for the treatment of other cancers that develop in the skeleton.” ![]()
From Revved Up to Banged Up
ANSWER
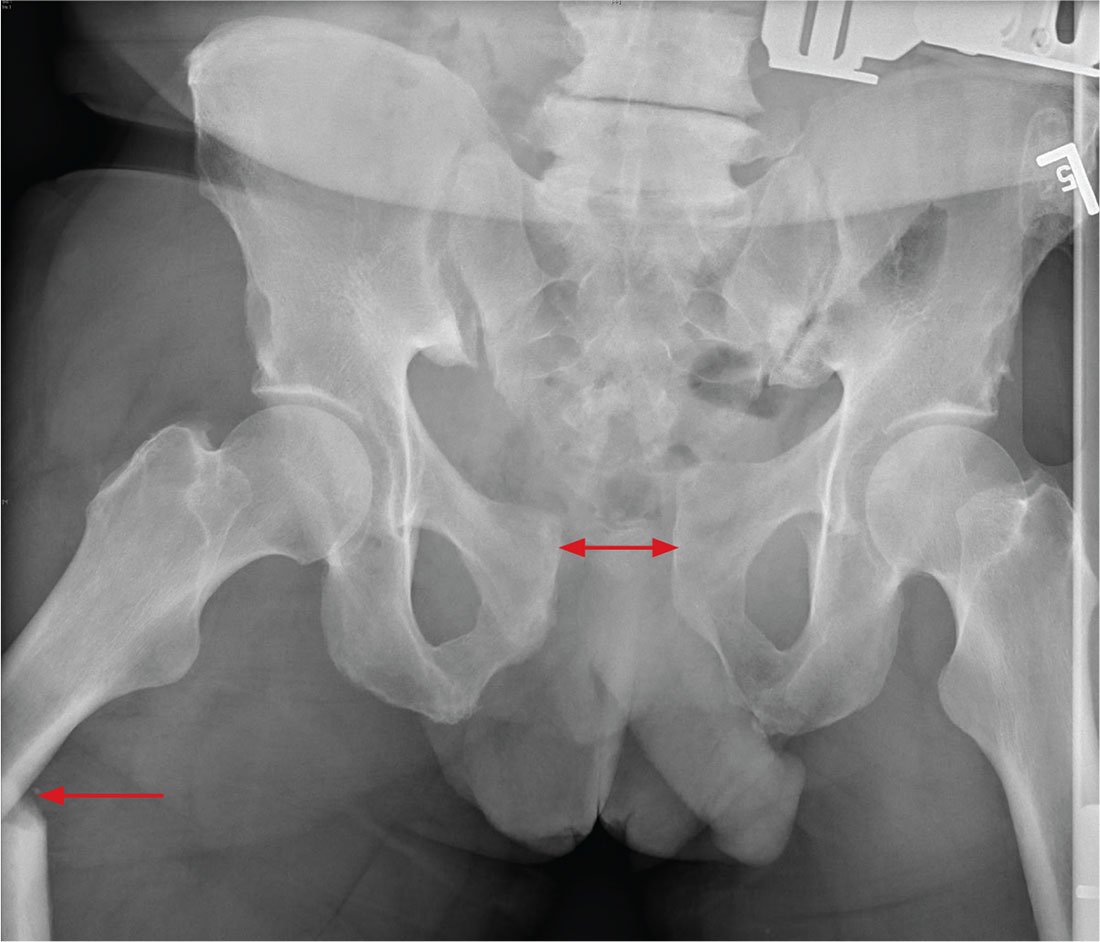
There is significant diastasis of the pubic symphysis, measuring nearly 4 cm. No obvious fractures of the hip or pelvis are seen, but there is malalignment of the rami. There is also evidence of a proximal right femur fracture, although this area is not fully imaged.
Such radiographic findings are typically referred to as an open book pelvic fracture. Usually the result of a shear injury, these fractures are not uncommon and carry an increased risk for pelvic vascular injury.
Emergent orthopedic and vascular consults were obtained, and the patient was placed in a pelvic binder to help reduce the distraction and tamponade any potential vascular injuries.
ANSWER
There is significant diastasis of the pubic symphysis, measuring nearly 4 cm. No obvious fractures of the hip or pelvis are seen, but there is malalignment of the rami. There is also evidence of a proximal right femur fracture, although this area is not fully imaged.
Such radiographic findings are typically referred to as an open book pelvic fracture. Usually the result of a shear injury, these fractures are not uncommon and carry an increased risk for pelvic vascular injury.
Emergent orthopedic and vascular consults were obtained, and the patient was placed in a pelvic binder to help reduce the distraction and tamponade any potential vascular injuries.
ANSWER
There is significant diastasis of the pubic symphysis, measuring nearly 4 cm. No obvious fractures of the hip or pelvis are seen, but there is malalignment of the rami. There is also evidence of a proximal right femur fracture, although this area is not fully imaged.
Such radiographic findings are typically referred to as an open book pelvic fracture. Usually the result of a shear injury, these fractures are not uncommon and carry an increased risk for pelvic vascular injury.
Emergent orthopedic and vascular consults were obtained, and the patient was placed in a pelvic binder to help reduce the distraction and tamponade any potential vascular injuries.
A man, approximately 60 years old, is brought to your facility as a trauma code following a vehicular accident. He was riding a motorcycle when he crashed into another vehicle and was thrown off. Emergency medical personnel report that the patient has obvious head, facial, and extremity trauma. Due to decreased level of consciousness, he was intubated en route. History is otherwise unknown.
Upon arrival, he has two large-bore IVs in place, with fluids going wide open. Despite that, his systolic blood pressure is 90 mm Hg and his heart rate is in the range of 150-160 beats/min. The massive transfusion protocol has been initiated to aid in the aggressive resuscitation efforts.
Portable radiographs of the chest and pelvis are obtained; the latter is shown. What is your impression?
Lithoplasty tames heavily calcified coronary lesions
PARIS – A novel therapeutic ultrasound-based technology known as lithoplasty is turning heads in interventional cardiology and vascular medicine because it addresses the bane of interventionalists’ existence: complex, heavily calcified coronary and peripheral artery lesions.
“Calcification is something we deal with every day in interventional cardiology. It makes the procedures more expensive, longer, and in fact several recent studies have shown that the complication rate for calcified lesions is higher than for any other lesion subtype. Calcification is the next big thing that we’re trying to take on in interventional cardiology,” Todd J. Brinton, MD, observed at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
At EuroPCR, he presented the results of DISRUPT CAD, a seven-center study in which 60 patients with heavily calcified coronary lesions underwent lithoplasty in order to facilitate stent placement. The study met all of its safety and performance endpoints. As a result, the week prior to EuroPCR the European regulatory agency granted marketing approval for Shockwave Medical’s coronary lithoplasty system; the indication is for coronary vessel preparation prior to stenting. A large phase III U.S. trial aimed at gaining FDA approval is planned.
Moreover, on the basis of the earlier favorable DISRUPT PAD trial, lithoplasty has already been approved for treatment of peripheral artery disease (PAD) in Europe since late 2015 and by the FDA since September 2016. Now underway is DISRUPT PAD III, a large postmarketing randomized trial comparing lithoplasty with conventional balloon angioplasty in patients with heavily calcified PAD, added Dr. Brinton, an interventional cardiologist at Stanford (Calif.) University and cofounder of Shockwave Medical.
Lithoplasty is a potentially transformative technology which he described as “lithotripsy inside a balloon.” Lithotripsy has an established 30-year track record for the safe treatment of kidney stones. However, lithotripsy utilizes focused ultrasound, while lithoplasty relies upon circumferential unfocused therapeutic ultrasound delivered by miniaturized emitters placed inside a 12-mm intravascular balloon. The balloon is crossed to the target lesion, inflated to a modest pressure of 4 atmospheres, then the operator delivers lithoplasty pulses lasting over 1 microsec in duration at a rate of 1/sec for 10 seconds in order to fracture the thick intramedial calcium plaque, allowing the lesion to open up and thereby normalize vessel compliance.
“Once you’ve cracked the calcium you can easily dilate the lesion. It’s the calcium that’s restricting the ability to dilate. The real fundamental need here is to maximize acute gain to get really good stent apposition. We’re trying to get expansion,” the cardiologist explained.
That was readily achieved in the DISRUPT CAD study. The 60 participants had reference vessel diameters of 2.5-4.0 mm, with an average target lesion length of 20 mm. The calcification was heavy, covering on average 270 degrees of the vessel circumference as measured by optical coherence tomography, with an average calcium thickness of 0.97 mm and a calcified segment length of 22.3 mm.
The mean stent expansion was 112%. The minimum luminal diameter improved from 0.9 mm pretreatment to 2.6 mm post treatment, for an acute gain of 1.7 mm. The amount of acute gain was similar across the full range of vessel diameters.
The mean diameter stenosis went from 68% pretreatment to 13% post-treatment.
