User login
Phenytoin trial in optic neuritis hints at neuroprotection
Patients with acute demyelinating optic neuritis who received the anticonvulsant drug phenytoin lost 30% less of their retinal nerve fiber layer than did placebo-treated patients in a randomized, phase II study.
“The results of this clinical trial support the concept of neuroprotection using phenytoin to inhibit voltage-gated sodium channels in patients with acute optic neuritis,” wrote Dr. Rhian Raftopoulos of the National Hospital for Neurology and Neurosurgery, London, and coauthors (Lancet Neurol. 2016 Jan 25. doi: 10.1016/S1474-4422(16)00004-1).

The study in 86 individuals with acute optic neuritis randomized 29 participants to receive 4 mg/kg per day of oral phenytoin, 13 to 6 mg/kg per day of oral phenytoin, and 44 to placebo for 3 months; all were randomized within 14 days of vision loss. One-third of the patients had previously been diagnosed with multiple sclerosis or were diagnosed at presentation, and 74% had at least one brain lesion on MRI.
Treatment with phenytoin resulted in a decline of mean retinal nerve fiber layer thickness in the affected eye from 130.62 mcm at baseline to 81.46 mcm at 6 months, compared with a decline from 125.20 mcm to 74.29 mcm in the placebo group, representing an adjusted mean difference of 7.15 mcm that reached statistical significance.
The researchers also noted a significant 34% reduction in macular volume loss in the treatment arm, compared with placebo, representing an adjusted mean difference of 0.20 mm3. However, the treatment had no significant effect on low-contrast visual acuity and visual evoked potentials.
The most common adverse event in the treatment arm was maculopapular rash, which was judged as severe in one patient treated with phenytoin.
The study was supported by the U.S. National Multiple Sclerosis Society, the Multiple Sclerosis Society of Great Britain and Northern Ireland, Novartis, the U.K. National Institute for Health Research, and the NIHR UCLH/UCL Biomedical Research Centre. Several authors declared personal fees, trial funding, grants, and consultancies for pharmaceutical companies, including Novartis.
The absence of regular, early outcome assessments around 1-2 months after initiation of treatment makes it hard to interpret the results because they would have helped to rule out a primarily anti-inflammatory effect of the treatment by tracking retinal nerve fiber layer (RNFL) swelling and possible optic nerve inflammation, especially given that there was higher baseline RNFL thickness and worse low-contrast visual acuity in the patients who received phenytoin. If the true RNFL thickness at baseline in the affected eye of patients in the phenytoin group was higher than those in the placebo group, it could have accounted for the findings even though the investigators made a prespecified adjustment for it.
Although the results of this study are a major advancement and undeniably encouraging, future studies need to include more frequent OCT sampling, as well as more detailed OCT-segmentation-derived retinal measures such as ganglion cell plus inner plexiform layer thickness, which do not swell during acute optic neuritis, mitigating the need for statistical corrections involving the unaffected eye.
Dr. Shiv Saidha and Dr. Peter A. Calabresi are from the division of neuroimmunology and neurological infections at Johns Hopkins University, Baltimore. These comments were taken from an accompanying editorial (Lancet Neurol. 2016 Jan 25. doi: 10.1016/S1474-4422(16)00024-7). Dr. Saidha declared receiving funding support, consulting fees, grant support, speaking honoraria, and advisory board positions with the pharmaceutical industry, including companies that market MS drugs. Dr. Calabresi declared consultancies, research funding, and advisory board positions with the pharmaceutical industry, including companies that market MS drugs.
The absence of regular, early outcome assessments around 1-2 months after initiation of treatment makes it hard to interpret the results because they would have helped to rule out a primarily anti-inflammatory effect of the treatment by tracking retinal nerve fiber layer (RNFL) swelling and possible optic nerve inflammation, especially given that there was higher baseline RNFL thickness and worse low-contrast visual acuity in the patients who received phenytoin. If the true RNFL thickness at baseline in the affected eye of patients in the phenytoin group was higher than those in the placebo group, it could have accounted for the findings even though the investigators made a prespecified adjustment for it.
Although the results of this study are a major advancement and undeniably encouraging, future studies need to include more frequent OCT sampling, as well as more detailed OCT-segmentation-derived retinal measures such as ganglion cell plus inner plexiform layer thickness, which do not swell during acute optic neuritis, mitigating the need for statistical corrections involving the unaffected eye.
Dr. Shiv Saidha and Dr. Peter A. Calabresi are from the division of neuroimmunology and neurological infections at Johns Hopkins University, Baltimore. These comments were taken from an accompanying editorial (Lancet Neurol. 2016 Jan 25. doi: 10.1016/S1474-4422(16)00024-7). Dr. Saidha declared receiving funding support, consulting fees, grant support, speaking honoraria, and advisory board positions with the pharmaceutical industry, including companies that market MS drugs. Dr. Calabresi declared consultancies, research funding, and advisory board positions with the pharmaceutical industry, including companies that market MS drugs.
The absence of regular, early outcome assessments around 1-2 months after initiation of treatment makes it hard to interpret the results because they would have helped to rule out a primarily anti-inflammatory effect of the treatment by tracking retinal nerve fiber layer (RNFL) swelling and possible optic nerve inflammation, especially given that there was higher baseline RNFL thickness and worse low-contrast visual acuity in the patients who received phenytoin. If the true RNFL thickness at baseline in the affected eye of patients in the phenytoin group was higher than those in the placebo group, it could have accounted for the findings even though the investigators made a prespecified adjustment for it.
Although the results of this study are a major advancement and undeniably encouraging, future studies need to include more frequent OCT sampling, as well as more detailed OCT-segmentation-derived retinal measures such as ganglion cell plus inner plexiform layer thickness, which do not swell during acute optic neuritis, mitigating the need for statistical corrections involving the unaffected eye.
Dr. Shiv Saidha and Dr. Peter A. Calabresi are from the division of neuroimmunology and neurological infections at Johns Hopkins University, Baltimore. These comments were taken from an accompanying editorial (Lancet Neurol. 2016 Jan 25. doi: 10.1016/S1474-4422(16)00024-7). Dr. Saidha declared receiving funding support, consulting fees, grant support, speaking honoraria, and advisory board positions with the pharmaceutical industry, including companies that market MS drugs. Dr. Calabresi declared consultancies, research funding, and advisory board positions with the pharmaceutical industry, including companies that market MS drugs.
Patients with acute demyelinating optic neuritis who received the anticonvulsant drug phenytoin lost 30% less of their retinal nerve fiber layer than did placebo-treated patients in a randomized, phase II study.
“The results of this clinical trial support the concept of neuroprotection using phenytoin to inhibit voltage-gated sodium channels in patients with acute optic neuritis,” wrote Dr. Rhian Raftopoulos of the National Hospital for Neurology and Neurosurgery, London, and coauthors (Lancet Neurol. 2016 Jan 25. doi: 10.1016/S1474-4422(16)00004-1).
The study in 86 individuals with acute optic neuritis randomized 29 participants to receive 4 mg/kg per day of oral phenytoin, 13 to 6 mg/kg per day of oral phenytoin, and 44 to placebo for 3 months; all were randomized within 14 days of vision loss. One-third of the patients had previously been diagnosed with multiple sclerosis or were diagnosed at presentation, and 74% had at least one brain lesion on MRI.
Treatment with phenytoin resulted in a decline of mean retinal nerve fiber layer thickness in the affected eye from 130.62 mcm at baseline to 81.46 mcm at 6 months, compared with a decline from 125.20 mcm to 74.29 mcm in the placebo group, representing an adjusted mean difference of 7.15 mcm that reached statistical significance.
The researchers also noted a significant 34% reduction in macular volume loss in the treatment arm, compared with placebo, representing an adjusted mean difference of 0.20 mm3. However, the treatment had no significant effect on low-contrast visual acuity and visual evoked potentials.
The most common adverse event in the treatment arm was maculopapular rash, which was judged as severe in one patient treated with phenytoin.
The study was supported by the U.S. National Multiple Sclerosis Society, the Multiple Sclerosis Society of Great Britain and Northern Ireland, Novartis, the U.K. National Institute for Health Research, and the NIHR UCLH/UCL Biomedical Research Centre. Several authors declared personal fees, trial funding, grants, and consultancies for pharmaceutical companies, including Novartis.
Patients with acute demyelinating optic neuritis who received the anticonvulsant drug phenytoin lost 30% less of their retinal nerve fiber layer than did placebo-treated patients in a randomized, phase II study.
“The results of this clinical trial support the concept of neuroprotection using phenytoin to inhibit voltage-gated sodium channels in patients with acute optic neuritis,” wrote Dr. Rhian Raftopoulos of the National Hospital for Neurology and Neurosurgery, London, and coauthors (Lancet Neurol. 2016 Jan 25. doi: 10.1016/S1474-4422(16)00004-1).
The study in 86 individuals with acute optic neuritis randomized 29 participants to receive 4 mg/kg per day of oral phenytoin, 13 to 6 mg/kg per day of oral phenytoin, and 44 to placebo for 3 months; all were randomized within 14 days of vision loss. One-third of the patients had previously been diagnosed with multiple sclerosis or were diagnosed at presentation, and 74% had at least one brain lesion on MRI.
Treatment with phenytoin resulted in a decline of mean retinal nerve fiber layer thickness in the affected eye from 130.62 mcm at baseline to 81.46 mcm at 6 months, compared with a decline from 125.20 mcm to 74.29 mcm in the placebo group, representing an adjusted mean difference of 7.15 mcm that reached statistical significance.
The researchers also noted a significant 34% reduction in macular volume loss in the treatment arm, compared with placebo, representing an adjusted mean difference of 0.20 mm3. However, the treatment had no significant effect on low-contrast visual acuity and visual evoked potentials.
The most common adverse event in the treatment arm was maculopapular rash, which was judged as severe in one patient treated with phenytoin.
The study was supported by the U.S. National Multiple Sclerosis Society, the Multiple Sclerosis Society of Great Britain and Northern Ireland, Novartis, the U.K. National Institute for Health Research, and the NIHR UCLH/UCL Biomedical Research Centre. Several authors declared personal fees, trial funding, grants, and consultancies for pharmaceutical companies, including Novartis.
FROM LANCET NEUROLOGY
Key clinical point: Phenytoin treatment is associated with a reduction in retinal nerve fiber layer loss in individuals with demyelinating optic neuritis.
Major finding: Treatment with phenytoin was associated with a 30% reduction in the extent of retinal nerve fiber layer loss, compared with placebo.
Data source: Randomized, placebo-controlled phase II trial in 86 individuals with acute demyelinating optic neuritis.
Disclosures: The study was supported by the U.S. National Multiple Sclerosis Society, the Multiple Sclerosis Society of Great Britain and Northern Ireland, Novartis, the U.K. National Institute for Health Research, and the NIHR UCLH/UCL Biomedical Research Centre. Several authors declared personal fees, trial funding, grants, and consultancies for pharmaceutical companies, including Novartis.

Flibanserin for hypoactive sexual desire disorder in premenopausal women
Flibanserin, FDA-approved in August 2015, is the first medication approved to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women (Table 1). In clinical trials,1-4 the drug has shown modest efficacy in improving symptoms of low sexual desire (number of satisfying sexual events [SSEs], sexual desire, and overall sexual function). Flibanserin is not indicated to enhance sexual performance, for HSDD in postmenopausal women, or in men.

Clinical implications
Flibanserin could help premenopausal women who have distressing low sexual desire, which must be acquired and generalized:
- “Acquired low sexual desire” means that a patient had an adequate sexual desire that decreased or ceased for an unknown reason.
- “Generalized low sexual desire” means that lack of sexual desire occurs all the time and in all situations, not only with a certain partner or in some situations.
Women taking flibanserin could experience gradually increased sexual desire, increase in SSEs, and decrease of sexual distress. Flibanserin is indicated for long-term use; however, it should be discontinued after 8 weeks if the patient does not report any improvement in symptoms.
The number needed to treat with flibanserin likely would be rather large, but it is not available because of complex outcome measures in clinical trials. Flibanserin was not approved at 2 previous FDA committee hearings—mainly because of safety issues but also because of concerns about efficacy. For example, during the 2013 FDA hearing, the results presented showed statistically significant, but numerically small, treatment differences at 24 weeks compared with placebo. In an FDA responder analysis of the Phase-III trials, after accounting for the placebo effect, approximately 8% to 13% women were at least “much improved” on at least 1 of the primary outcomes.5
Flibanserin is not indicated for women whose sexual desire is due to (1) coexisting medical or psychiatric condition, (2) effects of medication or substance abuse, or (3) a relationship problem. It is unknown whether supplemental treatment would help these patients; however, it seems reasonable that combining flibanserin with psychosocial treatment, such as sex therapy or individual therapy, could be beneficial because it may be difficult to disentangle sexual dysfunction and relationship issues—2 problems that often are interwoven.
How it works
Flibanserin is a serotonin 1A receptor agonist and serotonin 2A receptor antagonist. In vitro, flibanserin demonstrated high affinity for the following 5-HT receptors:
- agonist activity at 5-HT1A
- antagonist activity at 5-HT2A, mostly in the prefrontal cortex.
Flibanserin also has moderate antagonist activities at the 5-HT2B, 5-HT2C, and dopamine D4 receptors. Flibanserin presumably acts centrally in the CNS; it has been suggested that flibanserin could rebalance neural circuitry involved in processing sexual desire by reducing serotonin activity and enhancing dopamine and epinephrine activity. The exact mechanism of how flibanserin improves sexual desire in women is unknown.
Pharmacokinetics
Flibanserin has a mean termination half-life of approximately 11 hours. It is administered once a day (50 to 100 mg) at bedtime. Steady state in healthy women was achieved after 3 days. Based on clinical observations, onset of action seems to be gradual and reaches maximum efficacy in approximately 8 weeks. Patients should discontinue the drug if no improvement is reported after 8 weeks. Flibanserin is readily absorbed from the gastrointestinal tract; however, food slows its absorption. The drug is 98% protein (mostly albumin)-bound.
Flibanserin is primarily metabolized in the liver by cytochrome P450 (CYP) 3A4 and to a lesser extent by CYP2C19. Co-administration of moderate (diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil) or strong (eg, ketoconazole, clarithromycin, nefazodone, ritonavir) CYP3A4 inhibitors increases the concentration of flibanserin. This could lead to severe hypotension and syncope; therefore, co-administering flibanserin with a strong CYP3A4 inhibitor is contraindicated. Grapefruit juice is a moderate inhibitor of CYP3A4, and in a study of 26 healthy females, 240 mL of grapefruit juice increased flibanserin concentration 1.4-fold. Flibanserin is excreted though urine and feces. Flibanserin should be taken once a day at bedtime because of sedation, somnolence, and possible syncope.
Efficacy
The efficacy of flibanserin for treating HSDD was established in three 24-week, randomized, double-blind, placebo-controlled studies (Table 2). The target population in these studies was premenopausal women (mean age 36, range 19 to 55) with acquired HSDD lasting at least 6 months (mean duration, approximately 5 years). The 3 studies included 1,187 women who received flibanserin, 100 mg at bedtime, and 1,188 women who received placebo. Participants were mostly white (88.6%), and included black (9.6%) and Asian (1.5%) women. The completion rates were 69% for flibanserin and 78% for placebo. Some of the trials included arms with a lower dosage of flibanserin (25 mg and 50 mg), which are not included in this analysis.
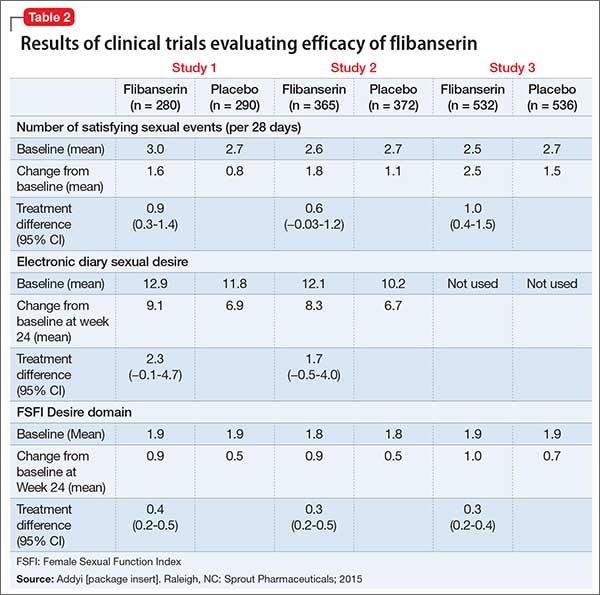
As noted in the package insert, these trials each had 2 co-primary efficacy endpoints, SSEs and sexual desire:
- change from baseline to Week 24 in the number of monthly SSEs (ie, sexual intercourse, oral sex, masturbation, or genital stimulation by the partner)
- change in sexual desire from baseline to 24-week endpoint.
