User login
Formerly Skin & Allergy News
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')]
The leading independent newspaper covering dermatology news and commentary.
Expert calls for paradigm shift in lab monitoring of some dermatology drugs
From time to time, Joslyn Kirby, MD, asks other physicians about their experience with certain medications used in dermatology, especially when something new hits the market.
“Sometimes I get an answer like, ‘The last time I used that medicine, my patient needed a liver transplant,’ ” Dr. Kirby, associate professor of dermatology, Penn State University, Hershey, said during the Orlando Dermatology Aesthetic and Clinical Conference. “It’s typically a story of something rare, uncommon, and awful. The challenge with an anecdote is that for all its power, it has a lower level of evidence. But it sticks with us and influences us more than a better level of evidence because it’s a situation and a story that we might relate to.”

Dr. Kirby said that when she thinks about managing side effects from drugs used in dermatology, it usually relates to something common and low-risk such as sore, dry skin with isotretinoin use. In contrast, if there is an uncommon but serious side effect, then mitigation rather than management is key. “I want to mitigate the risk – meaning warn my patient about it or be careful about how I select my patients when it is a serious side effect that happens infrequently,” she said. “The worst combination is a frequent and severe side effect. That is something we should avoid, for sure.”
Isotretinoin
But another aspect of prescribing a new drug for patients can be less clear-cut, Dr. Kirby continued, such as the rationale for routine lab monitoring. She began by discussing one of her male patients with moderate to severe acne. After he failed oral antibiotics and topical retinoids, she recommended isotretinoin, which carries a risk of hypertriglyceridemia-associated pancreatitis. “Early in my career, I was getting a lot of monthly labs in patients on this drug that were totally normal and not influencing my practice,” Dr. Kirby recalled. “We’ve seen studies coming out on isotretinoin lab monitoring, showing us that we can keep our patients safe and that we really don’t need to be checking labs as often, because lab changes are infrequent.”
In one of those studies, researchers evaluated 1,863 patients treated with isotretinoin for acne between Jan. 1, 2008, and June 30, 2017 (J Am Acad Dermatol. 2020 Jan;82[1]:72-9).Over time, fewer than 1% of patients screened developed grade 3 or greater triglyceride testing abnormalities, while fewer than 0.5% developed liver function testing (LFT) abnormalities. Authors of a separate systematic review concluded that for patients on isotretinoin therapy without elevated baseline triglycerides, or risk thereof, monitoring triglycerides is of little value (Br J Dermatol. 2017 Oct;177[4]:960-6). Of the 25 patients in the analysis who developed pancreatitis on isotretinoin, only 3 had elevated triglycerides at baseline.
“I was taught that I need to check triglycerides frequently due to the risk of pancreatitis developing with isotretinoin use,” Dr. Kirby said. “Lipid changes on therapy are expected, but they tend to peak early, meaning the first 3 months of treatment when we’re ramping up from a starting dose to a maintenance dose. It’s rare for somebody to be a late bloomer, meaning that they have totally normal labs in the first 3 months and then suddenly develop an abnormality. People are either going to demonstrate an abnormality early or not have one at all.”
When Dr. Kirby starts patients on isotretinoin, she orders baseline LFTs and a lipid panel and repeats them 60 days later. “If everything is fine or only mildly high, we don’t do more testing, only a review of systems,” she said. “This is valuable to our patients because fear of needles and fainting peak during adolescence.”
Spironolactone
The clinical use of regularly monitoring potassium levels in young women taking spironolactone for acne has also been questioned. The drug has been linked to an increased risk for hyperkalemia, but the prevalence is unclear. “I got a lot of normal potassium levels in these patients [when] I was in training and I really questioned, ‘Why am I doing this? What is the rationale?’ ” Dr. Kirby said.
In a study that informed her own practice, researchers reviewed the rate of hyperkalemia in 974 healthy young women taking spironolactone for acne or for an endocrine disorder with associated acne between Dec. 1, 2000, and March 31, 2014 (JAMA Dermatol. 2015 Sep;151[9]:941-4). Of the total of 1,802 serum potassium measurements taken during treatment, 13 (0.72%) were mildly elevated levels and none of the patients had a potassium level above 5.5 mEq/L. Retesting within 1 to 3 weeks in 6 of 13 patients with elevated levels found that potassium levels were normal. “The recommendation for spironolactone in healthy women is not to check the potassium level,” Dr. Kirby said, adding that she does counsel patients about the risk of breast tenderness (which can occur 5% to 40% of the time) and spotting (which can occur in 10% to 20% of patients). Gynecomastia can occur in 10% to 30% of men, which is one of the reasons she does not use spironolactone in male patients.