The primary safety endpoint was the 30-day rate of MACE, defined as cardiac death, MI, or target vessel revascularization. The rate was 5%, consisting of 3 patients with mild non–Q-wave MI defined by creatine kinase–MB elevations more than three times the upper limit of normal. The 6-month MACE rate was 8.5%, which included the three non–Q-wave MIs plus two cardiac deaths not related to the procedure or technology.
Final angiographic results adjudicated in a central core laboratory showed no perforations, abrupt closures, slow or no reflow events, or residual dissections. These are complications commonly seen with debulking devices such as rotational or orbital atherectomy, Dr. Brinton noted.
The primary performance endpoint in DISRUPT CAD was clinical success, defined as a residual stenosis of less than 50% post PCI with no in-hospital MACE. This was achieved in 57 of 60 patients, or 95%. The device was successfully delivered to the target lesion with subsequent performance of lithoplasty in 59 of 60 patients. An even more flexible and deliverable device will be released in the coming year, according to the cardiologist.
“I’d say the take-home is that the disease has changed,” Dr. Brinton commented. “It’s not the same disease that we had when Gruentzig did his first balloon angioplasty. These lesions are more calcified, more complex, yet for the most part we use the same balloon we’ve been using for the last 40 years. So lithoplasty is really an attempt to modernize the therapy in a new patient subset we now take care of who are much more complicated than the patients we originally took care of.”
“The reality is, we’re having difficulty taking care of these patients. For myself as an interventionalist, it’s not uncommon to look around the table and see a massive amount of tools when we’re doing these complex cases. Lithoplasty is intended to bring the simplicity. I would say it’s not necessarily to make the best operators better, it’s to bring all operators up to the ability to take on these complex lesions that are now usually reserved for high-volume centers that can do debulking,” he added.
Session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn., pronounced lithoplasty “tremendously exciting.” He and the other panelists focused on questions of safety and potential collateral damage: Where does the calcified debris go? What are the effects of the unfocused sonic pressure waves on noncalcified plaque? How hot does the vessel get?
Dr. Brinton replied that thick calcium plaque is located mostly in the medial vessel wall and stays there after fracturing. That’s why distal embolization wasn’t an issue in DISRUPT CAD. In animal studies, even at 20 times the energy dose used in clinical practice, lithoplasty had no effect on softer, noncalcified plaque or normal tissue. Vessel temperature increases by about 1.2 degrees C during lithoplasty, which isn’t sufficient to cause injury or drive restenosis.
Elsewhere at EuroPCR, Alberto Cremonesi, MD, who chaired a press conference where Dr. Brinton presented highlights of DISRUPT CAD, declared lithoplasty is “in my mind a real breakthrough, not only for coronary disease but also for PAD.”
Is it possible that stand-alone lithoplasty could reduce the need for multiple stents in longer coronary lesions, instead making possible more focal stenting? asked Dr. Cremonesi of Maria Cecilia Hospital in Cotignola, Italy.
That’s one of several possibilities worthy of future investigation, Dr. Brinton replied. Lithoplasty might also facilitate the results obtainable with bioresorbable coronary scaffolds or drug-coated balloons, he added.
He noted that as cofounder of and a consultant to Shockwave Medical, he has a sizable financial involvement with the company.
PARIS – A novel therapeutic ultrasound-based technology known as lithoplasty is turning heads in interventional cardiology and vascular medicine because it addresses the bane of interventionalists’ existence: complex, heavily calcified coronary and peripheral artery lesions.
“Calcification is something we deal with every day in interventional cardiology. It makes the procedures more expensive, longer, and in fact several recent studies have shown that the complication rate for calcified lesions is higher than for any other lesion subtype. Calcification is the next big thing that we’re trying to take on in interventional cardiology,” Todd J. Brinton, MD, observed at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
At EuroPCR, he presented the results of DISRUPT CAD, a seven-center study in which 60 patients with heavily calcified coronary lesions underwent lithoplasty in order to facilitate stent placement. The study met all of its safety and performance endpoints. As a result, the week prior to EuroPCR the European regulatory agency granted marketing approval for Shockwave Medical’s coronary lithoplasty system; the indication is for coronary vessel preparation prior to stenting. A large phase III U.S. trial aimed at gaining FDA approval is planned.
Moreover, on the basis of the earlier favorable DISRUPT PAD trial, lithoplasty has already been approved for treatment of peripheral artery disease (PAD) in Europe since late 2015 and by the FDA since September 2016. Now underway is DISRUPT PAD III, a large postmarketing randomized trial comparing lithoplasty with conventional balloon angioplasty in patients with heavily calcified PAD, added Dr. Brinton, an interventional cardiologist at Stanford (Calif.) University and cofounder of Shockwave Medical.
Lithoplasty is a potentially transformative technology which he described as “lithotripsy inside a balloon.” Lithotripsy has an established 30-year track record for the safe treatment of kidney stones. However, lithotripsy utilizes focused ultrasound, while lithoplasty relies upon circumferential unfocused therapeutic ultrasound delivered by miniaturized emitters placed inside a 12-mm intravascular balloon. The balloon is crossed to the target lesion, inflated to a modest pressure of 4 atmospheres, then the operator delivers lithoplasty pulses lasting over 1 microsec in duration at a rate of 1/sec for 10 seconds in order to fracture the thick intramedial calcium plaque, allowing the lesion to open up and thereby normalize vessel compliance.
“Once you’ve cracked the calcium you can easily dilate the lesion. It’s the calcium that’s restricting the ability to dilate. The real fundamental need here is to maximize acute gain to get really good stent apposition. We’re trying to get expansion,” the cardiologist explained.
That was readily achieved in the DISRUPT CAD study. The 60 participants had reference vessel diameters of 2.5-4.0 mm, with an average target lesion length of 20 mm. The calcification was heavy, covering on average 270 degrees of the vessel circumference as measured by optical coherence tomography, with an average calcium thickness of 0.97 mm and a calcified segment length of 22.3 mm.
The mean stent expansion was 112%. The minimum luminal diameter improved from 0.9 mm pretreatment to 2.6 mm post treatment, for an acute gain of 1.7 mm. The amount of acute gain was similar across the full range of vessel diameters.
The mean diameter stenosis went from 68% pretreatment to 13% post-treatment.
The primary safety endpoint was the 30-day rate of MACE, defined as cardiac death, MI, or target vessel revascularization. The rate was 5%, consisting of 3 patients with mild non–Q-wave MI defined by creatine kinase–MB elevations more than three times the upper limit of normal. The 6-month MACE rate was 8.5%, which included the three non–Q-wave MIs plus two cardiac deaths not related to the procedure or technology.
Final angiographic results adjudicated in a central core laboratory showed no perforations, abrupt closures, slow or no reflow events, or residual dissections. These are complications commonly seen with debulking devices such as rotational or orbital atherectomy, Dr. Brinton noted.
The primary performance endpoint in DISRUPT CAD was clinical success, defined as a residual stenosis of less than 50% post PCI with no in-hospital MACE. This was achieved in 57 of 60 patients, or 95%. The device was successfully delivered to the target lesion with subsequent performance of lithoplasty in 59 of 60 patients. An even more flexible and deliverable device will be released in the coming year, according to the cardiologist.
“I’d say the take-home is that the disease has changed,” Dr. Brinton commented. “It’s not the same disease that we had when Gruentzig did his first balloon angioplasty. These lesions are more calcified, more complex, yet for the most part we use the same balloon we’ve been using for the last 40 years. So lithoplasty is really an attempt to modernize the therapy in a new patient subset we now take care of who are much more complicated than the patients we originally took care of.”
“The reality is, we’re having difficulty taking care of these patients. For myself as an interventionalist, it’s not uncommon to look around the table and see a massive amount of tools when we’re doing these complex cases. Lithoplasty is intended to bring the simplicity. I would say it’s not necessarily to make the best operators better, it’s to bring all operators up to the ability to take on these complex lesions that are now usually reserved for high-volume centers that can do debulking,” he added.
Session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn., pronounced lithoplasty “tremendously exciting.” He and the other panelists focused on questions of safety and potential collateral damage: Where does the calcified debris go? What are the effects of the unfocused sonic pressure waves on noncalcified plaque? How hot does the vessel get?
Dr. Brinton replied that thick calcium plaque is located mostly in the medial vessel wall and stays there after fracturing. That’s why distal embolization wasn’t an issue in DISRUPT CAD. In animal studies, even at 20 times the energy dose used in clinical practice, lithoplasty had no effect on softer, noncalcified plaque or normal tissue. Vessel temperature increases by about 1.2 degrees C during lithoplasty, which isn’t sufficient to cause injury or drive restenosis.
Elsewhere at EuroPCR, Alberto Cremonesi, MD, who chaired a press conference where Dr. Brinton presented highlights of DISRUPT CAD, declared lithoplasty is “in my mind a real breakthrough, not only for coronary disease but also for PAD.”
Is it possible that stand-alone lithoplasty could reduce the need for multiple stents in longer coronary lesions, instead making possible more focal stenting? asked Dr. Cremonesi of Maria Cecilia Hospital in Cotignola, Italy.