In Study 1 and 2, change in sexual desire from baseline to Week 24 was measured daily by using an electronic diary. Every day, patients rated their sexual desire level by answering the question, “Indicate your most intense level of sexual desire” from 0 (no desire) to 3 (strong desire). These responses were totaled over a 28-day period to yield the monthly sexual desire score, which ranged from 0 to 84. These 2 studies also used the Female Sexual Function Index (FSFI) Desire domain as a secondary endpoint.
Study 3 used the FSFI Desire domain, comprising 2 questions, as the sexual desire co-primary endpoint:
- “Over the past 4 weeks, how often did you feel sexual desire or interest?” Responses ranged from 1 (almost never or never) to 5 (almost always or always).
- “Over the past 4 weeks, how would you rate your level (degree) of sexual desire or interest?” Responses ranged from 1 (very low or none at all) to 5 (very high).
In all 3 trials, flibanserin was associated with a small, yet statistically significant, improvement in change in monthly SSEs from baseline to Week 24 compared with placebo. In Study 1 and 2, there were no statistically significant differences between flibanserin and placebo for the electronic diary sexual desire endpoint. In the third study, there was statistically significant improvement in the change in sexual desire using the FSFI Desire domain with flibanserin compared with placebo. The FSFI Desire domain findings were consistent across all 3 trials. Flibanserin was associated with a decrease in sexual distress compared with placebo in all 3 studies.
Tolerability
Flibanserin was well tolerated in the 3 clinical trials. As the FDA noted, clinical trials are conducted under widely varying conditions and therefore adverse reaction rates observed in trials of flibanserin cannot be directly compared with those reported in clinical trials of another drug and might not reflect rates observed in clinical practice.
The discontinuation rate due to adverse reactions was 13% among patients treated with flibanserin, 100 mg at bedtime, and 6% among those taking placebo. The most common side effects were somnolence, dizziness, fatigue, nausea, insomnia, and dry mouth, which appear dose-dependent. Onset of most of these adverse events was within 14 days after the start of treatment.
Although hypotension and syncope rarely were seen with flibanserin alone in clinical trials, these adverse events occurred more frequently in the morning and when taken with alcohol and with some drugs (moderate or strong CYP3A4 inhibitors), and in patients with hepatic impairment. Therefore, women who drink alcohol or take a moderate or strong inhibitor of CYP3A4—both of which are contraindicated—and those with hepatic impairment should not take flibanserin.
Flibanserin should be taken at bedtime, because the risk of hypotension and syncope is higher when flibanserin is taken in the morning and because of associated sedation and somnolence.
Unique clinical issues
Flibanserin is the first FDA-approved medication for treating HSDD. It is important to note that the drug originally was developed as an antidepressant, but failed to show efficacy. Researchers noted that the drug was more effective than placebo when patients were asked, “How strong is your sexual desire?” The focus of development then shifted to a potential treatment of HSDD.
Flibanserin was not approved at 2 previous FDA hearings, mainly because of safety concerns. For the second hearing, the manufacturer, Boehringer Ingelheim, which sold the rights to the drug to Sprout Pharmaceuticals in 2011,6 did not present any new efficacy data, but provided additional safety data, such as research suggesting the absence of next-day driving impairment and data related to alcohol use (the study confirming hypotension associated with alcohol abuse used a small sample, and only 2 of 25 participants were women).
Contraindications
Flibanserin is contraindicated in patients using alcohol because of an increased risk of hypotension and syncope. A patient’s alcohol use should be evaluated before administering flibanserin, and patients should be counseled about the importance of abstaining from alcohol.
Similarly, concomitant use of flibanserin with a moderate or strong inhibitor of CYP3A4 increases the concentration of flibanserin and raises the risk of hypotension and syncope. Therefore, the use of a moderate or strong inhibitor of CYP3A4 in patients taking flibanserin is contraindicated. Similarly, patients with liver impairment should not take this drug.
Strong CYP2C19 inhibitors (proton-pump inhibitors, selective serotonin reuptake inhibitors, benzodiazepines, antifungals) could increase flibanserin exposure, which may increase risk of hypotension, syncope, and CNS depression. Discuss these risks with your patients; doing so is particularly important when treating women of Chinese heritage, and some other Asian women, because 20% of these populations are genotypic CYP2C19 poor metabolizers.
Because of the increased risk of hypotension and syncope with alcohol use, flibanserin is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Addyi REMS Program. Flibanserin can be prescribed or dispensed only by physicians and pharmacists who watch this program’s online slide presentation and passed a comprehension test.a
Pregnant women should not take flibanserin because the effect on the fetus is unknown. Also, because the interaction with some oral contraceptives is unknown, patients should be cautioned about unwanted pregnancy. Women who are breastfeeding also should avoid using flibanserin because it is not known whether the drug is excreted in breast milk.
Women taking flibanserin also should avoid grapefruit juice, which increases flibanserin levels, and avoid using herbal products, resveratrol, and some over-the-counter drugs such as cimetidine. Women who have a depressive disorder also should avoid using flibanserin because their low sexual desire is more likely due to depression, which is not a therapeutic target for the drug.
Dosing
Flibanserin is provided in 100-mg film-coated tablets. It should be taken once a day at bedtime; titration is unnecessary. Length of treatment has not been determined, but it is recommended that patients stop flibanserin if they do not experience any benefit after 8 weeks. Although there is no guidance in the prescribing information, the medication probably could be stopped without tapering because withdrawal effects have not been observed.
Bottom Line
Flibanserin is FDA-approved for treating generalized, acquired hypoactive sexual desire disorder in premenopausal women. In clinical trials, the drug increased the number of satisfying sexual events and sexual desire, as measured by a diary and rating scales. Alcohol use and use of any moderate or strong inhibitor of cytochrome P450 3A4 are contraindicated in patients taking flibanserin because of an increased risk of hypotension and syncope.
1. Goldfisher ER, Breaux J, Katz M, et al. Continued efficacy and safety of flibanserin in premenopausal women with Hypoactive Sexual desire Disorder (HSDD): results from a randomized withdrawal trial. J Sex Med. 2011;8(11):3160- 3172.
2. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793-804.
3. Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074-1085.
4. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807-1815.
5. Gellad WF, Flynn KE, Alexander GC. Evaluation of flibanserin: science and advocacy at the FDA. JAMA. 2015;314(9):869-870
6. Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med. 2016;374(2):101-104.
Flibanserin, FDA-approved in August 2015, is the first medication approved to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women (Table 1). In clinical trials,1-4 the drug has shown modest efficacy in improving symptoms of low sexual desire (number of satisfying sexual events [SSEs], sexual desire, and overall sexual function). Flibanserin is not indicated to enhance sexual performance, for HSDD in postmenopausal women, or in men.
Clinical implications
Flibanserin could help premenopausal women who have distressing low sexual desire, which must be acquired and generalized:
- “Acquired low sexual desire” means that a patient had an adequate sexual desire that decreased or ceased for an unknown reason.
- “Generalized low sexual desire” means that lack of sexual desire occurs all the time and in all situations, not only with a certain partner or in some situations.
Women taking flibanserin could experience gradually increased sexual desire, increase in SSEs, and decrease of sexual distress. Flibanserin is indicated for long-term use; however, it should be discontinued after 8 weeks if the patient does not report any improvement in symptoms.
The number needed to treat with flibanserin likely would be rather large, but it is not available because of complex outcome measures in clinical trials. Flibanserin was not approved at 2 previous FDA committee hearings—mainly because of safety issues but also because of concerns about efficacy. For example, during the 2013 FDA hearing, the results presented showed statistically significant, but numerically small, treatment differences at 24 weeks compared with placebo. In an FDA responder analysis of the Phase-III trials, after accounting for the placebo effect, approximately 8% to 13% women were at least “much improved” on at least 1 of the primary outcomes.5
Flibanserin is not indicated for women whose sexual desire is due to (1) coexisting medical or psychiatric condition, (2) effects of medication or substance abuse, or (3) a relationship problem. It is unknown whether supplemental treatment would help these patients; however, it seems reasonable that combining flibanserin with psychosocial treatment, such as sex therapy or individual therapy, could be beneficial because it may be difficult to disentangle sexual dysfunction and relationship issues—2 problems that often are interwoven.
How it works
Flibanserin is a serotonin 1A receptor agonist and serotonin 2A receptor antagonist. In vitro, flibanserin demonstrated high affinity for the following 5-HT receptors:
- agonist activity at 5-HT1A
- antagonist activity at 5-HT2A, mostly in the prefrontal cortex.
Flibanserin also has moderate antagonist activities at the 5-HT2B, 5-HT2C, and dopamine D4 receptors. Flibanserin presumably acts centrally in the CNS; it has been suggested that flibanserin could rebalance neural circuitry involved in processing sexual desire by reducing serotonin activity and enhancing dopamine and epinephrine activity. The exact mechanism of how flibanserin improves sexual desire in women is unknown.
Pharmacokinetics
Flibanserin has a mean termination half-life of approximately 11 hours. It is administered once a day (50 to 100 mg) at bedtime. Steady state in healthy women was achieved after 3 days. Based on clinical observations, onset of action seems to be gradual and reaches maximum efficacy in approximately 8 weeks. Patients should discontinue the drug if no improvement is reported after 8 weeks. Flibanserin is readily absorbed from the gastrointestinal tract; however, food slows its absorption. The drug is 98% protein (mostly albumin)-bound.
Flibanserin is primarily metabolized in the liver by cytochrome P450 (CYP) 3A4 and to a lesser extent by CYP2C19. Co-administration of moderate (diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil) or strong (eg, ketoconazole, clarithromycin, nefazodone, ritonavir) CYP3A4 inhibitors increases the concentration of flibanserin. This could lead to severe hypotension and syncope; therefore, co-administering flibanserin with a strong CYP3A4 inhibitor is contraindicated. Grapefruit juice is a moderate inhibitor of CYP3A4, and in a study of 26 healthy females, 240 mL of grapefruit juice increased flibanserin concentration 1.4-fold. Flibanserin is excreted though urine and feces. Flibanserin should be taken once a day at bedtime because of sedation, somnolence, and possible syncope.
Efficacy
The efficacy of flibanserin for treating HSDD was established in three 24-week, randomized, double-blind, placebo-controlled studies (Table 2). The target population in these studies was premenopausal women (mean age 36, range 19 to 55) with acquired HSDD lasting at least 6 months (mean duration, approximately 5 years). The 3 studies included 1,187 women who received flibanserin, 100 mg at bedtime, and 1,188 women who received placebo. Participants were mostly white (88.6%), and included black (9.6%) and Asian (1.5%) women. The completion rates were 69% for flibanserin and 78% for placebo. Some of the trials included arms with a lower dosage of flibanserin (25 mg and 50 mg), which are not included in this analysis.
As noted in the package insert, these trials each had 2 co-primary efficacy endpoints, SSEs and sexual desire:
- change from baseline to Week 24 in the number of monthly SSEs (ie, sexual intercourse, oral sex, masturbation, or genital stimulation by the partner)
- change in sexual desire from baseline to 24-week endpoint.
In Study 1 and 2, change in sexual desire from baseline to Week 24 was measured daily by using an electronic diary. Every day, patients rated their sexual desire level by answering the question, “Indicate your most intense level of sexual desire” from 0 (no desire) to 3 (strong desire). These responses were totaled over a 28-day period to yield the monthly sexual desire score, which ranged from 0 to 84. These 2 studies also used the Female Sexual Function Index (FSFI) Desire domain as a secondary endpoint.
Study 3 used the FSFI Desire domain, comprising 2 questions, as the sexual desire co-primary endpoint:
- “Over the past 4 weeks, how often did you feel sexual desire or interest?” Responses ranged from 1 (almost never or never) to 5 (almost always or always).
- “Over the past 4 weeks, how would you rate your level (degree) of sexual desire or interest?” Responses ranged from 1 (very low or none at all) to 5 (very high).
In all 3 trials, flibanserin was associated with a small, yet statistically significant, improvement in change in monthly SSEs from baseline to Week 24 compared with placebo. In Study 1 and 2, there were no statistically significant differences between flibanserin and placebo for the electronic diary sexual desire endpoint. In the third study, there was statistically significant improvement in the change in sexual desire using the FSFI Desire domain with flibanserin compared with placebo. The FSFI Desire domain findings were consistent across all 3 trials. Flibanserin was associated with a decrease in sexual distress compared with placebo in all 3 studies.
Tolerability
Flibanserin was well tolerated in the 3 clinical trials. As the FDA noted, clinical trials are conducted under widely varying conditions and therefore adverse reaction rates observed in trials of flibanserin cannot be directly compared with those reported in clinical trials of another drug and might not reflect rates observed in clinical practice.
The discontinuation rate due to adverse reactions was 13% among patients treated with flibanserin, 100 mg at bedtime, and 6% among those taking placebo. The most common side effects were somnolence, dizziness, fatigue, nausea, insomnia, and dry mouth, which appear dose-dependent. Onset of most of these adverse events was within 14 days after the start of treatment.
Although hypotension and syncope rarely were seen with flibanserin alone in clinical trials, these adverse events occurred more frequently in the morning and when taken with alcohol and with some drugs (moderate or strong CYP3A4 inhibitors), and in patients with hepatic impairment. Therefore, women who drink alcohol or take a moderate or strong inhibitor of CYP3A4—both of which are contraindicated—and those with hepatic impairment should not take flibanserin.
Flibanserin should be taken at bedtime, because the risk of hypotension and syncope is higher when flibanserin is taken in the morning and because of associated sedation and somnolence.
Unique clinical issues
Flibanserin is the first FDA-approved medication for treating HSDD. It is important to note that the drug originally was developed as an antidepressant, but failed to show efficacy. Researchers noted that the drug was more effective than placebo when patients were asked, “How strong is your sexual desire?” The focus of development then shifted to a potential treatment of HSDD.
Flibanserin was not approved at 2 previous FDA hearings, mainly because of safety concerns. For the second hearing, the manufacturer, Boehringer Ingelheim, which sold the rights to the drug to Sprout Pharmaceuticals in 2011,6 did not present any new efficacy data, but provided additional safety data, such as research suggesting the absence of next-day driving impairment and data related to alcohol use (the study confirming hypotension associated with alcohol abuse used a small sample, and only 2 of 25 participants were women).
Contraindications
Flibanserin is contraindicated in patients using alcohol because of an increased risk of hypotension and syncope. A patient’s alcohol use should be evaluated before administering flibanserin, and patients should be counseled about the importance of abstaining from alcohol.
Similarly, concomitant use of flibanserin with a moderate or strong inhibitor of CYP3A4 increases the concentration of flibanserin and raises the risk of hypotension and syncope. Therefore, the use of a moderate or strong inhibitor of CYP3A4 in patients taking flibanserin is contraindicated. Similarly, patients with liver impairment should not take this drug.
Strong CYP2C19 inhibitors (proton-pump inhibitors, selective serotonin reuptake inhibitors, benzodiazepines, antifungals) could increase flibanserin exposure, which may increase risk of hypotension, syncope, and CNS depression. Discuss these risks with your patients; doing so is particularly important when treating women of Chinese heritage, and some other Asian women, because 20% of these populations are genotypic CYP2C19 poor metabolizers.
Because of the increased risk of hypotension and syncope with alcohol use, flibanserin is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Addyi REMS Program. Flibanserin can be prescribed or dispensed only by physicians and pharmacists who watch this program’s online slide presentation and passed a comprehension test.a
Pregnant women should not take flibanserin because the effect on the fetus is unknown. Also, because the interaction with some oral contraceptives is unknown, patients should be cautioned about unwanted pregnancy. Women who are breastfeeding also should avoid using flibanserin because it is not known whether the drug is excreted in breast milk.
Women taking flibanserin also should avoid grapefruit juice, which increases flibanserin levels, and avoid using herbal products, resveratrol, and some over-the-counter drugs such as cimetidine. Women who have a depressive disorder also should avoid using flibanserin because their low sexual desire is more likely due to depression, which is not a therapeutic target for the drug.
Dosing
Flibanserin is provided in 100-mg film-coated tablets. It should be taken once a day at bedtime; titration is unnecessary. Length of treatment has not been determined, but it is recommended that patients stop flibanserin if they do not experience any benefit after 8 weeks. Although there is no guidance in the prescribing information, the medication probably could be stopped without tapering because withdrawal effects have not been observed.