TB testing and biologics
Whether or not to test for TB in patients with psoriasis taking biologic therapies represents another conundrum, she continued. Patients taking biologics are at risk of reactivation of latent TB infection, but in her experience, package inserts contain language like “perform TB testing at baseline, then periodically,” or “use at baseline, then with active TB symptoms,” and “after treatment is discontinued.”
“What the inserts didn’t recommend was to perform TB testing every year, which is what my routine had been,” Dr. Kirby said. “In the United States, thankfully we don’t have a lot of TB.” In a study that informed her own practice, researchers at a single academic medical center retrospectively reviewed the TB seroconversion rate among 316 patients treated with second-generation biologics (J Am Acad Dermatol. 2020 Oct 1;S0190-9622[20]32676-1. doi: 10.1016/j.jaad.2020.09.075). It found that only six patients (2%) converted and had a positive TB test later during treatment with the biologic. “Of these six people, all had grown up outside the U.S., had traveled outside of the U.S., or were in a group living situation,” said Dr. Kirby, who was not affiliated with the study.
“This informs our rationale for how we can do this testing. If insurance requires it every year, fine. But if they don’t, I ask patients about travel, about their living situation, and how they’re feeling. If everything’s going great, I don’t order TB testing. I do favor the interferon-gamma release assays because they’re a lot more effective than PPDs [purified protein derivative skin tests]. Also, PPDs are difficult for patients who have a low rate of returning to have that test read.”
Terbinafine for onychomycosis
Dr. Kirby also discussed the rationale for ordering regular LFTs in patients taking terbinafine for onychomycosis. “There is a risk of drug-induced liver injury from taking terbinafine, but it’s rare,” she said. “Can we be thoughtful about which patients we expose?”
Evidence suggests that patients with hyperkeratosis greater than 2 mm, with nail matrix involvement, with 50% or more of the nail involved, or having concomitant peripheral vascular disease and diabetes are recalcitrant to treatment with terbinafine
(J Am Acad Dermatol. 2019 Apr;80[4]:853-67). “If we can frame this risk, then we can frame it for our patients,” she said. “We’re more likely to cause liver injury with an antibiotic. When it comes to an oral antifungal, itraconazole is more likely than terbinafine to cause liver injury. The rate of liver injury with terbinafine is only about 2 out of 100,000. It’s five times more likely with itraconazole and 21 times more likely with Augmentin.”
She recommends obtaining a baseline LFT in patients starting terbinafine therapy “to make sure their liver is normal from the start.” In addition, she advised, “let them know that there is a TB seroconversion risk of about 1 in 50,000 people, and that if it happens there would be symptomatic changes. They would maybe notice pruritus and have a darkening in their urine, and they’d have some flu-like symptoms, which would mean stop the drug and get some care.”
Dr. Kirby emphasized that a patient’s propensity for developing drug-induced liver injury from terbinafine use is not predictable from LFT monitoring. “What you’re more likely to find is an asymptomatic LFT rise in about 1% of people,” she said.
She disclosed that she has received honoraria from AbbVie, ChemoCentryx, Incyte, Janssen, Novartis, and UCB Pharma.
From time to time, Joslyn Kirby, MD, asks other physicians about their experience with certain medications used in dermatology, especially when something new hits the market.
“Sometimes I get an answer like, ‘The last time I used that medicine, my patient needed a liver transplant,’ ” Dr. Kirby, associate professor of dermatology, Penn State University, Hershey, said during the Orlando Dermatology Aesthetic and Clinical Conference. “It’s typically a story of something rare, uncommon, and awful. The challenge with an anecdote is that for all its power, it has a lower level of evidence. But it sticks with us and influences us more than a better level of evidence because it’s a situation and a story that we might relate to.”
Dr. Kirby said that when she thinks about managing side effects from drugs used in dermatology, it usually relates to something common and low-risk such as sore, dry skin with isotretinoin use. In contrast, if there is an uncommon but serious side effect, then mitigation rather than management is key. “I want to mitigate the risk – meaning warn my patient about it or be careful about how I select my patients when it is a serious side effect that happens infrequently,” she said. “The worst combination is a frequent and severe side effect. That is something we should avoid, for sure.”
Isotretinoin
But another aspect of prescribing a new drug for patients can be less clear-cut, Dr. Kirby continued, such as the rationale for routine lab monitoring. She began by discussing one of her male patients with moderate to severe acne. After he failed oral antibiotics and topical retinoids, she recommended isotretinoin, which carries a risk of hypertriglyceridemia-associated pancreatitis. “Early in my career, I was getting a lot of monthly labs in patients on this drug that were totally normal and not influencing my practice,” Dr. Kirby recalled. “We’ve seen studies coming out on isotretinoin lab monitoring, showing us that we can keep our patients safe and that we really don’t need to be checking labs as often, because lab changes are infrequent.”