That’s one of several possibilities worthy of future investigation, Dr. Brinton replied. Lithoplasty might also facilitate the results obtainable with bioresorbable coronary scaffolds or drug-coated balloons, he added.
He noted that as cofounder of and a consultant to Shockwave Medical, he has a sizable financial involvement with the company.
PARIS – A novel therapeutic ultrasound-based technology known as lithoplasty is turning heads in interventional cardiology and vascular medicine because it addresses the bane of interventionalists’ existence: complex, heavily calcified coronary and peripheral artery lesions.
“Calcification is something we deal with every day in interventional cardiology. It makes the procedures more expensive, longer, and in fact several recent studies have shown that the complication rate for calcified lesions is higher than for any other lesion subtype. Calcification is the next big thing that we’re trying to take on in interventional cardiology,” Todd J. Brinton, MD, observed at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
At EuroPCR, he presented the results of DISRUPT CAD, a seven-center study in which 60 patients with heavily calcified coronary lesions underwent lithoplasty in order to facilitate stent placement. The study met all of its safety and performance endpoints. As a result, the week prior to EuroPCR the European regulatory agency granted marketing approval for Shockwave Medical’s coronary lithoplasty system; the indication is for coronary vessel preparation prior to stenting. A large phase III U.S. trial aimed at gaining FDA approval is planned.
Moreover, on the basis of the earlier favorable DISRUPT PAD trial, lithoplasty has already been approved for treatment of peripheral artery disease (PAD) in Europe since late 2015 and by the FDA since September 2016. Now underway is DISRUPT PAD III, a large postmarketing randomized trial comparing lithoplasty with conventional balloon angioplasty in patients with heavily calcified PAD, added Dr. Brinton, an interventional cardiologist at Stanford (Calif.) University and cofounder of Shockwave Medical.
Lithoplasty is a potentially transformative technology which he described as “lithotripsy inside a balloon.” Lithotripsy has an established 30-year track record for the safe treatment of kidney stones. However, lithotripsy utilizes focused ultrasound, while lithoplasty relies upon circumferential unfocused therapeutic ultrasound delivered by miniaturized emitters placed inside a 12-mm intravascular balloon. The balloon is crossed to the target lesion, inflated to a modest pressure of 4 atmospheres, then the operator delivers lithoplasty pulses lasting over 1 microsec in duration at a rate of 1/sec for 10 seconds in order to fracture the thick intramedial calcium plaque, allowing the lesion to open up and thereby normalize vessel compliance.
“Once you’ve cracked the calcium you can easily dilate the lesion. It’s the calcium that’s restricting the ability to dilate. The real fundamental need here is to maximize acute gain to get really good stent apposition. We’re trying to get expansion,” the cardiologist explained.
That was readily achieved in the DISRUPT CAD study. The 60 participants had reference vessel diameters of 2.5-4.0 mm, with an average target lesion length of 20 mm. The calcification was heavy, covering on average 270 degrees of the vessel circumference as measured by optical coherence tomography, with an average calcium thickness of 0.97 mm and a calcified segment length of 22.3 mm.
The mean stent expansion was 112%. The minimum luminal diameter improved from 0.9 mm pretreatment to 2.6 mm post treatment, for an acute gain of 1.7 mm. The amount of acute gain was similar across the full range of vessel diameters.
The mean diameter stenosis went from 68% pretreatment to 13% post-treatment.
The primary safety endpoint was the 30-day rate of MACE, defined as cardiac death, MI, or target vessel revascularization. The rate was 5%, consisting of 3 patients with mild non–Q-wave MI defined by creatine kinase–MB elevations more than three times the upper limit of normal. The 6-month MACE rate was 8.5%, which included the three non–Q-wave MIs plus two cardiac deaths not related to the procedure or technology.
Final angiographic results adjudicated in a central core laboratory showed no perforations, abrupt closures, slow or no reflow events, or residual dissections. These are complications commonly seen with debulking devices such as rotational or orbital atherectomy, Dr. Brinton noted.
The primary performance endpoint in DISRUPT CAD was clinical success, defined as a residual stenosis of less than 50% post PCI with no in-hospital MACE. This was achieved in 57 of 60 patients, or 95%. The device was successfully delivered to the target lesion with subsequent performance of lithoplasty in 59 of 60 patients. An even more flexible and deliverable device will be released in the coming year, according to the cardiologist.
“I’d say the take-home is that the disease has changed,” Dr. Brinton commented. “It’s not the same disease that we had when Gruentzig did his first balloon angioplasty. These lesions are more calcified, more complex, yet for the most part we use the same balloon we’ve been using for the last 40 years. So lithoplasty is really an attempt to modernize the therapy in a new patient subset we now take care of who are much more complicated than the patients we originally took care of.”
“The reality is, we’re having difficulty taking care of these patients. For myself as an interventionalist, it’s not uncommon to look around the table and see a massive amount of tools when we’re doing these complex cases. Lithoplasty is intended to bring the simplicity. I would say it’s not necessarily to make the best operators better, it’s to bring all operators up to the ability to take on these complex lesions that are now usually reserved for high-volume centers that can do debulking,” he added.
Session cochair David R. Holmes Jr., MD, of the Mayo Clinic in Rochester, Minn., pronounced lithoplasty “tremendously exciting.” He and the other panelists focused on questions of safety and potential collateral damage: Where does the calcified debris go? What are the effects of the unfocused sonic pressure waves on noncalcified plaque? How hot does the vessel get?
Dr. Brinton replied that thick calcium plaque is located mostly in the medial vessel wall and stays there after fracturing. That’s why distal embolization wasn’t an issue in DISRUPT CAD. In animal studies, even at 20 times the energy dose used in clinical practice, lithoplasty had no effect on softer, noncalcified plaque or normal tissue. Vessel temperature increases by about 1.2 degrees C during lithoplasty, which isn’t sufficient to cause injury or drive restenosis.
Elsewhere at EuroPCR, Alberto Cremonesi, MD, who chaired a press conference where Dr. Brinton presented highlights of DISRUPT CAD, declared lithoplasty is “in my mind a real breakthrough, not only for coronary disease but also for PAD.”
Is it possible that stand-alone lithoplasty could reduce the need for multiple stents in longer coronary lesions, instead making possible more focal stenting? asked Dr. Cremonesi of Maria Cecilia Hospital in Cotignola, Italy.
That’s one of several possibilities worthy of future investigation, Dr. Brinton replied. Lithoplasty might also facilitate the results obtainable with bioresorbable coronary scaffolds or drug-coated balloons, he added.
He noted that as cofounder of and a consultant to Shockwave Medical, he has a sizable financial involvement with the company.
AT EUROPCR
Key clinical point:
Major finding: Lithoplasty of heavily calcified coronary lesions improved the minimum luminal diameter from 0.9 mm pretreatment to 2.6 mm post-treatment, for an immediate gain of 1.7 mm prior to stent placement.
Data source: This study featured 6-month follow-up of 60 patients with heavily calcified coronary lesions who underwent lithoplasty followed by stenting.
Disclosures: The DISRUPT CAD study was sponsored by Shockwave Medical, which is developing lithoplasty. The presenter cofounded the company.
Common insurance plans exclude NCI, NCCN cancer centers

Narrow insurance plan coverage may prevent US cancer patients from receiving care at “high-quality” cancer centers, according to research published in the Journal of Clinical Oncology.
Researchers found that “narrow network” insurance plans—lower-premium plans with reduced access to certain providers—are more likely to exclude doctors associated with National Cancer Institute (NCI) and National Comprehensive Cancer Network (NCCN) cancer centers.
These centers are recognized for their scientific and research leadership, quality and safety initiatives, and access to expert physicians and clinical trials.
NCCN member institutions are particularly recognized for higher-quality care, and treatment at NCI-designated cancer centers is associated with lower mortality than other hospitals, particularly among more severely ill patients and those with more advanced disease.
For this study, researchers examined cancer provider networks offered on the 2014 individual health insurance exchanges and then determined which oncologists were affiliated with NCI-designated and NCCN cancer centers.
The researchers found that narrower networks were less likely to include physicians associated with NCI-designated and NCCN member institutions.
“To see such a robust result was surprising,” said study author Laura Yasaitis, PhD, of the University of Pennsylvania in Philadelphia.
“The finding that narrower networks were more likely to exclude NCI and NCCN oncologists was consistent no matter how we looked at it. This is not just a few networks. It’s a clear trend.”
The researchers said the results point to 2 major problems—transparency and access.
“Patients should be able to easily figure out whether the physicians they might need will be covered under a given plan,” said study author Justin E. Bekelman, MD, of the University of Pennsylvania.
The researchers suggested that insurers report doctor’s affiliations with NCI and NCCN cancer centers so that consumers can make more informed choices.
The team also suggested that insurers offer mechanisms that would allow patients to seek care out of network without incurring penalties in exceptional circumstances.
“If patients have narrow network plans and absolutely need the kind of complex cancer care that they can only receive from one of these providers, there should be a standard exception process to allow patients to access the care they need,” Dr Bekelman said. ![]()
Narrow insurance plan coverage may prevent US cancer patients from receiving care at “high-quality” cancer centers, according to research published in the Journal of Clinical Oncology.