Bottom Line
Flibanserin is FDA-approved for treating generalized, acquired hypoactive sexual desire disorder in premenopausal women. In clinical trials, the drug increased the number of satisfying sexual events and sexual desire, as measured by a diary and rating scales. Alcohol use and use of any moderate or strong inhibitor of cytochrome P450 3A4 are contraindicated in patients taking flibanserin because of an increased risk of hypotension and syncope.
Flibanserin, FDA-approved in August 2015, is the first medication approved to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women (Table 1). In clinical trials,1-4 the drug has shown modest efficacy in improving symptoms of low sexual desire (number of satisfying sexual events [SSEs], sexual desire, and overall sexual function). Flibanserin is not indicated to enhance sexual performance, for HSDD in postmenopausal women, or in men.
Clinical implications
Flibanserin could help premenopausal women who have distressing low sexual desire, which must be acquired and generalized:
- “Acquired low sexual desire” means that a patient had an adequate sexual desire that decreased or ceased for an unknown reason.
- “Generalized low sexual desire” means that lack of sexual desire occurs all the time and in all situations, not only with a certain partner or in some situations.
Women taking flibanserin could experience gradually increased sexual desire, increase in SSEs, and decrease of sexual distress. Flibanserin is indicated for long-term use; however, it should be discontinued after 8 weeks if the patient does not report any improvement in symptoms.
The number needed to treat with flibanserin likely would be rather large, but it is not available because of complex outcome measures in clinical trials. Flibanserin was not approved at 2 previous FDA committee hearings—mainly because of safety issues but also because of concerns about efficacy. For example, during the 2013 FDA hearing, the results presented showed statistically significant, but numerically small, treatment differences at 24 weeks compared with placebo. In an FDA responder analysis of the Phase-III trials, after accounting for the placebo effect, approximately 8% to 13% women were at least “much improved” on at least 1 of the primary outcomes.5
Flibanserin is not indicated for women whose sexual desire is due to (1) coexisting medical or psychiatric condition, (2) effects of medication or substance abuse, or (3) a relationship problem. It is unknown whether supplemental treatment would help these patients; however, it seems reasonable that combining flibanserin with psychosocial treatment, such as sex therapy or individual therapy, could be beneficial because it may be difficult to disentangle sexual dysfunction and relationship issues—2 problems that often are interwoven.
How it works
Flibanserin is a serotonin 1A receptor agonist and serotonin 2A receptor antagonist. In vitro, flibanserin demonstrated high affinity for the following 5-HT receptors:
- agonist activity at 5-HT1A
- antagonist activity at 5-HT2A, mostly in the prefrontal cortex.
Flibanserin also has moderate antagonist activities at the 5-HT2B, 5-HT2C, and dopamine D4 receptors. Flibanserin presumably acts centrally in the CNS; it has been suggested that flibanserin could rebalance neural circuitry involved in processing sexual desire by reducing serotonin activity and enhancing dopamine and epinephrine activity. The exact mechanism of how flibanserin improves sexual desire in women is unknown.
Pharmacokinetics
Flibanserin has a mean termination half-life of approximately 11 hours. It is administered once a day (50 to 100 mg) at bedtime. Steady state in healthy women was achieved after 3 days. Based on clinical observations, onset of action seems to be gradual and reaches maximum efficacy in approximately 8 weeks. Patients should discontinue the drug if no improvement is reported after 8 weeks. Flibanserin is readily absorbed from the gastrointestinal tract; however, food slows its absorption. The drug is 98% protein (mostly albumin)-bound.
Flibanserin is primarily metabolized in the liver by cytochrome P450 (CYP) 3A4 and to a lesser extent by CYP2C19. Co-administration of moderate (diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil) or strong (eg, ketoconazole, clarithromycin, nefazodone, ritonavir) CYP3A4 inhibitors increases the concentration of flibanserin. This could lead to severe hypotension and syncope; therefore, co-administering flibanserin with a strong CYP3A4 inhibitor is contraindicated. Grapefruit juice is a moderate inhibitor of CYP3A4, and in a study of 26 healthy females, 240 mL of grapefruit juice increased flibanserin concentration 1.4-fold. Flibanserin is excreted though urine and feces. Flibanserin should be taken once a day at bedtime because of sedation, somnolence, and possible syncope.
Efficacy
The efficacy of flibanserin for treating HSDD was established in three 24-week, randomized, double-blind, placebo-controlled studies (Table 2). The target population in these studies was premenopausal women (mean age 36, range 19 to 55) with acquired HSDD lasting at least 6 months (mean duration, approximately 5 years). The 3 studies included 1,187 women who received flibanserin, 100 mg at bedtime, and 1,188 women who received placebo. Participants were mostly white (88.6%), and included black (9.6%) and Asian (1.5%) women. The completion rates were 69% for flibanserin and 78% for placebo. Some of the trials included arms with a lower dosage of flibanserin (25 mg and 50 mg), which are not included in this analysis.
As noted in the package insert, these trials each had 2 co-primary efficacy endpoints, SSEs and sexual desire:
- change from baseline to Week 24 in the number of monthly SSEs (ie, sexual intercourse, oral sex, masturbation, or genital stimulation by the partner)
- change in sexual desire from baseline to 24-week endpoint.
In Study 1 and 2, change in sexual desire from baseline to Week 24 was measured daily by using an electronic diary. Every day, patients rated their sexual desire level by answering the question, “Indicate your most intense level of sexual desire” from 0 (no desire) to 3 (strong desire). These responses were totaled over a 28-day period to yield the monthly sexual desire score, which ranged from 0 to 84. These 2 studies also used the Female Sexual Function Index (FSFI) Desire domain as a secondary endpoint.
Study 3 used the FSFI Desire domain, comprising 2 questions, as the sexual desire co-primary endpoint:
- “Over the past 4 weeks, how often did you feel sexual desire or interest?” Responses ranged from 1 (almost never or never) to 5 (almost always or always).
- “Over the past 4 weeks, how would you rate your level (degree) of sexual desire or interest?” Responses ranged from 1 (very low or none at all) to 5 (very high).
In all 3 trials, flibanserin was associated with a small, yet statistically significant, improvement in change in monthly SSEs from baseline to Week 24 compared with placebo. In Study 1 and 2, there were no statistically significant differences between flibanserin and placebo for the electronic diary sexual desire endpoint. In the third study, there was statistically significant improvement in the change in sexual desire using the FSFI Desire domain with flibanserin compared with placebo. The FSFI Desire domain findings were consistent across all 3 trials. Flibanserin was associated with a decrease in sexual distress compared with placebo in all 3 studies.
Tolerability
Flibanserin was well tolerated in the 3 clinical trials. As the FDA noted, clinical trials are conducted under widely varying conditions and therefore adverse reaction rates observed in trials of flibanserin cannot be directly compared with those reported in clinical trials of another drug and might not reflect rates observed in clinical practice.
The discontinuation rate due to adverse reactions was 13% among patients treated with flibanserin, 100 mg at bedtime, and 6% among those taking placebo. The most common side effects were somnolence, dizziness, fatigue, nausea, insomnia, and dry mouth, which appear dose-dependent. Onset of most of these adverse events was within 14 days after the start of treatment.
Although hypotension and syncope rarely were seen with flibanserin alone in clinical trials, these adverse events occurred more frequently in the morning and when taken with alcohol and with some drugs (moderate or strong CYP3A4 inhibitors), and in patients with hepatic impairment. Therefore, women who drink alcohol or take a moderate or strong inhibitor of CYP3A4—both of which are contraindicated—and those with hepatic impairment should not take flibanserin.
Flibanserin should be taken at bedtime, because the risk of hypotension and syncope is higher when flibanserin is taken in the morning and because of associated sedation and somnolence.
Unique clinical issues
Flibanserin is the first FDA-approved medication for treating HSDD. It is important to note that the drug originally was developed as an antidepressant, but failed to show efficacy. Researchers noted that the drug was more effective than placebo when patients were asked, “How strong is your sexual desire?” The focus of development then shifted to a potential treatment of HSDD.
Flibanserin was not approved at 2 previous FDA hearings, mainly because of safety concerns. For the second hearing, the manufacturer, Boehringer Ingelheim, which sold the rights to the drug to Sprout Pharmaceuticals in 2011,6 did not present any new efficacy data, but provided additional safety data, such as research suggesting the absence of next-day driving impairment and data related to alcohol use (the study confirming hypotension associated with alcohol abuse used a small sample, and only 2 of 25 participants were women).
Contraindications
Flibanserin is contraindicated in patients using alcohol because of an increased risk of hypotension and syncope. A patient’s alcohol use should be evaluated before administering flibanserin, and patients should be counseled about the importance of abstaining from alcohol.
Similarly, concomitant use of flibanserin with a moderate or strong inhibitor of CYP3A4 increases the concentration of flibanserin and raises the risk of hypotension and syncope. Therefore, the use of a moderate or strong inhibitor of CYP3A4 in patients taking flibanserin is contraindicated. Similarly, patients with liver impairment should not take this drug.
Strong CYP2C19 inhibitors (proton-pump inhibitors, selective serotonin reuptake inhibitors, benzodiazepines, antifungals) could increase flibanserin exposure, which may increase risk of hypotension, syncope, and CNS depression. Discuss these risks with your patients; doing so is particularly important when treating women of Chinese heritage, and some other Asian women, because 20% of these populations are genotypic CYP2C19 poor metabolizers.
Because of the increased risk of hypotension and syncope with alcohol use, flibanserin is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Addyi REMS Program. Flibanserin can be prescribed or dispensed only by physicians and pharmacists who watch this program’s online slide presentation and passed a comprehension test.a
Pregnant women should not take flibanserin because the effect on the fetus is unknown. Also, because the interaction with some oral contraceptives is unknown, patients should be cautioned about unwanted pregnancy. Women who are breastfeeding also should avoid using flibanserin because it is not known whether the drug is excreted in breast milk.
Women taking flibanserin also should avoid grapefruit juice, which increases flibanserin levels, and avoid using herbal products, resveratrol, and some over-the-counter drugs such as cimetidine. Women who have a depressive disorder also should avoid using flibanserin because their low sexual desire is more likely due to depression, which is not a therapeutic target for the drug.
Dosing
Flibanserin is provided in 100-mg film-coated tablets. It should be taken once a day at bedtime; titration is unnecessary. Length of treatment has not been determined, but it is recommended that patients stop flibanserin if they do not experience any benefit after 8 weeks. Although there is no guidance in the prescribing information, the medication probably could be stopped without tapering because withdrawal effects have not been observed.
Bottom Line
Flibanserin is FDA-approved for treating generalized, acquired hypoactive sexual desire disorder in premenopausal women. In clinical trials, the drug increased the number of satisfying sexual events and sexual desire, as measured by a diary and rating scales. Alcohol use and use of any moderate or strong inhibitor of cytochrome P450 3A4 are contraindicated in patients taking flibanserin because of an increased risk of hypotension and syncope.
1. Goldfisher ER, Breaux J, Katz M, et al. Continued efficacy and safety of flibanserin in premenopausal women with Hypoactive Sexual desire Disorder (HSDD): results from a randomized withdrawal trial. J Sex Med. 2011;8(11):3160- 3172.
2. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793-804.
3. Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074-1085.
4. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807-1815.
5. Gellad WF, Flynn KE, Alexander GC. Evaluation of flibanserin: science and advocacy at the FDA. JAMA. 2015;314(9):869-870
6. Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med. 2016;374(2):101-104.
1. Goldfisher ER, Breaux J, Katz M, et al. Continued efficacy and safety of flibanserin in premenopausal women with Hypoactive Sexual desire Disorder (HSDD): results from a randomized withdrawal trial. J Sex Med. 2011;8(11):3160- 3172.
2. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793-804.
3. Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074-1085.
4. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807-1815.
5. Gellad WF, Flynn KE, Alexander GC. Evaluation of flibanserin: science and advocacy at the FDA. JAMA. 2015;314(9):869-870
6. Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med. 2016;374(2):101-104.
Erythematous Eruption on the Left Leg
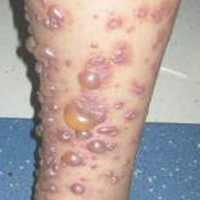
The Diagnosis: Bullous Henoch-Schönlein Purpura
Laboratory tests in this patient showed no abnormalities for complete blood cell count, immunoglobulins, anti–double-stranded DNA, antinuclear antibody, p–antineutrophil cytoplasmic antibodies, lupus anticoagulant, Sjögren antibodies, liver enzymes, and erythrocyte sedimentation rate. Urinalysis was normal. Punch biopsies were obtained and a histologic examination showed an intense inflammatory infiltrate of neutrophils around blood vessels within the dermis (Figure). These blood vessels showed swollen endothelium and narrowing of the vessel lumina with leukocytoclasia. Direct immunofluorescence revealed granular IgA, C3, fibrin, and weak IgM deposits in blood vessels in the papillary dermis consistent with Henoch-Schönlein purpura (HSP).
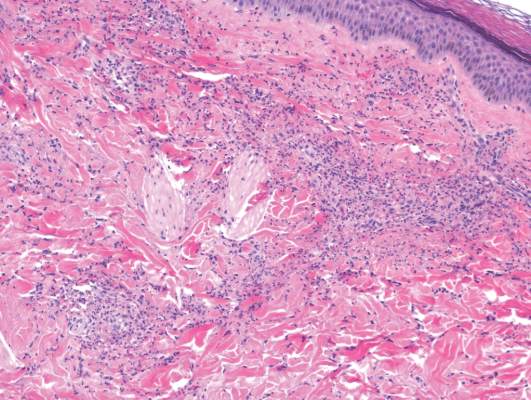
Henoch-Schönlein purpura is the most common vasculitis in children.1-6 However, its bullous variant is rare, with few pediatric cases reported. Bullous HSP affects arterioles through an IgA-mediated pathway.1-6 It is believed that the bullae are formed secondary to neutrophilic release of matrix metalloproteinase 9 (MMP-9), which degrades extracellular collagen.2 Additionally, bullous fluid from HSP has been noted to have markedly elevated levels of soluble CD23, a form of the CD23 B-cell surface receptor used in antibody feedback regulation and B-cell recruitment, which also has been found to be elevated in the fluid of bullous pemphigoid, suggesting a similar pathogenesis of exaggerated humoral immunity.3
The most common sign of HSP is palpable purpura; however, other cutaneous findings can be present including targetoid plaques, macules, papules, petechiae, and bullae that may become hemorrhagic, ulcerated, necrotic, or scarred.1-6 Bullae appear in the most dependent parts of the body, such as the feet and lower legs. Hydrostatic pressure may play a role in the pathogenesis of this phenomenon.1 When other classic signs of HSP are absent, the presence of bullae clouds the diagnosis and creates controversy regarding treatment, as there is a dearth of literature on proper therapy for severe cutaneous manifestations of HSP.6
Our patient was treated with morphine for pain management along with topical mupirocin and nonadherent dressings for wound care. She also received pulse intravenous methylprednisolone 2 mg/kg daily for 3 days and then was transitioned to oral prednisone 1 mg/kg daily, which was tapered over 3 weeks after discharge. This regimen resulted in resolution of symptoms with rapid regression of bullae and subsequent postinflammatory hyperpigmentation. Prior reports have noted that the presence of bullae does not alter the prognosis or predict probability of renal involvement of this self-limited disease, leading to controversy in determining if treatment offers more favorable outcomes.1,3 One study suggested that steroids only improve symptoms, arthralgia, and abdominal pain, but they do not aid in the resolution of cutaneous lesions or prevent the progression of renal disease.3 Contrarily, others have suggested that the presence of bullae and renal disease is an indication to start treatment.6 This claim is based on the mechanistic finding that immunosuppression with corticosteroids decreases inflammation by inhibiting activator protein 1, a transcription factor for MMP-9, thereby reducing MMP-9 activity and the formation of bullae.4 Clinical anecdotes, including our own, that demonstrate dramatic improvement of hemorrhagic bullae with the administration of corticosteroids substantiate this mechanism. Through the inhibition of neutrophil interactions and IgA production, other anti-inflammatory and immunosuppressive medications such as colchicine, dapsone, and azathioprine also have been reported to aid in resolution of the cutaneous lesions.1,5,6 Although there is a clear drawback to the lack of controlled trials and prospective studies regarding the treatment of bullous HSP, it is nearly impossible to expect such studies to be carried out given the rare and unpredictable nature of the disease. For now, claims derived from case series and case reports guide our understanding of treatment efficacy.
Acknowledgment—Quiz photograph courtesy of Steve Taylor, BS, Phoenix, Arizona.
- Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature [published online July 16, 2009]. Rheumatol Int. 2010;30:1355-1359. doi:10.1007/s00296-009-1055-8.
- Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiologic role of gelatinate (MMP-9). Dermatology. 1990;197:62-64.
- Bansal AS, Dwivedi N, Adsett M. Serum and blister fluid cytokines and complement proteins in a patient with Henoch Schönlein purpura associated with a bullous skin rash. Australas J Dermatol. 1997;38:190-192.
- Aljada A, Ghanim H, Mohanty P, et al. Hydrocortisone suppresses intranuclear activator-protein-1 (AP-1) binding activity in mononuclear cells and plasma matrix metalloproteinase 2 and 9 (MMP-2 and MMP-9). J Clin Endocrinol Metab. 2001;86:5988-5991.
- Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
- den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch Schönlein purpura as indication to start systemic prednisone [published online January 5, 2009]. Acta Paediatr. 2010;99:781-783. doi:10.1111/j.1651-2227.2009.01650.x.
The Diagnosis: Bullous Henoch-Schönlein Purpura
Laboratory tests in this patient showed no abnormalities for complete blood cell count, immunoglobulins, anti–double-stranded DNA, antinuclear antibody, p–antineutrophil cytoplasmic antibodies, lupus anticoagulant, Sjögren antibodies, liver enzymes, and erythrocyte sedimentation rate. Urinalysis was normal. Punch biopsies were obtained and a histologic examination showed an intense inflammatory infiltrate of neutrophils around blood vessels within the dermis (Figure). These blood vessels showed swollen endothelium and narrowing of the vessel lumina with leukocytoclasia. Direct immunofluorescence revealed granular IgA, C3, fibrin, and weak IgM deposits in blood vessels in the papillary dermis consistent with Henoch-Schönlein purpura (HSP).
Henoch-Schönlein purpura is the most common vasculitis in children.1-6 However, its bullous variant is rare, with few pediatric cases reported. Bullous HSP affects arterioles through an IgA-mediated pathway.1-6 It is believed that the bullae are formed secondary to neutrophilic release of matrix metalloproteinase 9 (MMP-9), which degrades extracellular collagen.2 Additionally, bullous fluid from HSP has been noted to have markedly elevated levels of soluble CD23, a form of the CD23 B-cell surface receptor used in antibody feedback regulation and B-cell recruitment, which also has been found to be elevated in the fluid of bullous pemphigoid, suggesting a similar pathogenesis of exaggerated humoral immunity.3
The most common sign of HSP is palpable purpura; however, other cutaneous findings can be present including targetoid plaques, macules, papules, petechiae, and bullae that may become hemorrhagic, ulcerated, necrotic, or scarred.1-6 Bullae appear in the most dependent parts of the body, such as the feet and lower legs. Hydrostatic pressure may play a role in the pathogenesis of this phenomenon.1 When other classic signs of HSP are absent, the presence of bullae clouds the diagnosis and creates controversy regarding treatment, as there is a dearth of literature on proper therapy for severe cutaneous manifestations of HSP.6
Our patient was treated with morphine for pain management along with topical mupirocin and nonadherent dressings for wound care. She also received pulse intravenous methylprednisolone 2 mg/kg daily for 3 days and then was transitioned to oral prednisone 1 mg/kg daily, which was tapered over 3 weeks after discharge. This regimen resulted in resolution of symptoms with rapid regression of bullae and subsequent postinflammatory hyperpigmentation. Prior reports have noted that the presence of bullae does not alter the prognosis or predict probability of renal involvement of this self-limited disease, leading to controversy in determining if treatment offers more favorable outcomes.1,3 One study suggested that steroids only improve symptoms, arthralgia, and abdominal pain, but they do not aid in the resolution of cutaneous lesions or prevent the progression of renal disease.3 Contrarily, others have suggested that the presence of bullae and renal disease is an indication to start treatment.6 This claim is based on the mechanistic finding that immunosuppression with corticosteroids decreases inflammation by inhibiting activator protein 1, a transcription factor for MMP-9, thereby reducing MMP-9 activity and the formation of bullae.4 Clinical anecdotes, including our own, that demonstrate dramatic improvement of hemorrhagic bullae with the administration of corticosteroids substantiate this mechanism. Through the inhibition of neutrophil interactions and IgA production, other anti-inflammatory and immunosuppressive medications such as colchicine, dapsone, and azathioprine also have been reported to aid in resolution of the cutaneous lesions.1,5,6 Although there is a clear drawback to the lack of controlled trials and prospective studies regarding the treatment of bullous HSP, it is nearly impossible to expect such studies to be carried out given the rare and unpredictable nature of the disease. For now, claims derived from case series and case reports guide our understanding of treatment efficacy.
Acknowledgment—Quiz photograph courtesy of Steve Taylor, BS, Phoenix, Arizona.
The Diagnosis: Bullous Henoch-Schönlein Purpura
Laboratory tests in this patient showed no abnormalities for complete blood cell count, immunoglobulins, anti–double-stranded DNA, antinuclear antibody, p–antineutrophil cytoplasmic antibodies, lupus anticoagulant, Sjögren antibodies, liver enzymes, and erythrocyte sedimentation rate. Urinalysis was normal. Punch biopsies were obtained and a histologic examination showed an intense inflammatory infiltrate of neutrophils around blood vessels within the dermis (Figure). These blood vessels showed swollen endothelium and narrowing of the vessel lumina with leukocytoclasia. Direct immunofluorescence revealed granular IgA, C3, fibrin, and weak IgM deposits in blood vessels in the papillary dermis consistent with Henoch-Schönlein purpura (HSP).
Henoch-Schönlein purpura is the most common vasculitis in children.1-6 However, its bullous variant is rare, with few pediatric cases reported. Bullous HSP affects arterioles through an IgA-mediated pathway.1-6 It is believed that the bullae are formed secondary to neutrophilic release of matrix metalloproteinase 9 (MMP-9), which degrades extracellular collagen.2 Additionally, bullous fluid from HSP has been noted to have markedly elevated levels of soluble CD23, a form of the CD23 B-cell surface receptor used in antibody feedback regulation and B-cell recruitment, which also has been found to be elevated in the fluid of bullous pemphigoid, suggesting a similar pathogenesis of exaggerated humoral immunity.3
The most common sign of HSP is palpable purpura; however, other cutaneous findings can be present including targetoid plaques, macules, papules, petechiae, and bullae that may become hemorrhagic, ulcerated, necrotic, or scarred.1-6 Bullae appear in the most dependent parts of the body, such as the feet and lower legs. Hydrostatic pressure may play a role in the pathogenesis of this phenomenon.1 When other classic signs of HSP are absent, the presence of bullae clouds the diagnosis and creates controversy regarding treatment, as there is a dearth of literature on proper therapy for severe cutaneous manifestations of HSP.6
Our patient was treated with morphine for pain management along with topical mupirocin and nonadherent dressings for wound care. She also received pulse intravenous methylprednisolone 2 mg/kg daily for 3 days and then was transitioned to oral prednisone 1 mg/kg daily, which was tapered over 3 weeks after discharge. This regimen resulted in resolution of symptoms with rapid regression of bullae and subsequent postinflammatory hyperpigmentation. Prior reports have noted that the presence of bullae does not alter the prognosis or predict probability of renal involvement of this self-limited disease, leading to controversy in determining if treatment offers more favorable outcomes.1,3 One study suggested that steroids only improve symptoms, arthralgia, and abdominal pain, but they do not aid in the resolution of cutaneous lesions or prevent the progression of renal disease.3 Contrarily, others have suggested that the presence of bullae and renal disease is an indication to start treatment.6 This claim is based on the mechanistic finding that immunosuppression with corticosteroids decreases inflammation by inhibiting activator protein 1, a transcription factor for MMP-9, thereby reducing MMP-9 activity and the formation of bullae.4 Clinical anecdotes, including our own, that demonstrate dramatic improvement of hemorrhagic bullae with the administration of corticosteroids substantiate this mechanism. Through the inhibition of neutrophil interactions and IgA production, other anti-inflammatory and immunosuppressive medications such as colchicine, dapsone, and azathioprine also have been reported to aid in resolution of the cutaneous lesions.1,5,6 Although there is a clear drawback to the lack of controlled trials and prospective studies regarding the treatment of bullous HSP, it is nearly impossible to expect such studies to be carried out given the rare and unpredictable nature of the disease. For now, claims derived from case series and case reports guide our understanding of treatment efficacy.
Acknowledgment—Quiz photograph courtesy of Steve Taylor, BS, Phoenix, Arizona.
- Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature [published online July 16, 2009]. Rheumatol Int. 2010;30:1355-1359. doi:10.1007/s00296-009-1055-8.
- Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiologic role of gelatinate (MMP-9). Dermatology. 1990;197:62-64.
- Bansal AS, Dwivedi N, Adsett M. Serum and blister fluid cytokines and complement proteins in a patient with Henoch Schönlein purpura associated with a bullous skin rash. Australas J Dermatol. 1997;38:190-192.
- Aljada A, Ghanim H, Mohanty P, et al. Hydrocortisone suppresses intranuclear activator-protein-1 (AP-1) binding activity in mononuclear cells and plasma matrix metalloproteinase 2 and 9 (MMP-2 and MMP-9). J Clin Endocrinol Metab. 2001;86:5988-5991.
- Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
- den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch Schönlein purpura as indication to start systemic prednisone [published online January 5, 2009]. Acta Paediatr. 2010;99:781-783. doi:10.1111/j.1651-2227.2009.01650.x.
- Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature [published online July 16, 2009]. Rheumatol Int. 2010;30:1355-1359. doi:10.1007/s00296-009-1055-8.
- Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiologic role of gelatinate (MMP-9). Dermatology. 1990;197:62-64.
- Bansal AS, Dwivedi N, Adsett M. Serum and blister fluid cytokines and complement proteins in a patient with Henoch Schönlein purpura associated with a bullous skin rash. Australas J Dermatol. 1997;38:190-192.
- Aljada A, Ghanim H, Mohanty P, et al. Hydrocortisone suppresses intranuclear activator-protein-1 (AP-1) binding activity in mononuclear cells and plasma matrix metalloproteinase 2 and 9 (MMP-2 and MMP-9). J Clin Endocrinol Metab. 2001;86:5988-5991.
- Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
- den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch Schönlein purpura as indication to start systemic prednisone [published online January 5, 2009]. Acta Paediatr. 2010;99:781-783. doi:10.1111/j.1651-2227.2009.01650.x.

A 12-year-old girl presented with an erythematous eruption that had started on the left leg approximately 1 week prior with subsequent spread to the abdomen and arms. She had associated knee pain, myalgia, abdominal pain, nausea, and nonbloody and nonbilious emesis. Her medical history was notable for methicillin-resistant Staphylococcus aureus abscesses, the most recent of which was treated with trimethoprim-sulfamethoxazole; treatment was completed 5 days before the onset of the rash. Family history was notable for her paternal aunt who died of systemic lupus erythematosus. Physical examination showed erythematous macules and purpuric papules with central vesiculation extending up the thighs and lower abdomen associated with edema of the lower extremities and pain after palpation. Tense bullae also were present.
IOM panel green-lights mitochondrial replacement techniques
Clinical study of mitochondrial replacement techniques for human reproduction is ethically permissible, concluded a special panel formed by the Institute of Medicine and charged by the Food and Drug Administration to study the issue.
If the FDA accepts the recommendations, issued Feb. 3, the agency could proceed with approving investigational new drug (IND) applications from U.S. research groups to test mitochondrial replacement techniques (MRTs) with the goal of allowing couples to bear children who would not inherit mitochondrial DNA diseases affecting the mother.
MRT, also termed mitochondrial transfer, involves creating an embryo with nuclear DNA from the intended mother and mitochondrial DNA from a healthy donor through modification of either an oocyte or zygote. The mitochondrial transfer techniques currently being considered are maternal spindle transfer, which uses oocytes, and pronuclear transfer, which uses zygotes. Critics of the controversial techniques have labeled MRTs as “three parent” IVF and assert that allowing their use will create a slippery slope toward enabling modification of nuclear DNA and germ-line modification.
While the IOM report, which had been in the works since late 2014, helps clear the ethical pathway to clinical testing of these novel methods, lawmakers created a new roadblock with the passage of the fiscal 2016 omnibus budget legislation, which specifically barred the FDA from using budgeted funds to evaluate proposals for therapies that involve embryo modification.
The IOM committee’s report noted that the FDA should consider approval of initial clinical investigations of MRTs “only if and when” several conditions are met, including establishment of safety and minimized risks, an adequate background of likely efficacy through preclinical research, limiting of research to women at risk for transmitting a mitochondrial DNA disease, and limiting of studies to investigators and centers with demonstrated expertise in relevant technologies.
Another key qualification the panel recommended is that initial studies be limited to male embryos so that when they become adults they would not pass their engineered mitochondrial genome to a next generation because mitochondrial inheritance occurs only via oocytes. The committee recommended that modification of mitochondrial DNA in oocytes or zygotes destined to become women be permitted in the future, but only after the procedure proved its safety in men.
The recommendations also stressed that the “health and well-being of any future children born as a result of clinical investigation protocols of MRT should have priority.”

“The main conclusion of the report is that it is ethically permissible to conduct investigations into MRT as long as significant conditions and parameters are met,” said Dr. Marni Falk, director of the mitochondrial-genetic disease clinic at the Children’s Hospital of Philadelphia and a member of the IOM committee.
Dr. Bruce H. Cohen, director of the neurodevelopmental science center and pediatric neurology at Akron (Ohio) Children’s Hospital, said he was pleased with the IOM report.
“There is nothing in the report that will stop this human research from going ahead in the United States. That’s the bottom line,” said Dr. Cohen, who has done clinical research in and drug development for children with mitochondrial disease. “They took everything into consideration, including the science and the pulse of the American public, and came up with a plan that will move this treatment forward.”
Dr. Cohen singled out the MRT research program at the Oregon Health & Science University (OHSU) as the U.S. effort closest to launching a clinical trial, and a senior research official at OHSU agreed that the panel’s recommendations represented a significant step forward.
“We feel the panel did a good job,” said Daniel Dorsa, Ph.D., senior vice president for research at OHSU in Portland. “They put in some restrictions, such as only using male embryos to start, but that is an understandable caution. We’re pleased the report is now out because we’ve been waiting for it. We’ve been in discussions with the FDA for an IND for mitochondrial transfer for quite a while, and the FDA said that we should wait for this report.”
The OHSU research team that works on MRT, led by Shoukhrat Mitalipov, Ph.D., already successfully used “maternal spindle transfer” to produce two macaque embryos that received donor mitochondrial DNA, said Dr. Dorsa. The two macaques remain in gestation with their delivery anticipated soon.
The next step for the OHSU group is to test the same technique with human oocytes, and they have identified several women with either Leigh syndrome or mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) who are interested in assisted reproduction using MRT.

“We’re anxiously waiting to see what the FDA will now do with the new recommendations in hand but with the congressional prohibition also in place,” Dr. Dorsa said. “With the recommendations now out, we anticipate that pressure will mount for Congress to change its position because for now U.S. women with mitochondrial DNA disease will be precluded from this cutting-edge technology that could ensure they give birth to healthy children.”
Dr. Dorsa said that OHSU holds several patents related to MRT, but these patents have not been licensed to any commercial entity.*
Dr. Falk cited results from a 2015 survey of 92 women with mitochondrial DNA disease or at-risk carriers of mutated mitochondrial DNA who expressed their interest in MRT. Among the 92 respondents, 21 said that they were interested in having children. Of that smaller group, 90% said they would be interested in MRT when becoming pregnant.
Dr. Falk also noted that roughly 30,000-60,000 Americans have a mitochondrial disease, although many others might be affected but have never been diagnosed or undergone confirmatory sequencing of their mitochondrial DNA.* Many affected patients have a mutation in 1 of the 200 nuclear genes that can produce mitochondrial dysfunction and not in 1 of the 37 genes contained in mitochondrial DNA. Only women with a mutation in their mitochondrial DNA can benefit from MRT.
*Correction, 2/5/2016: An earlier version of this story failed to correctly distinguish all forms of mitochondrial disease from mitochondrial DNA disease.
*This story was updated 2/8/2016.
On Twitter@mitchelzoler
Clinical study of mitochondrial replacement techniques for human reproduction is ethically permissible, concluded a special panel formed by the Institute of Medicine and charged by the Food and Drug Administration to study the issue.