Researchers found that “narrow network” insurance plans—lower-premium plans with reduced access to certain providers—are more likely to exclude doctors associated with National Cancer Institute (NCI) and National Comprehensive Cancer Network (NCCN) cancer centers.
These centers are recognized for their scientific and research leadership, quality and safety initiatives, and access to expert physicians and clinical trials.
NCCN member institutions are particularly recognized for higher-quality care, and treatment at NCI-designated cancer centers is associated with lower mortality than other hospitals, particularly among more severely ill patients and those with more advanced disease.
For this study, researchers examined cancer provider networks offered on the 2014 individual health insurance exchanges and then determined which oncologists were affiliated with NCI-designated and NCCN cancer centers.
The researchers found that narrower networks were less likely to include physicians associated with NCI-designated and NCCN member institutions.
“To see such a robust result was surprising,” said study author Laura Yasaitis, PhD, of the University of Pennsylvania in Philadelphia.
“The finding that narrower networks were more likely to exclude NCI and NCCN oncologists was consistent no matter how we looked at it. This is not just a few networks. It’s a clear trend.”
The researchers said the results point to 2 major problems—transparency and access.
“Patients should be able to easily figure out whether the physicians they might need will be covered under a given plan,” said study author Justin E. Bekelman, MD, of the University of Pennsylvania.
The researchers suggested that insurers report doctor’s affiliations with NCI and NCCN cancer centers so that consumers can make more informed choices.
The team also suggested that insurers offer mechanisms that would allow patients to seek care out of network without incurring penalties in exceptional circumstances.
“If patients have narrow network plans and absolutely need the kind of complex cancer care that they can only receive from one of these providers, there should be a standard exception process to allow patients to access the care they need,” Dr Bekelman said. ![]()
Narrow insurance plan coverage may prevent US cancer patients from receiving care at “high-quality” cancer centers, according to research published in the Journal of Clinical Oncology.
Researchers found that “narrow network” insurance plans—lower-premium plans with reduced access to certain providers—are more likely to exclude doctors associated with National Cancer Institute (NCI) and National Comprehensive Cancer Network (NCCN) cancer centers.
These centers are recognized for their scientific and research leadership, quality and safety initiatives, and access to expert physicians and clinical trials.
NCCN member institutions are particularly recognized for higher-quality care, and treatment at NCI-designated cancer centers is associated with lower mortality than other hospitals, particularly among more severely ill patients and those with more advanced disease.
For this study, researchers examined cancer provider networks offered on the 2014 individual health insurance exchanges and then determined which oncologists were affiliated with NCI-designated and NCCN cancer centers.
The researchers found that narrower networks were less likely to include physicians associated with NCI-designated and NCCN member institutions.
“To see such a robust result was surprising,” said study author Laura Yasaitis, PhD, of the University of Pennsylvania in Philadelphia.
“The finding that narrower networks were more likely to exclude NCI and NCCN oncologists was consistent no matter how we looked at it. This is not just a few networks. It’s a clear trend.”
The researchers said the results point to 2 major problems—transparency and access.
“Patients should be able to easily figure out whether the physicians they might need will be covered under a given plan,” said study author Justin E. Bekelman, MD, of the University of Pennsylvania.
The researchers suggested that insurers report doctor’s affiliations with NCI and NCCN cancer centers so that consumers can make more informed choices.
The team also suggested that insurers offer mechanisms that would allow patients to seek care out of network without incurring penalties in exceptional circumstances.
“If patients have narrow network plans and absolutely need the kind of complex cancer care that they can only receive from one of these providers, there should be a standard exception process to allow patients to access the care they need,” Dr Bekelman said. ![]()
New one-time treatment for head lice found safe for children
CHICAGO – A novel one-time topical treatment for head lice, abametapir, was well tolerated in children as young as 6 months, according to pooled results from 11 clinical trials.
The pooled safety data included results from 11 clinical trials including 1,372 patients. Of these, 700 were aged 6 months to 17 years, and patients were exposed to the novel metalloproteinase inhibitor for 10-20 minutes.
In examining safety data from the pooled trials, Lydie Hazan, MD, of Axis Clinical Trials and her collaborators found that for pediatric patients, most treatment-emergent adverse events (AEs) were skin and subcutaneous tissue-related. The most common AEs were erythema, rash, a burning sensation on the skin, and contact dermatitis.
Data for the three phase II pharmacokinetic trials, the phase II ovicidal efficacy trial, and the two phase III trials were reported separately by Dr. Hazan and her coauthors in a poster presentation at the World Congress of Pediatric Dermatology. The overall incidence of treatment-emergent AEs in the studies ranged from 20% to 29% for patients in the active arms of the trials. For patients who received the vehicle lotion only, the incidence of AEs ranged from 16% to 57%.
Of the 11 trials, 6 involved pediatric patients, with one phase IIB trial, one phase II ovicidal trial, two maximal-use open-label trials, and two phase III randomized, double-blind, vehicle-controlled trials. Of the 920 patients, in most of the trials they received a 10-minute exposure to the study drug (489 received abametapir lotion 0.74%, 431 received vehicle lotion).
Looking just at the phase III trials, 24% of patients in the abametapir arm reported AEs, while 19% of those receiving vehicle reported any AE.
In the two maximal-use pediatric trials, drug exposure ranged from 3.3 g to 200.8 g; AEs in these two trials occurred in 23% of participants.
Safety data collected for all studies also included vital signs, results of physical exams, and laboratory tests; no “clinically meaningful” changes were seen in any of the trials for any of these values, according to Dr. Hazan and her coauthors.
“AEs were mild, not age-related, and primarily in the system organ class of skin and subcutaneous tissue disorders,” said Dr. Hazan and her coauthors.
Abematapir 0.74% lotion had previously been shown to be an effective ovicidal treatment for head lice when used in a single application; the lotion is intended to be applied at home by the patient or caregiver (J Med Entomol. 2017. 54[1]:167-72).*
Dr. Hazan is employed by Axis clinical trials. Other study authors were employed by Hatchtech, which developed abametapir, and by Promius Pharma/Dr. Reddy’s Laboratories, which plans to market abametapir lotion.
*Correction, 8/7/17: An earlier version of this article had an incorrect citation.
koakes@frontlinemedcom.com
On Twitter @karioakes
CHICAGO – A novel one-time topical treatment for head lice, abametapir, was well tolerated in children as young as 6 months, according to pooled results from 11 clinical trials.
The pooled safety data included results from 11 clinical trials including 1,372 patients. Of these, 700 were aged 6 months to 17 years, and patients were exposed to the novel metalloproteinase inhibitor for 10-20 minutes.
In examining safety data from the pooled trials, Lydie Hazan, MD, of Axis Clinical Trials and her collaborators found that for pediatric patients, most treatment-emergent adverse events (AEs) were skin and subcutaneous tissue-related. The most common AEs were erythema, rash, a burning sensation on the skin, and contact dermatitis.
Data for the three phase II pharmacokinetic trials, the phase II ovicidal efficacy trial, and the two phase III trials were reported separately by Dr. Hazan and her coauthors in a poster presentation at the World Congress of Pediatric Dermatology. The overall incidence of treatment-emergent AEs in the studies ranged from 20% to 29% for patients in the active arms of the trials. For patients who received the vehicle lotion only, the incidence of AEs ranged from 16% to 57%.
Of the 11 trials, 6 involved pediatric patients, with one phase IIB trial, one phase II ovicidal trial, two maximal-use open-label trials, and two phase III randomized, double-blind, vehicle-controlled trials. Of the 920 patients, in most of the trials they received a 10-minute exposure to the study drug (489 received abametapir lotion 0.74%, 431 received vehicle lotion).
Looking just at the phase III trials, 24% of patients in the abametapir arm reported AEs, while 19% of those receiving vehicle reported any AE.
In the two maximal-use pediatric trials, drug exposure ranged from 3.3 g to 200.8 g; AEs in these two trials occurred in 23% of participants.
Safety data collected for all studies also included vital signs, results of physical exams, and laboratory tests; no “clinically meaningful” changes were seen in any of the trials for any of these values, according to Dr. Hazan and her coauthors.
“AEs were mild, not age-related, and primarily in the system organ class of skin and subcutaneous tissue disorders,” said Dr. Hazan and her coauthors.
Abematapir 0.74% lotion had previously been shown to be an effective ovicidal treatment for head lice when used in a single application; the lotion is intended to be applied at home by the patient or caregiver (J Med Entomol. 2017. 54[1]:167-72).*
Dr. Hazan is employed by Axis clinical trials. Other study authors were employed by Hatchtech, which developed abametapir, and by Promius Pharma/Dr. Reddy’s Laboratories, which plans to market abametapir lotion.
*Correction, 8/7/17: An earlier version of this article had an incorrect citation.
koakes@frontlinemedcom.com
On Twitter @karioakes
CHICAGO – A novel one-time topical treatment for head lice, abametapir, was well tolerated in children as young as 6 months, according to pooled results from 11 clinical trials.