If the FDA accepts the recommendations, issued Feb. 3, the agency could proceed with approving investigational new drug (IND) applications from U.S. research groups to test mitochondrial replacement techniques (MRTs) with the goal of allowing couples to bear children who would not inherit mitochondrial DNA diseases affecting the mother.
MRT, also termed mitochondrial transfer, involves creating an embryo with nuclear DNA from the intended mother and mitochondrial DNA from a healthy donor through modification of either an oocyte or zygote. The mitochondrial transfer techniques currently being considered are maternal spindle transfer, which uses oocytes, and pronuclear transfer, which uses zygotes. Critics of the controversial techniques have labeled MRTs as “three parent” IVF and assert that allowing their use will create a slippery slope toward enabling modification of nuclear DNA and germ-line modification.
While the IOM report, which had been in the works since late 2014, helps clear the ethical pathway to clinical testing of these novel methods, lawmakers created a new roadblock with the passage of the fiscal 2016 omnibus budget legislation, which specifically barred the FDA from using budgeted funds to evaluate proposals for therapies that involve embryo modification.
The IOM committee’s report noted that the FDA should consider approval of initial clinical investigations of MRTs “only if and when” several conditions are met, including establishment of safety and minimized risks, an adequate background of likely efficacy through preclinical research, limiting of research to women at risk for transmitting a mitochondrial DNA disease, and limiting of studies to investigators and centers with demonstrated expertise in relevant technologies.
Another key qualification the panel recommended is that initial studies be limited to male embryos so that when they become adults they would not pass their engineered mitochondrial genome to a next generation because mitochondrial inheritance occurs only via oocytes. The committee recommended that modification of mitochondrial DNA in oocytes or zygotes destined to become women be permitted in the future, but only after the procedure proved its safety in men.
The recommendations also stressed that the “health and well-being of any future children born as a result of clinical investigation protocols of MRT should have priority.”
“The main conclusion of the report is that it is ethically permissible to conduct investigations into MRT as long as significant conditions and parameters are met,” said Dr. Marni Falk, director of the mitochondrial-genetic disease clinic at the Children’s Hospital of Philadelphia and a member of the IOM committee.
Dr. Bruce H. Cohen, director of the neurodevelopmental science center and pediatric neurology at Akron (Ohio) Children’s Hospital, said he was pleased with the IOM report.
“There is nothing in the report that will stop this human research from going ahead in the United States. That’s the bottom line,” said Dr. Cohen, who has done clinical research in and drug development for children with mitochondrial disease. “They took everything into consideration, including the science and the pulse of the American public, and came up with a plan that will move this treatment forward.”
Dr. Cohen singled out the MRT research program at the Oregon Health & Science University (OHSU) as the U.S. effort closest to launching a clinical trial, and a senior research official at OHSU agreed that the panel’s recommendations represented a significant step forward.
“We feel the panel did a good job,” said Daniel Dorsa, Ph.D., senior vice president for research at OHSU in Portland. “They put in some restrictions, such as only using male embryos to start, but that is an understandable caution. We’re pleased the report is now out because we’ve been waiting for it. We’ve been in discussions with the FDA for an IND for mitochondrial transfer for quite a while, and the FDA said that we should wait for this report.”
The OHSU research team that works on MRT, led by Shoukhrat Mitalipov, Ph.D., already successfully used “maternal spindle transfer” to produce two macaque embryos that received donor mitochondrial DNA, said Dr. Dorsa. The two macaques remain in gestation with their delivery anticipated soon.
The next step for the OHSU group is to test the same technique with human oocytes, and they have identified several women with either Leigh syndrome or mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) who are interested in assisted reproduction using MRT.
“We’re anxiously waiting to see what the FDA will now do with the new recommendations in hand but with the congressional prohibition also in place,” Dr. Dorsa said. “With the recommendations now out, we anticipate that pressure will mount for Congress to change its position because for now U.S. women with mitochondrial DNA disease will be precluded from this cutting-edge technology that could ensure they give birth to healthy children.”
Dr. Dorsa said that OHSU holds several patents related to MRT, but these patents have not been licensed to any commercial entity.*
Dr. Falk cited results from a 2015 survey of 92 women with mitochondrial DNA disease or at-risk carriers of mutated mitochondrial DNA who expressed their interest in MRT. Among the 92 respondents, 21 said that they were interested in having children. Of that smaller group, 90% said they would be interested in MRT when becoming pregnant.
Dr. Falk also noted that roughly 30,000-60,000 Americans have a mitochondrial disease, although many others might be affected but have never been diagnosed or undergone confirmatory sequencing of their mitochondrial DNA.* Many affected patients have a mutation in 1 of the 200 nuclear genes that can produce mitochondrial dysfunction and not in 1 of the 37 genes contained in mitochondrial DNA. Only women with a mutation in their mitochondrial DNA can benefit from MRT.
*Correction, 2/5/2016: An earlier version of this story failed to correctly distinguish all forms of mitochondrial disease from mitochondrial DNA disease.
*This story was updated 2/8/2016.
On Twitter@mitchelzoler
Clinical study of mitochondrial replacement techniques for human reproduction is ethically permissible, concluded a special panel formed by the Institute of Medicine and charged by the Food and Drug Administration to study the issue.
If the FDA accepts the recommendations, issued Feb. 3, the agency could proceed with approving investigational new drug (IND) applications from U.S. research groups to test mitochondrial replacement techniques (MRTs) with the goal of allowing couples to bear children who would not inherit mitochondrial DNA diseases affecting the mother.
MRT, also termed mitochondrial transfer, involves creating an embryo with nuclear DNA from the intended mother and mitochondrial DNA from a healthy donor through modification of either an oocyte or zygote. The mitochondrial transfer techniques currently being considered are maternal spindle transfer, which uses oocytes, and pronuclear transfer, which uses zygotes. Critics of the controversial techniques have labeled MRTs as “three parent” IVF and assert that allowing their use will create a slippery slope toward enabling modification of nuclear DNA and germ-line modification.
While the IOM report, which had been in the works since late 2014, helps clear the ethical pathway to clinical testing of these novel methods, lawmakers created a new roadblock with the passage of the fiscal 2016 omnibus budget legislation, which specifically barred the FDA from using budgeted funds to evaluate proposals for therapies that involve embryo modification.
The IOM committee’s report noted that the FDA should consider approval of initial clinical investigations of MRTs “only if and when” several conditions are met, including establishment of safety and minimized risks, an adequate background of likely efficacy through preclinical research, limiting of research to women at risk for transmitting a mitochondrial DNA disease, and limiting of studies to investigators and centers with demonstrated expertise in relevant technologies.
Another key qualification the panel recommended is that initial studies be limited to male embryos so that when they become adults they would not pass their engineered mitochondrial genome to a next generation because mitochondrial inheritance occurs only via oocytes. The committee recommended that modification of mitochondrial DNA in oocytes or zygotes destined to become women be permitted in the future, but only after the procedure proved its safety in men.
The recommendations also stressed that the “health and well-being of any future children born as a result of clinical investigation protocols of MRT should have priority.”
“The main conclusion of the report is that it is ethically permissible to conduct investigations into MRT as long as significant conditions and parameters are met,” said Dr. Marni Falk, director of the mitochondrial-genetic disease clinic at the Children’s Hospital of Philadelphia and a member of the IOM committee.
Dr. Bruce H. Cohen, director of the neurodevelopmental science center and pediatric neurology at Akron (Ohio) Children’s Hospital, said he was pleased with the IOM report.
“There is nothing in the report that will stop this human research from going ahead in the United States. That’s the bottom line,” said Dr. Cohen, who has done clinical research in and drug development for children with mitochondrial disease. “They took everything into consideration, including the science and the pulse of the American public, and came up with a plan that will move this treatment forward.”
Dr. Cohen singled out the MRT research program at the Oregon Health & Science University (OHSU) as the U.S. effort closest to launching a clinical trial, and a senior research official at OHSU agreed that the panel’s recommendations represented a significant step forward.
“We feel the panel did a good job,” said Daniel Dorsa, Ph.D., senior vice president for research at OHSU in Portland. “They put in some restrictions, such as only using male embryos to start, but that is an understandable caution. We’re pleased the report is now out because we’ve been waiting for it. We’ve been in discussions with the FDA for an IND for mitochondrial transfer for quite a while, and the FDA said that we should wait for this report.”
The OHSU research team that works on MRT, led by Shoukhrat Mitalipov, Ph.D., already successfully used “maternal spindle transfer” to produce two macaque embryos that received donor mitochondrial DNA, said Dr. Dorsa. The two macaques remain in gestation with their delivery anticipated soon.
The next step for the OHSU group is to test the same technique with human oocytes, and they have identified several women with either Leigh syndrome or mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) who are interested in assisted reproduction using MRT.
“We’re anxiously waiting to see what the FDA will now do with the new recommendations in hand but with the congressional prohibition also in place,” Dr. Dorsa said. “With the recommendations now out, we anticipate that pressure will mount for Congress to change its position because for now U.S. women with mitochondrial DNA disease will be precluded from this cutting-edge technology that could ensure they give birth to healthy children.”
Dr. Dorsa said that OHSU holds several patents related to MRT, but these patents have not been licensed to any commercial entity.*
Dr. Falk cited results from a 2015 survey of 92 women with mitochondrial DNA disease or at-risk carriers of mutated mitochondrial DNA who expressed their interest in MRT. Among the 92 respondents, 21 said that they were interested in having children. Of that smaller group, 90% said they would be interested in MRT when becoming pregnant.
Dr. Falk also noted that roughly 30,000-60,000 Americans have a mitochondrial disease, although many others might be affected but have never been diagnosed or undergone confirmatory sequencing of their mitochondrial DNA.* Many affected patients have a mutation in 1 of the 200 nuclear genes that can produce mitochondrial dysfunction and not in 1 of the 37 genes contained in mitochondrial DNA. Only women with a mutation in their mitochondrial DNA can benefit from MRT.
*Correction, 2/5/2016: An earlier version of this story failed to correctly distinguish all forms of mitochondrial disease from mitochondrial DNA disease.
*This story was updated 2/8/2016.
On Twitter@mitchelzoler
Venetoclax shows promise for relapsed CLL, SLL
Daily oral treatment with venetoclax induced substantial responses with manageable adverse effects in patients with relapsed chronic lymphocytic leukemia or small lymphocytic lymphoma in a first-in-human phase I dose-escalation study.
The promising effects of the highly selective investigational inhibitor of BCL2 – a protein central to the survival of CLL cells – were noted even in patients with poor prognostic features, who comprised 89% of the cohort, reported Dr. Andrew W. Roberts of Royal Melbourne Hospital, Australia, and his colleagues. The study was published online Jan. 28 in The New England Journal of Medicine.
In the dose escalation phase of the study, 56 patients received active treatment at doses raging from 150 to 1,200 mg daily, and 60 additional patients received weekly stepwise ramp-up with doses beginning at 20 mg daily with weekly increases to 50 mg, 100 mg, and 200 mg daily up to the target dose of 400 mg daily. The patients had received a median of 3 previous therapies (range, 1-11).
Of 116 treated patients, 92 (79%) had a response, and 20% achieved complete remission, including 5% with no minimal residual disease on flow cytometry, the investigators said (N Engl J Med. 2016;374:311-22).
Venetoclax was active at all doses used in the study, and no maximum tolerated dose was identified.
Tumor lysis syndrome occurred in 10 patients, but clinically important sequelae occurred in only 3 of those patients, 2 of whom had severe sequelae. After adjustments were made to dosing schedule, no further cases occurred.
Other side effects included mild diarrhea, upper respiratory tract infection, nausea, and grade 3 or 4 neutropenia, which occurred in 41%-52% of patients. Serious adverse events included febrile neutropenia in 6% of patients, pneumonia in 4%, upper respiratory tract infection in 3%, and immune thrombocytopenia in 3%.
Among the patients with an adverse prognosis, treatment response rates ranged from 71% to 79%, depending on the subgroup. For example, the response rate was 79% in 70 patients with resistance to fludarabine, and 71% in 31 patients with chromosome 17p deletions.
New treatments, including ibrutinib monotherapy and idelalisib in combination with rituximab, have improved outcomes for patients with relapsed CLL, but despite these advances, complete remissions remain uncommon, the authors said.
“This first trial of venetoclax showed the potential of BCL2 antagonism as an additional therapeutic avenue for patients with relapsed CLL,” they wrote, adding that the 79% overall response rate in this study – including deep responses and complete responses without minimal residual disease in patients up to age 86 years and patients with poor prognostic factors – “provides support for further development of venetoclax as a treatment option for patients with heavily pretreated relapsed or refractory CLL or SLL.”
Of note, the Food and Drug Administration on Jan. 28 – the date this study was released – granted venetoclax Breakthrough Therapy Designation for use in combination with hypomethylating agents for the treatment of acute myeloid leukemia patients who aren’t eligible for standard induction chemotherapy. The designation – the third for the agent – is supported by data from a single study of untreated patients aged 65 years or older with AML. Prior venetoclax Breakthrough Therapy Designations were granted in April 2015 for its use as monotherapy in patients with refractory CLL who have the 17p deletion genetic mutation, and in January for its use with rituximab for the treatment of relapsed/refractory CLL.
AbbVie and Genentech supported the study. Dr. Roberts reported receiving grant support and study drugs form AbbVie, serving as an investigator in trials sponsored by Genentech, AbbVie, Janssen, and Beigene, and receiving institutional research funding from Genentech for the development of venetoclax. His coauthors reported ties to various pharmaceutical companies.
Targeted therapies have fundamentally changed the management and outcomes of patients with CLL in recent years, and new findings for second-generation drugs offer even more promise, according to Dr. Wyndham H. Wilson.
Taken together with the recent finding that acalabrutinib has a high degree of Bruton’s tyrosine kinase (BTK) inhibition with lower toxicity than ibrutinib, the findings of Roberts et al. with respect to venetoclax suggest a possible new avenue for combination treatment, Dr. Wilson wrote in an editorial (N Engl J Med. 2016;374;4:386-8).
“The transformative characteristics of acalabrutinib and venetoclax arise from effective targeting of important survival pathways in CLL. Indeed, BTK inhibition produces durable responses, improves survival, and selects for mutations in the BTK-binding domain,” Dr. Wilson said, adding that BCL2 also plays an important role in CLL survival, as indicated by the activity of venetoclax.
While neither venetoclax nor acalabrutinib regularly induce complete remission, in vitro findings show that venetoclax and BTK inhibitors are synergistic, which suggests that combining the two might “further transform the targeted treatment of CLL,” he explained.
Dr. Wilson is with the National Cancer Institute, Bethesda, Md.
Targeted therapies have fundamentally changed the management and outcomes of patients with CLL in recent years, and new findings for second-generation drugs offer even more promise, according to Dr. Wyndham H. Wilson.
Taken together with the recent finding that acalabrutinib has a high degree of Bruton’s tyrosine kinase (BTK) inhibition with lower toxicity than ibrutinib, the findings of Roberts et al. with respect to venetoclax suggest a possible new avenue for combination treatment, Dr. Wilson wrote in an editorial (N Engl J Med. 2016;374;4:386-8).
“The transformative characteristics of acalabrutinib and venetoclax arise from effective targeting of important survival pathways in CLL. Indeed, BTK inhibition produces durable responses, improves survival, and selects for mutations in the BTK-binding domain,” Dr. Wilson said, adding that BCL2 also plays an important role in CLL survival, as indicated by the activity of venetoclax.
While neither venetoclax nor acalabrutinib regularly induce complete remission, in vitro findings show that venetoclax and BTK inhibitors are synergistic, which suggests that combining the two might “further transform the targeted treatment of CLL,” he explained.
Dr. Wilson is with the National Cancer Institute, Bethesda, Md.
Targeted therapies have fundamentally changed the management and outcomes of patients with CLL in recent years, and new findings for second-generation drugs offer even more promise, according to Dr. Wyndham H. Wilson.
Taken together with the recent finding that acalabrutinib has a high degree of Bruton’s tyrosine kinase (BTK) inhibition with lower toxicity than ibrutinib, the findings of Roberts et al. with respect to venetoclax suggest a possible new avenue for combination treatment, Dr. Wilson wrote in an editorial (N Engl J Med. 2016;374;4:386-8).
“The transformative characteristics of acalabrutinib and venetoclax arise from effective targeting of important survival pathways in CLL. Indeed, BTK inhibition produces durable responses, improves survival, and selects for mutations in the BTK-binding domain,” Dr. Wilson said, adding that BCL2 also plays an important role in CLL survival, as indicated by the activity of venetoclax.
While neither venetoclax nor acalabrutinib regularly induce complete remission, in vitro findings show that venetoclax and BTK inhibitors are synergistic, which suggests that combining the two might “further transform the targeted treatment of CLL,” he explained.
Dr. Wilson is with the National Cancer Institute, Bethesda, Md.
Daily oral treatment with venetoclax induced substantial responses with manageable adverse effects in patients with relapsed chronic lymphocytic leukemia or small lymphocytic lymphoma in a first-in-human phase I dose-escalation study.
The promising effects of the highly selective investigational inhibitor of BCL2 – a protein central to the survival of CLL cells – were noted even in patients with poor prognostic features, who comprised 89% of the cohort, reported Dr. Andrew W. Roberts of Royal Melbourne Hospital, Australia, and his colleagues. The study was published online Jan. 28 in The New England Journal of Medicine.
In the dose escalation phase of the study, 56 patients received active treatment at doses raging from 150 to 1,200 mg daily, and 60 additional patients received weekly stepwise ramp-up with doses beginning at 20 mg daily with weekly increases to 50 mg, 100 mg, and 200 mg daily up to the target dose of 400 mg daily. The patients had received a median of 3 previous therapies (range, 1-11).
Of 116 treated patients, 92 (79%) had a response, and 20% achieved complete remission, including 5% with no minimal residual disease on flow cytometry, the investigators said (N Engl J Med. 2016;374:311-22).
Venetoclax was active at all doses used in the study, and no maximum tolerated dose was identified.
Tumor lysis syndrome occurred in 10 patients, but clinically important sequelae occurred in only 3 of those patients, 2 of whom had severe sequelae. After adjustments were made to dosing schedule, no further cases occurred.
Other side effects included mild diarrhea, upper respiratory tract infection, nausea, and grade 3 or 4 neutropenia, which occurred in 41%-52% of patients. Serious adverse events included febrile neutropenia in 6% of patients, pneumonia in 4%, upper respiratory tract infection in 3%, and immune thrombocytopenia in 3%.
Among the patients with an adverse prognosis, treatment response rates ranged from 71% to 79%, depending on the subgroup. For example, the response rate was 79% in 70 patients with resistance to fludarabine, and 71% in 31 patients with chromosome 17p deletions.
New treatments, including ibrutinib monotherapy and idelalisib in combination with rituximab, have improved outcomes for patients with relapsed CLL, but despite these advances, complete remissions remain uncommon, the authors said.
“This first trial of venetoclax showed the potential of BCL2 antagonism as an additional therapeutic avenue for patients with relapsed CLL,” they wrote, adding that the 79% overall response rate in this study – including deep responses and complete responses without minimal residual disease in patients up to age 86 years and patients with poor prognostic factors – “provides support for further development of venetoclax as a treatment option for patients with heavily pretreated relapsed or refractory CLL or SLL.”
Of note, the Food and Drug Administration on Jan. 28 – the date this study was released – granted venetoclax Breakthrough Therapy Designation for use in combination with hypomethylating agents for the treatment of acute myeloid leukemia patients who aren’t eligible for standard induction chemotherapy. The designation – the third for the agent – is supported by data from a single study of untreated patients aged 65 years or older with AML. Prior venetoclax Breakthrough Therapy Designations were granted in April 2015 for its use as monotherapy in patients with refractory CLL who have the 17p deletion genetic mutation, and in January for its use with rituximab for the treatment of relapsed/refractory CLL.
AbbVie and Genentech supported the study. Dr. Roberts reported receiving grant support and study drugs form AbbVie, serving as an investigator in trials sponsored by Genentech, AbbVie, Janssen, and Beigene, and receiving institutional research funding from Genentech for the development of venetoclax. His coauthors reported ties to various pharmaceutical companies.
Daily oral treatment with venetoclax induced substantial responses with manageable adverse effects in patients with relapsed chronic lymphocytic leukemia or small lymphocytic lymphoma in a first-in-human phase I dose-escalation study.
The promising effects of the highly selective investigational inhibitor of BCL2 – a protein central to the survival of CLL cells – were noted even in patients with poor prognostic features, who comprised 89% of the cohort, reported Dr. Andrew W. Roberts of Royal Melbourne Hospital, Australia, and his colleagues. The study was published online Jan. 28 in The New England Journal of Medicine.
In the dose escalation phase of the study, 56 patients received active treatment at doses raging from 150 to 1,200 mg daily, and 60 additional patients received weekly stepwise ramp-up with doses beginning at 20 mg daily with weekly increases to 50 mg, 100 mg, and 200 mg daily up to the target dose of 400 mg daily. The patients had received a median of 3 previous therapies (range, 1-11).
Of 116 treated patients, 92 (79%) had a response, and 20% achieved complete remission, including 5% with no minimal residual disease on flow cytometry, the investigators said (N Engl J Med. 2016;374:311-22).
Venetoclax was active at all doses used in the study, and no maximum tolerated dose was identified.
Tumor lysis syndrome occurred in 10 patients, but clinically important sequelae occurred in only 3 of those patients, 2 of whom had severe sequelae. After adjustments were made to dosing schedule, no further cases occurred.
Other side effects included mild diarrhea, upper respiratory tract infection, nausea, and grade 3 or 4 neutropenia, which occurred in 41%-52% of patients. Serious adverse events included febrile neutropenia in 6% of patients, pneumonia in 4%, upper respiratory tract infection in 3%, and immune thrombocytopenia in 3%.
Among the patients with an adverse prognosis, treatment response rates ranged from 71% to 79%, depending on the subgroup. For example, the response rate was 79% in 70 patients with resistance to fludarabine, and 71% in 31 patients with chromosome 17p deletions.
New treatments, including ibrutinib monotherapy and idelalisib in combination with rituximab, have improved outcomes for patients with relapsed CLL, but despite these advances, complete remissions remain uncommon, the authors said.
“This first trial of venetoclax showed the potential of BCL2 antagonism as an additional therapeutic avenue for patients with relapsed CLL,” they wrote, adding that the 79% overall response rate in this study – including deep responses and complete responses without minimal residual disease in patients up to age 86 years and patients with poor prognostic factors – “provides support for further development of venetoclax as a treatment option for patients with heavily pretreated relapsed or refractory CLL or SLL.”
Of note, the Food and Drug Administration on Jan. 28 – the date this study was released – granted venetoclax Breakthrough Therapy Designation for use in combination with hypomethylating agents for the treatment of acute myeloid leukemia patients who aren’t eligible for standard induction chemotherapy. The designation – the third for the agent – is supported by data from a single study of untreated patients aged 65 years or older with AML. Prior venetoclax Breakthrough Therapy Designations were granted in April 2015 for its use as monotherapy in patients with refractory CLL who have the 17p deletion genetic mutation, and in January for its use with rituximab for the treatment of relapsed/refractory CLL.
AbbVie and Genentech supported the study. Dr. Roberts reported receiving grant support and study drugs form AbbVie, serving as an investigator in trials sponsored by Genentech, AbbVie, Janssen, and Beigene, and receiving institutional research funding from Genentech for the development of venetoclax. His coauthors reported ties to various pharmaceutical companies.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Daily oral treatment with venetoclax induced substantial responses with manageable adverse effects in patients with relapsed chronic lymphocytic leukemia or small lymphocytic lymphoma in a first-in-human phase I dose-escalation study.
Major finding: Of 116 treated patients, 92 (79%) had a response, and 20% achieved complete remission, including 5% with no minimal residual disease on flow cytometry.
Data source: A phase I dose-escalation study involving 116 patients.
Disclosures: AbbVie and Genentech supported the study. Dr. Roberts reported receiving grant support and study drugs form AbbVie, serving as an investigator in trials sponsored by Genentech, AbbVie, Janssen, and Beigene, and receiving institutional research funding from Genentech for the development of venetoclax. His coauthors reported ties to various pharmaceutical companies.
Tourniquets overemphasized in mass shooting response planning
SAN ANTONIO – Tourniquets are being overemphasized in preparations for mass shootings in the United States, according to a review by the Committee for Tactical Emergency Casualty Care of wounding patterns in 12 mass shootings.
Since the 2012 shootings in Newtown, Conn., efforts to improve survival have tended to emphasize tourniquets to stop bleeding from wounded limbs. The recently-launched federal “Stop the Bleed” campaign promotes public awareness of compression and tourniquets, and the American College of Surgeons and other groups recently called for public education and use of tourniquets as one of many responses to mass-casualty and active-shooter events, stating that “the most significant preventable cause of death in the prehospital environment is external hemorrhage.” There are thoughts of making tourniquets at least as common as automated external defibrillators in public settings.
The efforts are driven by combat experience in Afghanistan and Iraq, where about half of wounds were to arms and legs, and tourniquets saved many lives.

Tourniquets are important especially for civilian bombings, but fatalities in civilian mass shootings – shootings with at least four deaths in which killing is the primary aim – are from head and chest wounds, not extremity wounds. The emphasis on bleeding control overshadows public education about other simple interventions that could be more useful in shootings, according to emergency physician Dr. Reed Smith, medical director of the Arlington County (Va.) Fire Department and cofounder and cochairman of the Committee for Tactical Emergency Casualty Care (C-TECC).
“We need a civilian perspective on civilian incidents, not a military perspective,” he said at the Eastern Association for the Surgery of Trauma scientific assembly.
Dr. Smith and other C-TECC investigators reviewed autopsy reports from 12 U.S. civilian mass shootings from the period of 1996-2012. They tried to get data for more, but ran into significant resistance from authorities.
There were 371 bullet wounds and 139 deaths, an average of 2.7 wounds per victim but a range of 1-10. About 40% of deaths were from shots to the head; another 40% from shots to the chest or upper back; and the rest from shots to the face/neck, abdomen/lower back, or multiple regions.

“We looked at every one of these events, and with the exception of the Gabby Giffords Tucson shooting, no tourniquets were used. There were no cases of major peripheral vascular injury, and no fatalities from extremity exsanguination,” Dr. Smith said.
The investigators estimated that nine fatalities were potentially survivable, without injuries to the heart, brain, or major blood vessels, and definitive care available within 60 minutes. The victims died from face and torso wounds, not wounds to the limbs.
“Wounding in active-shooter events differs from combat and may require different therapeutic emphasis. Although tourniquets and external hemorrhage control techniques hold value, their role in active-shooter events may be overemphasized as a means to decrease fatality. The focus on external hemorrhage is important, but it’s not enough,” Dr. Smith said.
Additional interventions could be taught for every step in the chain of contact, from citizen to trauma surgeon, that could have greater benefit. Teachers, for instance, could be taught to roll victims on their sides and clear out their mouths to keep airways open, and to keep them warm to prevent hypothermia until help arrives. Police could learn to insert nasal airways, and occlude sucking chest wounds. Fire/EMS could start initial damage control. “Everything builds,” Dr. Smith said.
C-TECC made many recommendations for enhanced mass-causality response its 2015 Tactical Emergency Casualty Care Guidelines.
Dr. Smith had no disclosures.
SAN ANTONIO – Tourniquets are being overemphasized in preparations for mass shootings in the United States, according to a review by the Committee for Tactical Emergency Casualty Care of wounding patterns in 12 mass shootings.
Since the 2012 shootings in Newtown, Conn., efforts to improve survival have tended to emphasize tourniquets to stop bleeding from wounded limbs. The recently-launched federal “Stop the Bleed” campaign promotes public awareness of compression and tourniquets, and the American College of Surgeons and other groups recently called for public education and use of tourniquets as one of many responses to mass-casualty and active-shooter events, stating that “the most significant preventable cause of death in the prehospital environment is external hemorrhage.” There are thoughts of making tourniquets at least as common as automated external defibrillators in public settings.
The efforts are driven by combat experience in Afghanistan and Iraq, where about half of wounds were to arms and legs, and tourniquets saved many lives.
Tourniquets are important especially for civilian bombings, but fatalities in civilian mass shootings – shootings with at least four deaths in which killing is the primary aim – are from head and chest wounds, not extremity wounds. The emphasis on bleeding control overshadows public education about other simple interventions that could be more useful in shootings, according to emergency physician Dr. Reed Smith, medical director of the Arlington County (Va.) Fire Department and cofounder and cochairman of the Committee for Tactical Emergency Casualty Care (C-TECC).
“We need a civilian perspective on civilian incidents, not a military perspective,” he said at the Eastern Association for the Surgery of Trauma scientific assembly.
Dr. Smith and other C-TECC investigators reviewed autopsy reports from 12 U.S. civilian mass shootings from the period of 1996-2012. They tried to get data for more, but ran into significant resistance from authorities.
There were 371 bullet wounds and 139 deaths, an average of 2.7 wounds per victim but a range of 1-10. About 40% of deaths were from shots to the head; another 40% from shots to the chest or upper back; and the rest from shots to the face/neck, abdomen/lower back, or multiple regions.
“We looked at every one of these events, and with the exception of the Gabby Giffords Tucson shooting, no tourniquets were used. There were no cases of major peripheral vascular injury, and no fatalities from extremity exsanguination,” Dr. Smith said.
The investigators estimated that nine fatalities were potentially survivable, without injuries to the heart, brain, or major blood vessels, and definitive care available within 60 minutes. The victims died from face and torso wounds, not wounds to the limbs.
“Wounding in active-shooter events differs from combat and may require different therapeutic emphasis. Although tourniquets and external hemorrhage control techniques hold value, their role in active-shooter events may be overemphasized as a means to decrease fatality. The focus on external hemorrhage is important, but it’s not enough,” Dr. Smith said.
Additional interventions could be taught for every step in the chain of contact, from citizen to trauma surgeon, that could have greater benefit. Teachers, for instance, could be taught to roll victims on their sides and clear out their mouths to keep airways open, and to keep them warm to prevent hypothermia until help arrives. Police could learn to insert nasal airways, and occlude sucking chest wounds. Fire/EMS could start initial damage control. “Everything builds,” Dr. Smith said.
C-TECC made many recommendations for enhanced mass-causality response its 2015 Tactical Emergency Casualty Care Guidelines.
Dr. Smith had no disclosures.
SAN ANTONIO – Tourniquets are being overemphasized in preparations for mass shootings in the United States, according to a review by the Committee for Tactical Emergency Casualty Care of wounding patterns in 12 mass shootings.
Since the 2012 shootings in Newtown, Conn., efforts to improve survival have tended to emphasize tourniquets to stop bleeding from wounded limbs. The recently-launched federal “Stop the Bleed” campaign promotes public awareness of compression and tourniquets, and the American College of Surgeons and other groups recently called for public education and use of tourniquets as one of many responses to mass-casualty and active-shooter events, stating that “the most significant preventable cause of death in the prehospital environment is external hemorrhage.” There are thoughts of making tourniquets at least as common as automated external defibrillators in public settings.
The efforts are driven by combat experience in Afghanistan and Iraq, where about half of wounds were to arms and legs, and tourniquets saved many lives.
Tourniquets are important especially for civilian bombings, but fatalities in civilian mass shootings – shootings with at least four deaths in which killing is the primary aim – are from head and chest wounds, not extremity wounds. The emphasis on bleeding control overshadows public education about other simple interventions that could be more useful in shootings, according to emergency physician Dr. Reed Smith, medical director of the Arlington County (Va.) Fire Department and cofounder and cochairman of the Committee for Tactical Emergency Casualty Care (C-TECC).
“We need a civilian perspective on civilian incidents, not a military perspective,” he said at the Eastern Association for the Surgery of Trauma scientific assembly.
Dr. Smith and other C-TECC investigators reviewed autopsy reports from 12 U.S. civilian mass shootings from the period of 1996-2012. They tried to get data for more, but ran into significant resistance from authorities.
There were 371 bullet wounds and 139 deaths, an average of 2.7 wounds per victim but a range of 1-10. About 40% of deaths were from shots to the head; another 40% from shots to the chest or upper back; and the rest from shots to the face/neck, abdomen/lower back, or multiple regions.
“We looked at every one of these events, and with the exception of the Gabby Giffords Tucson shooting, no tourniquets were used. There were no cases of major peripheral vascular injury, and no fatalities from extremity exsanguination,” Dr. Smith said.
The investigators estimated that nine fatalities were potentially survivable, without injuries to the heart, brain, or major blood vessels, and definitive care available within 60 minutes. The victims died from face and torso wounds, not wounds to the limbs.