The pooled safety data included results from 11 clinical trials including 1,372 patients. Of these, 700 were aged 6 months to 17 years, and patients were exposed to the novel metalloproteinase inhibitor for 10-20 minutes.
In examining safety data from the pooled trials, Lydie Hazan, MD, of Axis Clinical Trials and her collaborators found that for pediatric patients, most treatment-emergent adverse events (AEs) were skin and subcutaneous tissue-related. The most common AEs were erythema, rash, a burning sensation on the skin, and contact dermatitis.
Data for the three phase II pharmacokinetic trials, the phase II ovicidal efficacy trial, and the two phase III trials were reported separately by Dr. Hazan and her coauthors in a poster presentation at the World Congress of Pediatric Dermatology. The overall incidence of treatment-emergent AEs in the studies ranged from 20% to 29% for patients in the active arms of the trials. For patients who received the vehicle lotion only, the incidence of AEs ranged from 16% to 57%.
Of the 11 trials, 6 involved pediatric patients, with one phase IIB trial, one phase II ovicidal trial, two maximal-use open-label trials, and two phase III randomized, double-blind, vehicle-controlled trials. Of the 920 patients, in most of the trials they received a 10-minute exposure to the study drug (489 received abametapir lotion 0.74%, 431 received vehicle lotion).
Looking just at the phase III trials, 24% of patients in the abametapir arm reported AEs, while 19% of those receiving vehicle reported any AE.
In the two maximal-use pediatric trials, drug exposure ranged from 3.3 g to 200.8 g; AEs in these two trials occurred in 23% of participants.
Safety data collected for all studies also included vital signs, results of physical exams, and laboratory tests; no “clinically meaningful” changes were seen in any of the trials for any of these values, according to Dr. Hazan and her coauthors.
“AEs were mild, not age-related, and primarily in the system organ class of skin and subcutaneous tissue disorders,” said Dr. Hazan and her coauthors.
Abematapir 0.74% lotion had previously been shown to be an effective ovicidal treatment for head lice when used in a single application; the lotion is intended to be applied at home by the patient or caregiver (J Med Entomol. 2017. 54[1]:167-72).*
Dr. Hazan is employed by Axis clinical trials. Other study authors were employed by Hatchtech, which developed abametapir, and by Promius Pharma/Dr. Reddy’s Laboratories, which plans to market abametapir lotion.
*Correction, 8/7/17: An earlier version of this article had an incorrect citation.
koakes@frontlinemedcom.com
On Twitter @karioakes
AT WCPD 2017
Key clinical point:
Major finding: In pooled clinical trial data, pediatric patients had adverse events at the same rate as adult patients, with overall rates ranging from 20% to 57%.
Data source: Pooled data from 11 clinical trials including 1,372 patients, 700 of whom were aged 6 months to 17 years.
Disclosures: Dr. Hazan is employed by Axis Clinical Trials. Other study authors were employed by Hatchtech, which developed abametapir, and by Promius Pharma/Dr. Reddy’s Laboratories, which plans to market abametapir lotion.
Isotretinoin patients need not postpone skin surgery
Skin procedures including superficial chemical peels, laser hair removal, minor cutaneous surgery, manual dermabrasion, and fractional ablative and fractional nonablative laser procedures can be performed safely on patients who have recently been or are currently being treated with isotretinoin, according to new recommendations from a consensus panel.
The recommendations were published online in JAMA Dermatology.
Postponing surgical procedures in patients taking isotretinoin because of the potential for keloid formation and delayed wound healing “has persisted despite increasing reports to the contrary,” wrote Leah K. Spring, DO, of Naval Hospital Camp Lejeune, Camp Lejeune, N.C., and her colleagues (JAMA Dermatol. 2017. doi: 10.1001/jamadermatol.2017.2077).
This protocol is based on 11 patients with delayed healing and keloids, the researchers noted. “In our consensus-based assessment, these initial cases presented a hypothesis to be tested, rather than the foundation for medical dogma on which more than 30 years of clinical practice was built,” they wrote.
To establish the current level of evidence for delaying procedures in isotretinoin patients and to make recommendations, an expert panel reviewed data from 32 publications and more than 1,485 procedures. The literature was divided into five areas: dermabrasion, chemical peels, cutaneous surgery, laser hair removal, and ablative/nonablative laser treatments.
The researchers determined that evidence does not support the safety of mechanical dermabrasion or fully ablative laser surgeries for current or recent isotretinoin users. Manual dermabrasion and microdermabrasion were deemed safe for isotretinoin patients based on the latest evidence, however, as were fractional ablative and fractional nonablative procedures.
In addition, the evidence did not support refraining from chemical peels, laser hair removal, or cutaneous surgery for current and recent isotretinoin patients, although the panel recommended additional prospective, controlled clinical trials in these areas.
In the area of cutaneous surgery, the consensus panel also noted the need for “a rigorous evaluation of the aforementioned specific warning that muscle flap insertion should be delayed until the patient displays normal [creatine phosphokinase (CPK)] levels or, at least, CPK levels below twofold of normal.”
The recommendations can be a resource for discussions with patients about the risks of procedures concurrent with isotretinoin, Dr. Spring and her associates emphasized. “For some patients and some conditions, an informed decision may lead to earlier and potentially more effective interventions.”
Lead author Dr. Spring had no relevant financial conflicts to disclose. Several members of the consensus group disclosed relationships with multiple companies including Allergan, Merz, Leo, Promius, Lumenis, Cynosure, and Valeant.
Skin procedures including superficial chemical peels, laser hair removal, minor cutaneous surgery, manual dermabrasion, and fractional ablative and fractional nonablative laser procedures can be performed safely on patients who have recently been or are currently being treated with isotretinoin, according to new recommendations from a consensus panel.
The recommendations were published online in JAMA Dermatology.
Postponing surgical procedures in patients taking isotretinoin because of the potential for keloid formation and delayed wound healing “has persisted despite increasing reports to the contrary,” wrote Leah K. Spring, DO, of Naval Hospital Camp Lejeune, Camp Lejeune, N.C., and her colleagues (JAMA Dermatol. 2017. doi: 10.1001/jamadermatol.2017.2077).
This protocol is based on 11 patients with delayed healing and keloids, the researchers noted. “In our consensus-based assessment, these initial cases presented a hypothesis to be tested, rather than the foundation for medical dogma on which more than 30 years of clinical practice was built,” they wrote.
To establish the current level of evidence for delaying procedures in isotretinoin patients and to make recommendations, an expert panel reviewed data from 32 publications and more than 1,485 procedures. The literature was divided into five areas: dermabrasion, chemical peels, cutaneous surgery, laser hair removal, and ablative/nonablative laser treatments.
The researchers determined that evidence does not support the safety of mechanical dermabrasion or fully ablative laser surgeries for current or recent isotretinoin users. Manual dermabrasion and microdermabrasion were deemed safe for isotretinoin patients based on the latest evidence, however, as were fractional ablative and fractional nonablative procedures.
In addition, the evidence did not support refraining from chemical peels, laser hair removal, or cutaneous surgery for current and recent isotretinoin patients, although the panel recommended additional prospective, controlled clinical trials in these areas.
In the area of cutaneous surgery, the consensus panel also noted the need for “a rigorous evaluation of the aforementioned specific warning that muscle flap insertion should be delayed until the patient displays normal [creatine phosphokinase (CPK)] levels or, at least, CPK levels below twofold of normal.”
The recommendations can be a resource for discussions with patients about the risks of procedures concurrent with isotretinoin, Dr. Spring and her associates emphasized. “For some patients and some conditions, an informed decision may lead to earlier and potentially more effective interventions.”
Lead author Dr. Spring had no relevant financial conflicts to disclose. Several members of the consensus group disclosed relationships with multiple companies including Allergan, Merz, Leo, Promius, Lumenis, Cynosure, and Valeant.
Skin procedures including superficial chemical peels, laser hair removal, minor cutaneous surgery, manual dermabrasion, and fractional ablative and fractional nonablative laser procedures can be performed safely on patients who have recently been or are currently being treated with isotretinoin, according to new recommendations from a consensus panel.
The recommendations were published online in JAMA Dermatology.
Postponing surgical procedures in patients taking isotretinoin because of the potential for keloid formation and delayed wound healing “has persisted despite increasing reports to the contrary,” wrote Leah K. Spring, DO, of Naval Hospital Camp Lejeune, Camp Lejeune, N.C., and her colleagues (JAMA Dermatol. 2017. doi: 10.1001/jamadermatol.2017.2077).
This protocol is based on 11 patients with delayed healing and keloids, the researchers noted. “In our consensus-based assessment, these initial cases presented a hypothesis to be tested, rather than the foundation for medical dogma on which more than 30 years of clinical practice was built,” they wrote.
To establish the current level of evidence for delaying procedures in isotretinoin patients and to make recommendations, an expert panel reviewed data from 32 publications and more than 1,485 procedures. The literature was divided into five areas: dermabrasion, chemical peels, cutaneous surgery, laser hair removal, and ablative/nonablative laser treatments.