“Wounding in active-shooter events differs from combat and may require different therapeutic emphasis. Although tourniquets and external hemorrhage control techniques hold value, their role in active-shooter events may be overemphasized as a means to decrease fatality. The focus on external hemorrhage is important, but it’s not enough,” Dr. Smith said.
Additional interventions could be taught for every step in the chain of contact, from citizen to trauma surgeon, that could have greater benefit. Teachers, for instance, could be taught to roll victims on their sides and clear out their mouths to keep airways open, and to keep them warm to prevent hypothermia until help arrives. Police could learn to insert nasal airways, and occlude sucking chest wounds. Fire/EMS could start initial damage control. “Everything builds,” Dr. Smith said.
C-TECC made many recommendations for enhanced mass-causality response its 2015 Tactical Emergency Casualty Care Guidelines.
Dr. Smith had no disclosures.
AT THE EAST SCIENTIFIC ASSEMBLY
Key clinical point: Tourniquets aren’t much help in civilian mass shootings.
Major finding: There were no cases of major peripheral vascular injury, and no fatalities from extremity exsanguination in a review of 12 mass shootings in the United States from 1996-2012.
Data source: Autopsy reports.
Disclosures: The lead investigator had no disclosures.

Majority of children aged 6-23 months are not vaccinated for flu
Less than half of children aged 6-23 months are vaccinated for influenza in the United States, according to an analysis of data obtained via the 2003-2013 National Immunization Survey.
The researchers analyzed providers’ reports of influenza vaccinations, received as one or two doses by children aged 6-23 months. The age group studied is at highest risk of influenza-related complications and was the first group of children for which the Advisory Committee on Immunization Practices recommended influenza vaccination, regardless of an individual’s medical condition.
A child’s age was defined by his or her age on Nov. 1 of each influenza season under study. Two full calendar years of data files were combined to enable analysis of full influenza seasons, which cover parts of 2 consecutive calendar years. The percentages of children requiring two doses to be considered fully vaccinated were based on the dosage recommendations for each flu season.
Overall, flu vaccination coverage increased, reaching 45% in the 2011-2012 flu season, up from 5% during the 2002-2003 flu season. Within each racial/ethnic group examined, influenza vaccination coverage also grew; however, lower percentages of non-Hispanic black children and Hispanic children were vaccinated than of non-Hispanic white children during all 10 of the flu seasons studied. Coverage ranged from 24% in Mississippi to 72% in Massachusetts.
“Despite the increase, the majority of children 6-23 months in the United States were not fully vaccinated against influenza,” said Tammy A. Santibanez, Ph.D., of the Centers for Disease Control and Prevention, and her colleagues.
Among other findings consistent throughout each flu season examined was that full influenza vaccination coverage was higher among children requiring only one dose of a flu vaccine, compared with those requiring two doses of a flu vaccine.
“Prevention of influenza among infants and young children is a public health priority because of their high risk for influenza-related complications,” wrote Dr. Santibanez and her colleagues. “Appropriate implementation of evidence-based strategies is needed to increase the percentage of children who are fully vaccinated.”
Read the study in Pediatrics (doi: 10.1542/peds.2015.3280).
Less than half of children aged 6-23 months are vaccinated for influenza in the United States, according to an analysis of data obtained via the 2003-2013 National Immunization Survey.
The researchers analyzed providers’ reports of influenza vaccinations, received as one or two doses by children aged 6-23 months. The age group studied is at highest risk of influenza-related complications and was the first group of children for which the Advisory Committee on Immunization Practices recommended influenza vaccination, regardless of an individual’s medical condition.
A child’s age was defined by his or her age on Nov. 1 of each influenza season under study. Two full calendar years of data files were combined to enable analysis of full influenza seasons, which cover parts of 2 consecutive calendar years. The percentages of children requiring two doses to be considered fully vaccinated were based on the dosage recommendations for each flu season.
Overall, flu vaccination coverage increased, reaching 45% in the 2011-2012 flu season, up from 5% during the 2002-2003 flu season. Within each racial/ethnic group examined, influenza vaccination coverage also grew; however, lower percentages of non-Hispanic black children and Hispanic children were vaccinated than of non-Hispanic white children during all 10 of the flu seasons studied. Coverage ranged from 24% in Mississippi to 72% in Massachusetts.
“Despite the increase, the majority of children 6-23 months in the United States were not fully vaccinated against influenza,” said Tammy A. Santibanez, Ph.D., of the Centers for Disease Control and Prevention, and her colleagues.
Among other findings consistent throughout each flu season examined was that full influenza vaccination coverage was higher among children requiring only one dose of a flu vaccine, compared with those requiring two doses of a flu vaccine.
“Prevention of influenza among infants and young children is a public health priority because of their high risk for influenza-related complications,” wrote Dr. Santibanez and her colleagues. “Appropriate implementation of evidence-based strategies is needed to increase the percentage of children who are fully vaccinated.”
Read the study in Pediatrics (doi: 10.1542/peds.2015.3280).
Less than half of children aged 6-23 months are vaccinated for influenza in the United States, according to an analysis of data obtained via the 2003-2013 National Immunization Survey.
The researchers analyzed providers’ reports of influenza vaccinations, received as one or two doses by children aged 6-23 months. The age group studied is at highest risk of influenza-related complications and was the first group of children for which the Advisory Committee on Immunization Practices recommended influenza vaccination, regardless of an individual’s medical condition.
A child’s age was defined by his or her age on Nov. 1 of each influenza season under study. Two full calendar years of data files were combined to enable analysis of full influenza seasons, which cover parts of 2 consecutive calendar years. The percentages of children requiring two doses to be considered fully vaccinated were based on the dosage recommendations for each flu season.
Overall, flu vaccination coverage increased, reaching 45% in the 2011-2012 flu season, up from 5% during the 2002-2003 flu season. Within each racial/ethnic group examined, influenza vaccination coverage also grew; however, lower percentages of non-Hispanic black children and Hispanic children were vaccinated than of non-Hispanic white children during all 10 of the flu seasons studied. Coverage ranged from 24% in Mississippi to 72% in Massachusetts.
“Despite the increase, the majority of children 6-23 months in the United States were not fully vaccinated against influenza,” said Tammy A. Santibanez, Ph.D., of the Centers for Disease Control and Prevention, and her colleagues.
Among other findings consistent throughout each flu season examined was that full influenza vaccination coverage was higher among children requiring only one dose of a flu vaccine, compared with those requiring two doses of a flu vaccine.
“Prevention of influenza among infants and young children is a public health priority because of their high risk for influenza-related complications,” wrote Dr. Santibanez and her colleagues. “Appropriate implementation of evidence-based strategies is needed to increase the percentage of children who are fully vaccinated.”
Read the study in Pediatrics (doi: 10.1542/peds.2015.3280).
FROM PEDIATRICS
Practice Expanding: The Rising Trend in Hospitalist Co-Management
As the practice of medicine continues to transition to performance-based payment systems, the number of mergers of hospitalists and specialists has surged. Payment models that focus on clinical outcomes and best practices link payment to the ability of physicians to provide efficient, quality healthcare and improve patient outcomes. These payment systems are changing the way healthcare services are delivered by demanding better patient care at a lower cost. The result is increasing pressure on physicians to meet operational and quality goals, or receive less reimbursement for their services.

Studies have shown that the effective use of hospitalists can improve standardized patient care for surgical patients. Hospitalists also provide value to specialists by freeing up time so they can focus on their area of expertise. As a result, co-management arrangements between hospitalists and specialists have become a popular tool to define working relationships and improve the quality of care patients receive.
Hospitalist Evolution

When hospitalists first debuted, they were seen as a threat to primary care physicians and specialists. Over time, they were criticized for performing routine work for specialized physicians. To overcome these negative connotations and prove their worth, hospitalists began co-managing patients for surgical specialists, who soon realized the significant value hospitalist services provided. Not only do they share in the responsibility of care provided to patients, but they also reduce readmissions and costs associated with providing healthcare.
Now there are even specialty hospitalists who specialize in a particular field, such as orthopedics or obstetrics.
Why Co-Management?
Hospitalists add value by helping to alleviate the burden on specialists—providing ED coverage, assisting in the operating room, and rounding on patients. They evaluate surgical patients for medical issues, reconcile medications across the spectrum of a patient’s care, and standardize the patient discharge and communication processes.
Providing these services frees specialists from rounding and allows them to concentrate on their specialty. Hospitalists do not have office-based practices, which allows them to spend their time in the hospital caring for admitted, pre-operative, and post-operative patients.
It is in the pre-operative and post-operative environments where hospitalists have established their extreme value to specialists. Under co-management arrangements, hospitalists are able to ensure that all pre-operative tests are conducted, reports are dictated, and the patient’s medical history is available. Pre-operative evaluations allow the hospitalist to develop a post-operative plan of care and proactively address many medical concerns. Also, the hospitalist is available to see patients immediately after surgery, allowing immediate evaluation and treatment for high blood pressure, diabetic issues, or other medical issues.
In sum, the hospitalist is responsible for the medical care of the specialist’s patients, and the specialist is able to focus on the specialty services he or she provides. Providing these services gives hospitalists the opportunity to anticipate problems and overcome issues, which results in more efficient care, shorter lengths of stay in the hospital, and improved patient satisfaction. Such results make hospitalists critical to success in performance-based payment systems.
Successful Co-Management Arrangements
A key to success in establishing a co-management arrangement between a hospitalist and a specialist is setting forth the parameters of the relationship in a written agreement. It is particularly important that the relationship foster equality among the parties, regardless of who is the attending physician of record. The parties should be jointly responsible for patient care, with the hospitalist treating the patient’s general medical concerns and the specialist focusing on techniques within his specialty to improve the patient’s issues.
The agreement should clearly state the responsibilities of each party, including delineating the party responsible for decisions such as admission and discharge. It should address resources and set forth the standardized processes and protocols to be used when treating patients.
Specialists can vary in their treatment of patients, so it is best to document their expectations at the onset of the relationship. Also, successful co-management is contingent upon regular communication between the hospitalist and the specialist. It is important to establish those boundaries in advance to prevent miscommunication down the road.
In particular, the agreement should explicitly describe the lines of authority and how conflicts will be addressed.
Final Thoughts
Co-management is a growing trend that can provide an opportunity for hospitalists to expand their practice and reinforce their value to both specialists and the hospital. The improved quality of care and patient satisfaction that is associated with hospitalist services can be crucial to maximizing reimbursement under a value-based reimbursement system. TH
As the practice of medicine continues to transition to performance-based payment systems, the number of mergers of hospitalists and specialists has surged. Payment models that focus on clinical outcomes and best practices link payment to the ability of physicians to provide efficient, quality healthcare and improve patient outcomes. These payment systems are changing the way healthcare services are delivered by demanding better patient care at a lower cost. The result is increasing pressure on physicians to meet operational and quality goals, or receive less reimbursement for their services.
Studies have shown that the effective use of hospitalists can improve standardized patient care for surgical patients. Hospitalists also provide value to specialists by freeing up time so they can focus on their area of expertise. As a result, co-management arrangements between hospitalists and specialists have become a popular tool to define working relationships and improve the quality of care patients receive.
Hospitalist Evolution
When hospitalists first debuted, they were seen as a threat to primary care physicians and specialists. Over time, they were criticized for performing routine work for specialized physicians. To overcome these negative connotations and prove their worth, hospitalists began co-managing patients for surgical specialists, who soon realized the significant value hospitalist services provided. Not only do they share in the responsibility of care provided to patients, but they also reduce readmissions and costs associated with providing healthcare.
Now there are even specialty hospitalists who specialize in a particular field, such as orthopedics or obstetrics.
Why Co-Management?
Hospitalists add value by helping to alleviate the burden on specialists—providing ED coverage, assisting in the operating room, and rounding on patients. They evaluate surgical patients for medical issues, reconcile medications across the spectrum of a patient’s care, and standardize the patient discharge and communication processes.
Providing these services frees specialists from rounding and allows them to concentrate on their specialty. Hospitalists do not have office-based practices, which allows them to spend their time in the hospital caring for admitted, pre-operative, and post-operative patients.
It is in the pre-operative and post-operative environments where hospitalists have established their extreme value to specialists. Under co-management arrangements, hospitalists are able to ensure that all pre-operative tests are conducted, reports are dictated, and the patient’s medical history is available. Pre-operative evaluations allow the hospitalist to develop a post-operative plan of care and proactively address many medical concerns. Also, the hospitalist is available to see patients immediately after surgery, allowing immediate evaluation and treatment for high blood pressure, diabetic issues, or other medical issues.
In sum, the hospitalist is responsible for the medical care of the specialist’s patients, and the specialist is able to focus on the specialty services he or she provides. Providing these services gives hospitalists the opportunity to anticipate problems and overcome issues, which results in more efficient care, shorter lengths of stay in the hospital, and improved patient satisfaction. Such results make hospitalists critical to success in performance-based payment systems.
Successful Co-Management Arrangements
A key to success in establishing a co-management arrangement between a hospitalist and a specialist is setting forth the parameters of the relationship in a written agreement. It is particularly important that the relationship foster equality among the parties, regardless of who is the attending physician of record. The parties should be jointly responsible for patient care, with the hospitalist treating the patient’s general medical concerns and the specialist focusing on techniques within his specialty to improve the patient’s issues.
The agreement should clearly state the responsibilities of each party, including delineating the party responsible for decisions such as admission and discharge. It should address resources and set forth the standardized processes and protocols to be used when treating patients.
Specialists can vary in their treatment of patients, so it is best to document their expectations at the onset of the relationship. Also, successful co-management is contingent upon regular communication between the hospitalist and the specialist. It is important to establish those boundaries in advance to prevent miscommunication down the road.
In particular, the agreement should explicitly describe the lines of authority and how conflicts will be addressed.
Final Thoughts
Co-management is a growing trend that can provide an opportunity for hospitalists to expand their practice and reinforce their value to both specialists and the hospital. The improved quality of care and patient satisfaction that is associated with hospitalist services can be crucial to maximizing reimbursement under a value-based reimbursement system. TH
As the practice of medicine continues to transition to performance-based payment systems, the number of mergers of hospitalists and specialists has surged. Payment models that focus on clinical outcomes and best practices link payment to the ability of physicians to provide efficient, quality healthcare and improve patient outcomes. These payment systems are changing the way healthcare services are delivered by demanding better patient care at a lower cost. The result is increasing pressure on physicians to meet operational and quality goals, or receive less reimbursement for their services.
Studies have shown that the effective use of hospitalists can improve standardized patient care for surgical patients. Hospitalists also provide value to specialists by freeing up time so they can focus on their area of expertise. As a result, co-management arrangements between hospitalists and specialists have become a popular tool to define working relationships and improve the quality of care patients receive.
Hospitalist Evolution
When hospitalists first debuted, they were seen as a threat to primary care physicians and specialists. Over time, they were criticized for performing routine work for specialized physicians. To overcome these negative connotations and prove their worth, hospitalists began co-managing patients for surgical specialists, who soon realized the significant value hospitalist services provided. Not only do they share in the responsibility of care provided to patients, but they also reduce readmissions and costs associated with providing healthcare.
Now there are even specialty hospitalists who specialize in a particular field, such as orthopedics or obstetrics.
Why Co-Management?
Hospitalists add value by helping to alleviate the burden on specialists—providing ED coverage, assisting in the operating room, and rounding on patients. They evaluate surgical patients for medical issues, reconcile medications across the spectrum of a patient’s care, and standardize the patient discharge and communication processes.
Providing these services frees specialists from rounding and allows them to concentrate on their specialty. Hospitalists do not have office-based practices, which allows them to spend their time in the hospital caring for admitted, pre-operative, and post-operative patients.
It is in the pre-operative and post-operative environments where hospitalists have established their extreme value to specialists. Under co-management arrangements, hospitalists are able to ensure that all pre-operative tests are conducted, reports are dictated, and the patient’s medical history is available. Pre-operative evaluations allow the hospitalist to develop a post-operative plan of care and proactively address many medical concerns. Also, the hospitalist is available to see patients immediately after surgery, allowing immediate evaluation and treatment for high blood pressure, diabetic issues, or other medical issues.
In sum, the hospitalist is responsible for the medical care of the specialist’s patients, and the specialist is able to focus on the specialty services he or she provides. Providing these services gives hospitalists the opportunity to anticipate problems and overcome issues, which results in more efficient care, shorter lengths of stay in the hospital, and improved patient satisfaction. Such results make hospitalists critical to success in performance-based payment systems.
Successful Co-Management Arrangements
A key to success in establishing a co-management arrangement between a hospitalist and a specialist is setting forth the parameters of the relationship in a written agreement. It is particularly important that the relationship foster equality among the parties, regardless of who is the attending physician of record. The parties should be jointly responsible for patient care, with the hospitalist treating the patient’s general medical concerns and the specialist focusing on techniques within his specialty to improve the patient’s issues.
The agreement should clearly state the responsibilities of each party, including delineating the party responsible for decisions such as admission and discharge. It should address resources and set forth the standardized processes and protocols to be used when treating patients.
Specialists can vary in their treatment of patients, so it is best to document their expectations at the onset of the relationship. Also, successful co-management is contingent upon regular communication between the hospitalist and the specialist. It is important to establish those boundaries in advance to prevent miscommunication down the road.
In particular, the agreement should explicitly describe the lines of authority and how conflicts will be addressed.
Final Thoughts
Co-management is a growing trend that can provide an opportunity for hospitalists to expand their practice and reinforce their value to both specialists and the hospital. The improved quality of care and patient satisfaction that is associated with hospitalist services can be crucial to maximizing reimbursement under a value-based reimbursement system. TH

Study Shows ICDs Benefit Women in Heart Failure Prevention
NEW YORK (Reuters Health)—Women with heart failure (HF) derive as much benefit from implantable cardiac defibrillators (ICDs) for primary prevention as men, according to a comparative effectiveness study.
"Randomized clinical trials demonstrated that the ICD confers survival benefit to many patients with heart failure; however, data on ICDs in women were inadequate due to the relatively small number of women enrolled in those trials," study investigator Dr. Sana Al-Khatib, heart rhythm specialist from Duke Clinical Research Institute in Durham, North Carolina, told Reuters Health by email.
This analysis, online January 12 in Circulation: Heart Failure, showed that women with heart failure and an ICD had "significantly better survival than women with no ICD," Dr. Al-Khatib said.
"Our findings support the gender-neutral guideline recommendations regarding the use of primary prevention ICDs in eligible patients. As a result, women with heart failure should be equally considered for a primary prevention ICD as men," she added.
For the analysis, researchers linked data from 264 hospitals participating in the Get With The Guidelines Heart Failure registry with data from the Centers for Medicare and Medicaid
Services.
Using propensity score matching, they created a cohort of 430 women with heart failure and a preventive ICD and 430 similar women with heart failure but no ICD. For comparison, they propensity score matched 859 men with heart failure and an ICD to 859 men without an ICD.
After three years, 40.2% of women with an ICD had died, compared with 48.7% of women without an ICD. The corresponding mortality rates in men were 42.9% and 52.9%.
In the matched cohorts, an ICD was associated with "similarly better survival" in women and men (hazard ratios, 0.78 and 0.76, respectively). There was no interaction between sex and presence of an ICD with regard to survival (p=0.79).
"Despite the survival benefits of primary prevention ICDs in patients with HF demonstrated in randomized clinical trials, benefit in the subgroup of women from these trials has not been
definitively proved. This uncertainty on survival benefit may be one of several contributing factors to the lower rates of ICD referral and implantation in eligible women," the investigators note in their article.
The current data, they conclude, "support current guideline recommendations" for the implantation of a primary prevention ICD in eligible women and men with HF and reduced left ventricular ejection fraction.
The Agency for Healthcare Research and Quality funded the study. One author reported consulting for Medtronic.
NEW YORK (Reuters Health)—Women with heart failure (HF) derive as much benefit from implantable cardiac defibrillators (ICDs) for primary prevention as men, according to a comparative effectiveness study.
"Randomized clinical trials demonstrated that the ICD confers survival benefit to many patients with heart failure; however, data on ICDs in women were inadequate due to the relatively small number of women enrolled in those trials," study investigator Dr. Sana Al-Khatib, heart rhythm specialist from Duke Clinical Research Institute in Durham, North Carolina, told Reuters Health by email.
This analysis, online January 12 in Circulation: Heart Failure, showed that women with heart failure and an ICD had "significantly better survival than women with no ICD," Dr. Al-Khatib said.
"Our findings support the gender-neutral guideline recommendations regarding the use of primary prevention ICDs in eligible patients. As a result, women with heart failure should be equally considered for a primary prevention ICD as men," she added.
For the analysis, researchers linked data from 264 hospitals participating in the Get With The Guidelines Heart Failure registry with data from the Centers for Medicare and Medicaid
Services.
Using propensity score matching, they created a cohort of 430 women with heart failure and a preventive ICD and 430 similar women with heart failure but no ICD. For comparison, they propensity score matched 859 men with heart failure and an ICD to 859 men without an ICD.
After three years, 40.2% of women with an ICD had died, compared with 48.7% of women without an ICD. The corresponding mortality rates in men were 42.9% and 52.9%.
In the matched cohorts, an ICD was associated with "similarly better survival" in women and men (hazard ratios, 0.78 and 0.76, respectively). There was no interaction between sex and presence of an ICD with regard to survival (p=0.79).
"Despite the survival benefits of primary prevention ICDs in patients with HF demonstrated in randomized clinical trials, benefit in the subgroup of women from these trials has not been
definitively proved. This uncertainty on survival benefit may be one of several contributing factors to the lower rates of ICD referral and implantation in eligible women," the investigators note in their article.
The current data, they conclude, "support current guideline recommendations" for the implantation of a primary prevention ICD in eligible women and men with HF and reduced left ventricular ejection fraction.
The Agency for Healthcare Research and Quality funded the study. One author reported consulting for Medtronic.
NEW YORK (Reuters Health)—Women with heart failure (HF) derive as much benefit from implantable cardiac defibrillators (ICDs) for primary prevention as men, according to a comparative effectiveness study.
"Randomized clinical trials demonstrated that the ICD confers survival benefit to many patients with heart failure; however, data on ICDs in women were inadequate due to the relatively small number of women enrolled in those trials," study investigator Dr. Sana Al-Khatib, heart rhythm specialist from Duke Clinical Research Institute in Durham, North Carolina, told Reuters Health by email.
This analysis, online January 12 in Circulation: Heart Failure, showed that women with heart failure and an ICD had "significantly better survival than women with no ICD," Dr. Al-Khatib said.
"Our findings support the gender-neutral guideline recommendations regarding the use of primary prevention ICDs in eligible patients. As a result, women with heart failure should be equally considered for a primary prevention ICD as men," she added.
For the analysis, researchers linked data from 264 hospitals participating in the Get With The Guidelines Heart Failure registry with data from the Centers for Medicare and Medicaid
Services.
Using propensity score matching, they created a cohort of 430 women with heart failure and a preventive ICD and 430 similar women with heart failure but no ICD. For comparison, they propensity score matched 859 men with heart failure and an ICD to 859 men without an ICD.
After three years, 40.2% of women with an ICD had died, compared with 48.7% of women without an ICD. The corresponding mortality rates in men were 42.9% and 52.9%.
In the matched cohorts, an ICD was associated with "similarly better survival" in women and men (hazard ratios, 0.78 and 0.76, respectively). There was no interaction between sex and presence of an ICD with regard to survival (p=0.79).
"Despite the survival benefits of primary prevention ICDs in patients with HF demonstrated in randomized clinical trials, benefit in the subgroup of women from these trials has not been
definitively proved. This uncertainty on survival benefit may be one of several contributing factors to the lower rates of ICD referral and implantation in eligible women," the investigators note in their article.
The current data, they conclude, "support current guideline recommendations" for the implantation of a primary prevention ICD in eligible women and men with HF and reduced left ventricular ejection fraction.
The Agency for Healthcare Research and Quality funded the study. One author reported consulting for Medtronic.
Mutations may impact response to HDACis in PTCL-NOS

Photo by Larry Young
SAN FRANCISCO—Preclinical research has revealed mutations that may affect the performance of histone deacetylase inhibitors (HDACis) in patients with peripheral T-cell lymphoma-not otherwise specified (PTCL-NOS).
The researchers identified histone-modifying gene mutations in patients with PTCL-NOS and found evidence to suggest these mutations confer shorter survival.
The team also conducted experiments in Jurkat cells showing that certain HDACis could counteract loss-of-function mutations, while others could not.
Meng-Meng Ji, MD, PhD, of the Shanghai Institute of Hematology in China, presented this research at the 8th Annual T-cell Lymphoma Forum.
Dr Ji and her colleagues first performed targeted sequencing in tumor samples from 105 patients newly diagnosed with PTCL-NOS.
The team discovered 62 mutations of “important” lymphoma-associated histone-modifying genes in 31 patients. They found mutations in MLL2 (n=21), TET2 (n=17), EP300 (n=8), CREBBP (n=8), and SETD2 (n=6).
Clinical data revealed a significant difference in overall survival between patients who had these mutations and those who did not (P=0.0038).
Because most of the mutations they identified are loss-of-function mutations, Dr Ji and her colleagues wanted to determine whether HDACis could restore the expression of the mutated genes. So they tested 4 HDACis—valproic acid, vorinostat, romidepsin, and chidamide—in Jurkat cells.
All 4 HDACis upregulated expression of EP300 and CREBBP. However, only romidepsin and chidamide upregulated MLL2, and only valproic acid and vorinostat upregulated SETD2. Dr Ji said there was no obvious change in TET2 expression with any of the HDACis.
The researchers then took a closer look at the EP300, MLL2, and SETD2 mutations. They found that most EP300 mutations were located on the HAT domain. MLL2 mutations could be found in a variety of locations, but some were located on the SET domain. And most SETD2 mutations were located on the SET domain.
Based on the crystal structure of each gene, the team found that EP300 mutations on the HAT domain and both MLL2 mutations and SETD2 mutations located on the SET domain induce loss of function.
So the researchers constructed a mutant for EP300 (p.H1377R), MLL2 (p.V5389M), and SETD2 (p.R1598_) and transfected Jurkat cells with each mutant.
The mutants reduced gene expression significantly when compared to wild-type cells.
Like in the previous experiments, all 4 HDACis could restore the expression of EP300. But only romidepsin and chidamide could restore MLL2 expression, and only valproic acid and vorinostat could restore SETD2 expression.
In addition, all 4 HDACis restored H3K18 hypoacetylation, which was inhibited in Jurkat cells transfected with the EP300 mutant.
Romidepsin and chidamide restored H3K4me3 expression, which was inhibited by the MLL2 mutant. And valproic acid and vorinostat restored H3K36me3 expression, which was inhibited by the SETD2 mutant.
“HDACis targeted differently histone H3 acetylation or methylation modulated by the mutations, suggesting their distinct therapeutic efficiency in PTCL-NOS,” Dr Ji noted.
She said she and her colleagues are continuing this research in samples from patients with PTCL-NOS. ![]()
Photo by Larry Young
SAN FRANCISCO—Preclinical research has revealed mutations that may affect the performance of histone deacetylase inhibitors (HDACis) in patients with peripheral T-cell lymphoma-not otherwise specified (PTCL-NOS).
The researchers identified histone-modifying gene mutations in patients with PTCL-NOS and found evidence to suggest these mutations confer shorter survival.
The team also conducted experiments in Jurkat cells showing that certain HDACis could counteract loss-of-function mutations, while others could not.
Meng-Meng Ji, MD, PhD, of the Shanghai Institute of Hematology in China, presented this research at the 8th Annual T-cell Lymphoma Forum.
Dr Ji and her colleagues first performed targeted sequencing in tumor samples from 105 patients newly diagnosed with PTCL-NOS.
The team discovered 62 mutations of “important” lymphoma-associated histone-modifying genes in 31 patients. They found mutations in MLL2 (n=21), TET2 (n=17), EP300 (n=8), CREBBP (n=8), and SETD2 (n=6).
Clinical data revealed a significant difference in overall survival between patients who had these mutations and those who did not (P=0.0038).
Because most of the mutations they identified are loss-of-function mutations, Dr Ji and her colleagues wanted to determine whether HDACis could restore the expression of the mutated genes. So they tested 4 HDACis—valproic acid, vorinostat, romidepsin, and chidamide—in Jurkat cells.
All 4 HDACis upregulated expression of EP300 and CREBBP. However, only romidepsin and chidamide upregulated MLL2, and only valproic acid and vorinostat upregulated SETD2. Dr Ji said there was no obvious change in TET2 expression with any of the HDACis.
The researchers then took a closer look at the EP300, MLL2, and SETD2 mutations. They found that most EP300 mutations were located on the HAT domain. MLL2 mutations could be found in a variety of locations, but some were located on the SET domain. And most SETD2 mutations were located on the SET domain.
Based on the crystal structure of each gene, the team found that EP300 mutations on the HAT domain and both MLL2 mutations and SETD2 mutations located on the SET domain induce loss of function.
So the researchers constructed a mutant for EP300 (p.H1377R), MLL2 (p.V5389M), and SETD2 (p.R1598_) and transfected Jurkat cells with each mutant.
The mutants reduced gene expression significantly when compared to wild-type cells.
Like in the previous experiments, all 4 HDACis could restore the expression of EP300. But only romidepsin and chidamide could restore MLL2 expression, and only valproic acid and vorinostat could restore SETD2 expression.
In addition, all 4 HDACis restored H3K18 hypoacetylation, which was inhibited in Jurkat cells transfected with the EP300 mutant.
Romidepsin and chidamide restored H3K4me3 expression, which was inhibited by the MLL2 mutant. And valproic acid and vorinostat restored H3K36me3 expression, which was inhibited by the SETD2 mutant.
“HDACis targeted differently histone H3 acetylation or methylation modulated by the mutations, suggesting their distinct therapeutic efficiency in PTCL-NOS,” Dr Ji noted.
She said she and her colleagues are continuing this research in samples from patients with PTCL-NOS. ![]()
Photo by Larry Young
SAN FRANCISCO—Preclinical research has revealed mutations that may affect the performance of histone deacetylase inhibitors (HDACis) in patients with peripheral T-cell lymphoma-not otherwise specified (PTCL-NOS).
The researchers identified histone-modifying gene mutations in patients with PTCL-NOS and found evidence to suggest these mutations confer shorter survival.
The team also conducted experiments in Jurkat cells showing that certain HDACis could counteract loss-of-function mutations, while others could not.
Meng-Meng Ji, MD, PhD, of the Shanghai Institute of Hematology in China, presented this research at the 8th Annual T-cell Lymphoma Forum.
Dr Ji and her colleagues first performed targeted sequencing in tumor samples from 105 patients newly diagnosed with PTCL-NOS.
The team discovered 62 mutations of “important” lymphoma-associated histone-modifying genes in 31 patients. They found mutations in MLL2 (n=21), TET2 (n=17), EP300 (n=8), CREBBP (n=8), and SETD2 (n=6).
Clinical data revealed a significant difference in overall survival between patients who had these mutations and those who did not (P=0.0038).
Because most of the mutations they identified are loss-of-function mutations, Dr Ji and her colleagues wanted to determine whether HDACis could restore the expression of the mutated genes. So they tested 4 HDACis—valproic acid, vorinostat, romidepsin, and chidamide—in Jurkat cells.
All 4 HDACis upregulated expression of EP300 and CREBBP. However, only romidepsin and chidamide upregulated MLL2, and only valproic acid and vorinostat upregulated SETD2. Dr Ji said there was no obvious change in TET2 expression with any of the HDACis.
The researchers then took a closer look at the EP300, MLL2, and SETD2 mutations. They found that most EP300 mutations were located on the HAT domain. MLL2 mutations could be found in a variety of locations, but some were located on the SET domain. And most SETD2 mutations were located on the SET domain.
Based on the crystal structure of each gene, the team found that EP300 mutations on the HAT domain and both MLL2 mutations and SETD2 mutations located on the SET domain induce loss of function.
So the researchers constructed a mutant for EP300 (p.H1377R), MLL2 (p.V5389M), and SETD2 (p.R1598_) and transfected Jurkat cells with each mutant.
The mutants reduced gene expression significantly when compared to wild-type cells.
Like in the previous experiments, all 4 HDACis could restore the expression of EP300. But only romidepsin and chidamide could restore MLL2 expression, and only valproic acid and vorinostat could restore SETD2 expression.
In addition, all 4 HDACis restored H3K18 hypoacetylation, which was inhibited in Jurkat cells transfected with the EP300 mutant.
Romidepsin and chidamide restored H3K4me3 expression, which was inhibited by the MLL2 mutant. And valproic acid and vorinostat restored H3K36me3 expression, which was inhibited by the SETD2 mutant.
“HDACis targeted differently histone H3 acetylation or methylation modulated by the mutations, suggesting their distinct therapeutic efficiency in PTCL-NOS,” Dr Ji noted.
She said she and her colleagues are continuing this research in samples from patients with PTCL-NOS. ![]()