The researchers determined that evidence does not support the safety of mechanical dermabrasion or fully ablative laser surgeries for current or recent isotretinoin users. Manual dermabrasion and microdermabrasion were deemed safe for isotretinoin patients based on the latest evidence, however, as were fractional ablative and fractional nonablative procedures.
In addition, the evidence did not support refraining from chemical peels, laser hair removal, or cutaneous surgery for current and recent isotretinoin patients, although the panel recommended additional prospective, controlled clinical trials in these areas.
In the area of cutaneous surgery, the consensus panel also noted the need for “a rigorous evaluation of the aforementioned specific warning that muscle flap insertion should be delayed until the patient displays normal [creatine phosphokinase (CPK)] levels or, at least, CPK levels below twofold of normal.”
The recommendations can be a resource for discussions with patients about the risks of procedures concurrent with isotretinoin, Dr. Spring and her associates emphasized. “For some patients and some conditions, an informed decision may lead to earlier and potentially more effective interventions.”
Lead author Dr. Spring had no relevant financial conflicts to disclose. Several members of the consensus group disclosed relationships with multiple companies including Allergan, Merz, Leo, Promius, Lumenis, Cynosure, and Valeant.
FROM JAMA DERMATOLOGY
Key clinical point:
Major finding: Experts found insufficient evidence to postpone manual dermabrasion, superficial chemical peels, fractional ablative or nonablative laser resurfacing, laser hair removal, or cutaneous surgery in patients taking isotretinoin or those who have taken it within the past 6 to 12 months.
Data source: The data come from 32 clinical publications and a total of 1,485 procedures.
Disclosures: Lead author Dr. Spring had no relevant financial conflicts to disclose. Several members of the consensus group disclosed relationships with multiple companies, including Allergan, Merz, Leo, Promius, Lumenis, Cynosure, and Valeant.
Comorbidities in psoriatic arthritis flag worse prognosis
MADRID – Comorbidities are relatively common in psoriatic arthritis patients, and they are more prevalent in patients with a worse disease course while on initial treatment with a tumor necrosis factor inhibitor, based on data from more than 1,700 Danish patients.
The presence of comorbidities in psoriatic arthritis (PsA) patients on initial tumor necrosis factor inhibitor (TNFi) treatment “was associated with higher disease activity, shorter adherence to the first TNFi, and reduced clinical response,” Lars Erik Kristensen, MD, said at the European Congress of Rheumatology.
To better understand the possible impact of comorbidities on PsA, he and his associates reviewed 1,750 Danish patients with PsA enrolled in a national registry at the time they began treatment with a TNFi. At the time they started treatment, 1,066 (61%) had no comorbidities, 493 (28%) had one comorbidity, and 191 (11%) had two or more comorbidities.
A comparison of the subgroups with no comorbidities and those with two or more showed several important and statistically significant differences in their baseline characteristics. Patients with at least two comorbidities had longer disease duration, and they had more active disease as measured by parameters including the Disease Activity Score 28 and the Health Assessment Questionnaire. Patients with two or more comorbidities also were older and had a higher average body mass index.
Further analyses showed that patients with two or more comorbidities were 72% more like to discontinue their TNFi treatment, compared with patients with no comorbidities – a statistically significant difference, Dr. Kristensen reported.
After 6 months of TNFi treatment, patients with two or more comorbidities had lower rates of achieving the American College of Rheumatology 20%, 50%, or 70% improvement criteria compared with patients with no comorbidities. For example, an ACR20 response occurred in 40% of patients with no comorbidities and in 31% of patients with two or more comorbidities after 6 months in an adjusted analysis.
Dr. Kristensen has been a consultant to or a speaker for several drug companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
MADRID – Comorbidities are relatively common in psoriatic arthritis patients, and they are more prevalent in patients with a worse disease course while on initial treatment with a tumor necrosis factor inhibitor, based on data from more than 1,700 Danish patients.
The presence of comorbidities in psoriatic arthritis (PsA) patients on initial tumor necrosis factor inhibitor (TNFi) treatment “was associated with higher disease activity, shorter adherence to the first TNFi, and reduced clinical response,” Lars Erik Kristensen, MD, said at the European Congress of Rheumatology.
To better understand the possible impact of comorbidities on PsA, he and his associates reviewed 1,750 Danish patients with PsA enrolled in a national registry at the time they began treatment with a TNFi. At the time they started treatment, 1,066 (61%) had no comorbidities, 493 (28%) had one comorbidity, and 191 (11%) had two or more comorbidities.
A comparison of the subgroups with no comorbidities and those with two or more showed several important and statistically significant differences in their baseline characteristics. Patients with at least two comorbidities had longer disease duration, and they had more active disease as measured by parameters including the Disease Activity Score 28 and the Health Assessment Questionnaire. Patients with two or more comorbidities also were older and had a higher average body mass index.
Further analyses showed that patients with two or more comorbidities were 72% more like to discontinue their TNFi treatment, compared with patients with no comorbidities – a statistically significant difference, Dr. Kristensen reported.
After 6 months of TNFi treatment, patients with two or more comorbidities had lower rates of achieving the American College of Rheumatology 20%, 50%, or 70% improvement criteria compared with patients with no comorbidities. For example, an ACR20 response occurred in 40% of patients with no comorbidities and in 31% of patients with two or more comorbidities after 6 months in an adjusted analysis.
Dr. Kristensen has been a consultant to or a speaker for several drug companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
MADRID – Comorbidities are relatively common in psoriatic arthritis patients, and they are more prevalent in patients with a worse disease course while on initial treatment with a tumor necrosis factor inhibitor, based on data from more than 1,700 Danish patients.
The presence of comorbidities in psoriatic arthritis (PsA) patients on initial tumor necrosis factor inhibitor (TNFi) treatment “was associated with higher disease activity, shorter adherence to the first TNFi, and reduced clinical response,” Lars Erik Kristensen, MD, said at the European Congress of Rheumatology.
To better understand the possible impact of comorbidities on PsA, he and his associates reviewed 1,750 Danish patients with PsA enrolled in a national registry at the time they began treatment with a TNFi. At the time they started treatment, 1,066 (61%) had no comorbidities, 493 (28%) had one comorbidity, and 191 (11%) had two or more comorbidities.
A comparison of the subgroups with no comorbidities and those with two or more showed several important and statistically significant differences in their baseline characteristics. Patients with at least two comorbidities had longer disease duration, and they had more active disease as measured by parameters including the Disease Activity Score 28 and the Health Assessment Questionnaire. Patients with two or more comorbidities also were older and had a higher average body mass index.
Further analyses showed that patients with two or more comorbidities were 72% more like to discontinue their TNFi treatment, compared with patients with no comorbidities – a statistically significant difference, Dr. Kristensen reported.
After 6 months of TNFi treatment, patients with two or more comorbidities had lower rates of achieving the American College of Rheumatology 20%, 50%, or 70% improvement criteria compared with patients with no comorbidities. For example, an ACR20 response occurred in 40% of patients with no comorbidities and in 31% of patients with two or more comorbidities after 6 months in an adjusted analysis.
Dr. Kristensen has been a consultant to or a speaker for several drug companies.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AT THE EULAR 2017 CONGRESS
Key clinical point: , compared with patients with no comorbidities.
Major finding: An ACR20 response occurred in 40% of patients with no comorbidities but only 31% of those with two or more comorbidities.
Data source: Review of national registry data for 1,750 Danish psoriatic arthritis patients.
Disclosures: Dr. Kristensen has been a consultant to or a speaker for several drug companies.
Look for comorbidities associated with hidradenitis suppurativa
CHICAGO – Hidradenitis suppurativa in children is often associated with comorbidities, especially obesity and endocrine abnormalities, a retrospective review of cases showed.
“When treating hidradenitis suppurativa, it is imperative to not only treat the skin but also to look for associated comorbidities,” Maria del Carmen Liy-Wong, MD, said in an interview in advance of the World Congress for Pediatric Dermatology.
Of the 41 patients, 78% were girls; the mean age of onset was 11 years, and the mean age at diagnosis was 14 years. A positive family history was found in 24% of cases. The most common cutaneous lesions were papules and pustules (51%), followed by scars (39%), and 88% of patients reported associated tenderness and pain.
After using the Hurley severity grade to classify disease severity, the researchers found that 56% of cases were mild, 32% were moderate, and 12% were severe. Comorbidities were identified in 92% of the cases; the most common was obesity (73%), followed by endocrine abnormalities (29%) and menstrual irregularities (20%). The researchers also found that 70% of patients were treated with a combination of topical and systemic antibiotics, and that early onset of disease correlated with more severe disease (P = .03).
Dr. Liy-Wong acknowledged that the study’s retrospective design is a limitation of the analysis, but she said that a prospective evaluation in planned for the near future.
The study was supported in part by a grant from AbbVie. Dr. Liy-Wong reported having no relevant financial disclosures.
dbrunk@frontlinemedcom.com
CHICAGO – Hidradenitis suppurativa in children is often associated with comorbidities, especially obesity and endocrine abnormalities, a retrospective review of cases showed.
“When treating hidradenitis suppurativa, it is imperative to not only treat the skin but also to look for associated comorbidities,” Maria del Carmen Liy-Wong, MD, said in an interview in advance of the World Congress for Pediatric Dermatology.
Of the 41 patients, 78% were girls; the mean age of onset was 11 years, and the mean age at diagnosis was 14 years. A positive family history was found in 24% of cases. The most common cutaneous lesions were papules and pustules (51%), followed by scars (39%), and 88% of patients reported associated tenderness and pain.
After using the Hurley severity grade to classify disease severity, the researchers found that 56% of cases were mild, 32% were moderate, and 12% were severe. Comorbidities were identified in 92% of the cases; the most common was obesity (73%), followed by endocrine abnormalities (29%) and menstrual irregularities (20%). The researchers also found that 70% of patients were treated with a combination of topical and systemic antibiotics, and that early onset of disease correlated with more severe disease (P = .03).
Dr. Liy-Wong acknowledged that the study’s retrospective design is a limitation of the analysis, but she said that a prospective evaluation in planned for the near future.
The study was supported in part by a grant from AbbVie. Dr. Liy-Wong reported having no relevant financial disclosures.
dbrunk@frontlinemedcom.com
CHICAGO – Hidradenitis suppurativa in children is often associated with comorbidities, especially obesity and endocrine abnormalities, a retrospective review of cases showed.
“When treating hidradenitis suppurativa, it is imperative to not only treat the skin but also to look for associated comorbidities,” Maria del Carmen Liy-Wong, MD, said in an interview in advance of the World Congress for Pediatric Dermatology.
Of the 41 patients, 78% were girls; the mean age of onset was 11 years, and the mean age at diagnosis was 14 years. A positive family history was found in 24% of cases. The most common cutaneous lesions were papules and pustules (51%), followed by scars (39%), and 88% of patients reported associated tenderness and pain.
After using the Hurley severity grade to classify disease severity, the researchers found that 56% of cases were mild, 32% were moderate, and 12% were severe. Comorbidities were identified in 92% of the cases; the most common was obesity (73%), followed by endocrine abnormalities (29%) and menstrual irregularities (20%). The researchers also found that 70% of patients were treated with a combination of topical and systemic antibiotics, and that early onset of disease correlated with more severe disease (P = .03).
Dr. Liy-Wong acknowledged that the study’s retrospective design is a limitation of the analysis, but she said that a prospective evaluation in planned for the near future.
The study was supported in part by a grant from AbbVie. Dr. Liy-Wong reported having no relevant financial disclosures.
dbrunk@frontlinemedcom.com
AT WCPD 2017
Key clinical point:
Major finding: Comorbidities were identified in 92% of the cases, with obesity (73%) the most common.
Data source: A retrospective review of clinical characteristics, degree of severity, comorbidities, and management of hidradenitis suppurativa in 41 patients followed between January 1995 and January 2015.
Disclosures: Dr. Liy-Wong reported having no relevant financial disclosures.
As designer drugs multiply, toxicologists spring into action
SAN DIEGO – Forensic toxicologist Donna Papsun spends her days with drugs, but she doesn’t see patients or try to make anyone better. Still, her work is crucial to every medical professional who needs to know which new illicit drugs their patients have been taking.
In Willow Grove, Pa., a suburb of Philadelphia, Ms. Papsun and her colleagues at NMS Labs develop screening tests for designer drugs that have just appeared on the black market or crossed the Drug Enforcement Administration’s (DEA) radar.
As Ms. Papsun told an audience at the annual meeting of the American Psychiatric Association, she faced a unique obstacle last summer, when an elephant tranquilizer called carfentanil, a derivative of fentanyl, began to make headlines. The obstacle? The U.S.-Canada border.
She wanted to develop a test for the opioid but couldn’t start until she got a reference sample of the controlled substance from carfentanil’s only manufacturer, a firm in Canada. It took months. “Crossing an international border caused all sorts of problems,” she said.
New designer drugs are constantly hitting the market. They’re often especially appealing – and especially risky – because routine drug tests can’t detect them, at least not yet.
In an interview, Ms. Papsun talked about the challenges of trying to keep up with the drug makers – and users.
“Designer drug testing developed back in 2008 will not catch anything that is seen in today’s designer drug market,” she said. “Designer drug testing requires constant attention, assessment, resources, and updating.”
Question: How long does it typically take to create a test for a new strain of illicit drug?
Answer: This can take anywhere from 3 to 9 months, and potentially longer, and it depends on many factors. Once a new drug has hit the market, we check to see if there is certified reference material available. If there is, then we can start to develop a test. Development includes identifying a successful chemical extraction technique – isolating the drug in question from biological matrix such as blood or urine – as well as a platform that reliably detects the drug without falsely reporting positives.
After development, the test has to go through a process called validation, which is a series of experiments to prove that the developed method works rigorously, day after day, and provides the same results. This is very important in forensic toxicology, because our results may be involved in criminal and civil litigation and must stand up to the rigors of court.
Q: What are some examples of the types of drugs that you’ve had to develop tests for?
A: Just in the past year, we have developed tests for new designer opioids (including carfentanil, furanylfentanyl, acrylfentanyl, and U-47700), designer benzodiazepines (including etizolam, diclazepam, flubromazolam, and flubromazepam) and new designer stimulants (including n-ethyl pentylone and dibutylone).
We have a synthetic cannabinoid test that was developed for the first time back in 2010. That test has been redeveloped several times since then, because we constantly have to update the test to keep up with the rapid changes in market availability of substances.
Q: What are some of the challenges that you face in terms of getting samples of human fluids that you can test for the drugs?
A: Most of the samples we see are from either death investigation cases or driving-under-the-influence cases. Samples from intoxications at hospitals are important, because those data help [us] understand the concentrations of drugs at which people can survive. But often, if the patients survive, their biological specimens are not forwarded for specialized toxicology testing. Most hospital systems do not have the analytical capabilities to detect designer drugs, and most lack the resources to seek out the causal agent for an intoxication or apparent overdose.
Q: At the APA meeting, you talked about the risk that you’ll hear about a strain from the DEA, develop a test and find out it’s obsolete because the drug isn’t used anymore. Does that happen very often?
A: Yes. The problem with designer drugs is that there are so many, so you can spend a lot of time, money, and other resources dedicated to developing and validating a test for a drug that may or may not even be popular.
As a business, you have to make decisions regarding prioritization: Do we build a test for a drug that has only been reported once, or do we focus our efforts on a substance that has been reported dozens of times?
We certainly have spent time and resources developing a test that became obsolete, or never reported a positive case. For example, we developed a test for desomorphine, and we have never chemically confirmed desomorphine in a biological specimen. It definitely is hit or miss, but we spend a lot of time and research a lot of different avenues to make educated decisions regarding the substances we develop tests for.
SAN DIEGO – Forensic toxicologist Donna Papsun spends her days with drugs, but she doesn’t see patients or try to make anyone better. Still, her work is crucial to every medical professional who needs to know which new illicit drugs their patients have been taking.
In Willow Grove, Pa., a suburb of Philadelphia, Ms. Papsun and her colleagues at NMS Labs develop screening tests for designer drugs that have just appeared on the black market or crossed the Drug Enforcement Administration’s (DEA) radar.
As Ms. Papsun told an audience at the annual meeting of the American Psychiatric Association, she faced a unique obstacle last summer, when an elephant tranquilizer called carfentanil, a derivative of fentanyl, began to make headlines. The obstacle? The U.S.-Canada border.
She wanted to develop a test for the opioid but couldn’t start until she got a reference sample of the controlled substance from carfentanil’s only manufacturer, a firm in Canada. It took months. “Crossing an international border caused all sorts of problems,” she said.
New designer drugs are constantly hitting the market. They’re often especially appealing – and especially risky – because routine drug tests can’t detect them, at least not yet.
In an interview, Ms. Papsun talked about the challenges of trying to keep up with the drug makers – and users.
“Designer drug testing developed back in 2008 will not catch anything that is seen in today’s designer drug market,” she said. “Designer drug testing requires constant attention, assessment, resources, and updating.”
Question: How long does it typically take to create a test for a new strain of illicit drug?
Answer: This can take anywhere from 3 to 9 months, and potentially longer, and it depends on many factors. Once a new drug has hit the market, we check to see if there is certified reference material available. If there is, then we can start to develop a test. Development includes identifying a successful chemical extraction technique – isolating the drug in question from biological matrix such as blood or urine – as well as a platform that reliably detects the drug without falsely reporting positives.
After development, the test has to go through a process called validation, which is a series of experiments to prove that the developed method works rigorously, day after day, and provides the same results. This is very important in forensic toxicology, because our results may be involved in criminal and civil litigation and must stand up to the rigors of court.
Q: What are some examples of the types of drugs that you’ve had to develop tests for?
A: Just in the past year, we have developed tests for new designer opioids (including carfentanil, furanylfentanyl, acrylfentanyl, and U-47700), designer benzodiazepines (including etizolam, diclazepam, flubromazolam, and flubromazepam) and new designer stimulants (including n-ethyl pentylone and dibutylone).
We have a synthetic cannabinoid test that was developed for the first time back in 2010. That test has been redeveloped several times since then, because we constantly have to update the test to keep up with the rapid changes in market availability of substances.
Q: What are some of the challenges that you face in terms of getting samples of human fluids that you can test for the drugs?
A: Most of the samples we see are from either death investigation cases or driving-under-the-influence cases. Samples from intoxications at hospitals are important, because those data help [us] understand the concentrations of drugs at which people can survive. But often, if the patients survive, their biological specimens are not forwarded for specialized toxicology testing. Most hospital systems do not have the analytical capabilities to detect designer drugs, and most lack the resources to seek out the causal agent for an intoxication or apparent overdose.
Q: At the APA meeting, you talked about the risk that you’ll hear about a strain from the DEA, develop a test and find out it’s obsolete because the drug isn’t used anymore. Does that happen very often?
A: Yes. The problem with designer drugs is that there are so many, so you can spend a lot of time, money, and other resources dedicated to developing and validating a test for a drug that may or may not even be popular.
As a business, you have to make decisions regarding prioritization: Do we build a test for a drug that has only been reported once, or do we focus our efforts on a substance that has been reported dozens of times?
We certainly have spent time and resources developing a test that became obsolete, or never reported a positive case. For example, we developed a test for desomorphine, and we have never chemically confirmed desomorphine in a biological specimen. It definitely is hit or miss, but we spend a lot of time and research a lot of different avenues to make educated decisions regarding the substances we develop tests for.
SAN DIEGO – Forensic toxicologist Donna Papsun spends her days with drugs, but she doesn’t see patients or try to make anyone better. Still, her work is crucial to every medical professional who needs to know which new illicit drugs their patients have been taking.
In Willow Grove, Pa., a suburb of Philadelphia, Ms. Papsun and her colleagues at NMS Labs develop screening tests for designer drugs that have just appeared on the black market or crossed the Drug Enforcement Administration’s (DEA) radar.
As Ms. Papsun told an audience at the annual meeting of the American Psychiatric Association, she faced a unique obstacle last summer, when an elephant tranquilizer called carfentanil, a derivative of fentanyl, began to make headlines. The obstacle? The U.S.-Canada border.
She wanted to develop a test for the opioid but couldn’t start until she got a reference sample of the controlled substance from carfentanil’s only manufacturer, a firm in Canada. It took months. “Crossing an international border caused all sorts of problems,” she said.
New designer drugs are constantly hitting the market. They’re often especially appealing – and especially risky – because routine drug tests can’t detect them, at least not yet.
In an interview, Ms. Papsun talked about the challenges of trying to keep up with the drug makers – and users.
“Designer drug testing developed back in 2008 will not catch anything that is seen in today’s designer drug market,” she said. “Designer drug testing requires constant attention, assessment, resources, and updating.”
Question: How long does it typically take to create a test for a new strain of illicit drug?
Answer: This can take anywhere from 3 to 9 months, and potentially longer, and it depends on many factors. Once a new drug has hit the market, we check to see if there is certified reference material available. If there is, then we can start to develop a test. Development includes identifying a successful chemical extraction technique – isolating the drug in question from biological matrix such as blood or urine – as well as a platform that reliably detects the drug without falsely reporting positives.
After development, the test has to go through a process called validation, which is a series of experiments to prove that the developed method works rigorously, day after day, and provides the same results. This is very important in forensic toxicology, because our results may be involved in criminal and civil litigation and must stand up to the rigors of court.
Q: What are some examples of the types of drugs that you’ve had to develop tests for?
A: Just in the past year, we have developed tests for new designer opioids (including carfentanil, furanylfentanyl, acrylfentanyl, and U-47700), designer benzodiazepines (including etizolam, diclazepam, flubromazolam, and flubromazepam) and new designer stimulants (including n-ethyl pentylone and dibutylone).
We have a synthetic cannabinoid test that was developed for the first time back in 2010. That test has been redeveloped several times since then, because we constantly have to update the test to keep up with the rapid changes in market availability of substances.
Q: What are some of the challenges that you face in terms of getting samples of human fluids that you can test for the drugs?
A: Most of the samples we see are from either death investigation cases or driving-under-the-influence cases. Samples from intoxications at hospitals are important, because those data help [us] understand the concentrations of drugs at which people can survive. But often, if the patients survive, their biological specimens are not forwarded for specialized toxicology testing. Most hospital systems do not have the analytical capabilities to detect designer drugs, and most lack the resources to seek out the causal agent for an intoxication or apparent overdose.
Q: At the APA meeting, you talked about the risk that you’ll hear about a strain from the DEA, develop a test and find out it’s obsolete because the drug isn’t used anymore. Does that happen very often?
A: Yes. The problem with designer drugs is that there are so many, so you can spend a lot of time, money, and other resources dedicated to developing and validating a test for a drug that may or may not even be popular.
As a business, you have to make decisions regarding prioritization: Do we build a test for a drug that has only been reported once, or do we focus our efforts on a substance that has been reported dozens of times?
We certainly have spent time and resources developing a test that became obsolete, or never reported a positive case. For example, we developed a test for desomorphine, and we have never chemically confirmed desomorphine in a biological specimen. It definitely is hit or miss, but we spend a lot of time and research a lot of different avenues to make educated decisions regarding the substances we develop tests for.
EXPERT ANALYSIS FROM APA

FDA approves new treatment for sickle cell disease

The US Food and Drug Administration (FDA) has granted approval for L-glutamine oral powder (Endari), the first treatment approved to treat sickle cell disease (SCD) in the US in nearly 20 years.
L-glutamine oral powder, a product developed by Emmaus Medical Inc., is intended to reduce severe complications of SCD in patients age 5 and older.
The FDA’s approval of L-glutamine was supported by efficacy data from a phase 3 trial.
The trial enrolled 230 adults and children with SCD, and they were randomized to receive L-glutamine or placebo.
Patients who received L-glutamine had fewer sickle cell crises, hospitalizations, cumulative hospital days, and cases of acute chest syndrome than patients who received placebo.
Results from this trial were presented at the 2014 ASH Annual Meeting.
The FDA approval of L-glutamine was also supported by safety data from 298 patients treated with L-glutamine and 111 patients treated with placebo in the phase 2 and phase 3 studies.
Based on these data, L-glutamine was considered well-tolerated in pediatric and adult patients. The most common adverse events (occurring in more than 10% of patients receiving L-glutamine) were constipation, nausea, headache, abdominal pain, cough, pain in extremity, back pain, and chest pain (non-cardiac). ![]()
The US Food and Drug Administration (FDA) has granted approval for L-glutamine oral powder (Endari), the first treatment approved to treat sickle cell disease (SCD) in the US in nearly 20 years.
L-glutamine oral powder, a product developed by Emmaus Medical Inc., is intended to reduce severe complications of SCD in patients age 5 and older.
The FDA’s approval of L-glutamine was supported by efficacy data from a phase 3 trial.
The trial enrolled 230 adults and children with SCD, and they were randomized to receive L-glutamine or placebo.
Patients who received L-glutamine had fewer sickle cell crises, hospitalizations, cumulative hospital days, and cases of acute chest syndrome than patients who received placebo.
Results from this trial were presented at the 2014 ASH Annual Meeting.
The FDA approval of L-glutamine was also supported by safety data from 298 patients treated with L-glutamine and 111 patients treated with placebo in the phase 2 and phase 3 studies.
Based on these data, L-glutamine was considered well-tolerated in pediatric and adult patients. The most common adverse events (occurring in more than 10% of patients receiving L-glutamine) were constipation, nausea, headache, abdominal pain, cough, pain in extremity, back pain, and chest pain (non-cardiac). ![]()
The US Food and Drug Administration (FDA) has granted approval for L-glutamine oral powder (Endari), the first treatment approved to treat sickle cell disease (SCD) in the US in nearly 20 years.
L-glutamine oral powder, a product developed by Emmaus Medical Inc., is intended to reduce severe complications of SCD in patients age 5 and older.
The FDA’s approval of L-glutamine was supported by efficacy data from a phase 3 trial.
The trial enrolled 230 adults and children with SCD, and they were randomized to receive L-glutamine or placebo.
Patients who received L-glutamine had fewer sickle cell crises, hospitalizations, cumulative hospital days, and cases of acute chest syndrome than patients who received placebo.
Results from this trial were presented at the 2014 ASH Annual Meeting.
The FDA approval of L-glutamine was also supported by safety data from 298 patients treated with L-glutamine and 111 patients treated with placebo in the phase 2 and phase 3 studies.
Based on these data, L-glutamine was considered well-tolerated in pediatric and adult patients. The most common adverse events (occurring in more than 10% of patients receiving L-glutamine) were constipation, nausea, headache, abdominal pain, cough, pain in extremity, back pain, and chest pain (non-cardiac). ![]()