In one of those studies, researchers evaluated 1,863 patients treated with isotretinoin for acne between Jan. 1, 2008, and June 30, 2017 (J Am Acad Dermatol. 2020 Jan;82[1]:72-9).Over time, fewer than 1% of patients screened developed grade 3 or greater triglyceride testing abnormalities, while fewer than 0.5% developed liver function testing (LFT) abnormalities. Authors of a separate systematic review concluded that for patients on isotretinoin therapy without elevated baseline triglycerides, or risk thereof, monitoring triglycerides is of little value (Br J Dermatol. 2017 Oct;177[4]:960-6). Of the 25 patients in the analysis who developed pancreatitis on isotretinoin, only 3 had elevated triglycerides at baseline.
“I was taught that I need to check triglycerides frequently due to the risk of pancreatitis developing with isotretinoin use,” Dr. Kirby said. “Lipid changes on therapy are expected, but they tend to peak early, meaning the first 3 months of treatment when we’re ramping up from a starting dose to a maintenance dose. It’s rare for somebody to be a late bloomer, meaning that they have totally normal labs in the first 3 months and then suddenly develop an abnormality. People are either going to demonstrate an abnormality early or not have one at all.”
When Dr. Kirby starts patients on isotretinoin, she orders baseline LFTs and a lipid panel and repeats them 60 days later. “If everything is fine or only mildly high, we don’t do more testing, only a review of systems,” she said. “This is valuable to our patients because fear of needles and fainting peak during adolescence.”
Spironolactone
The clinical use of regularly monitoring potassium levels in young women taking spironolactone for acne has also been questioned. The drug has been linked to an increased risk for hyperkalemia, but the prevalence is unclear. “I got a lot of normal potassium levels in these patients [when] I was in training and I really questioned, ‘Why am I doing this? What is the rationale?’ ” Dr. Kirby said.
In a study that informed her own practice, researchers reviewed the rate of hyperkalemia in 974 healthy young women taking spironolactone for acne or for an endocrine disorder with associated acne between Dec. 1, 2000, and March 31, 2014 (JAMA Dermatol. 2015 Sep;151[9]:941-4). Of the total of 1,802 serum potassium measurements taken during treatment, 13 (0.72%) were mildly elevated levels and none of the patients had a potassium level above 5.5 mEq/L. Retesting within 1 to 3 weeks in 6 of 13 patients with elevated levels found that potassium levels were normal. “The recommendation for spironolactone in healthy women is not to check the potassium level,” Dr. Kirby said, adding that she does counsel patients about the risk of breast tenderness (which can occur 5% to 40% of the time) and spotting (which can occur in 10% to 20% of patients). Gynecomastia can occur in 10% to 30% of men, which is one of the reasons she does not use spironolactone in male patients.
TB testing and biologics
Whether or not to test for TB in patients with psoriasis taking biologic therapies represents another conundrum, she continued. Patients taking biologics are at risk of reactivation of latent TB infection, but in her experience, package inserts contain language like “perform TB testing at baseline, then periodically,” or “use at baseline, then with active TB symptoms,” and “after treatment is discontinued.”
“What the inserts didn’t recommend was to perform TB testing every year, which is what my routine had been,” Dr. Kirby said. “In the United States, thankfully we don’t have a lot of TB.” In a study that informed her own practice, researchers at a single academic medical center retrospectively reviewed the TB seroconversion rate among 316 patients treated with second-generation biologics (J Am Acad Dermatol. 2020 Oct 1;S0190-9622[20]32676-1. doi: 10.1016/j.jaad.2020.09.075). It found that only six patients (2%) converted and had a positive TB test later during treatment with the biologic. “Of these six people, all had grown up outside the U.S., had traveled outside of the U.S., or were in a group living situation,” said Dr. Kirby, who was not affiliated with the study.
“This informs our rationale for how we can do this testing. If insurance requires it every year, fine. But if they don’t, I ask patients about travel, about their living situation, and how they’re feeling. If everything’s going great, I don’t order TB testing. I do favor the interferon-gamma release assays because they’re a lot more effective than PPDs [purified protein derivative skin tests]. Also, PPDs are difficult for patients who have a low rate of returning to have that test read.”
Terbinafine for onychomycosis
Dr. Kirby also discussed the rationale for ordering regular LFTs in patients taking terbinafine for onychomycosis. “There is a risk of drug-induced liver injury from taking terbinafine, but it’s rare,” she said. “Can we be thoughtful about which patients we expose?”
Evidence suggests that patients with hyperkeratosis greater than 2 mm, with nail matrix involvement, with 50% or more of the nail involved, or having concomitant peripheral vascular disease and diabetes are recalcitrant to treatment with terbinafine
(J Am Acad Dermatol. 2019 Apr;80[4]:853-67). “If we can frame this risk, then we can frame it for our patients,” she said. “We’re more likely to cause liver injury with an antibiotic. When it comes to an oral antifungal, itraconazole is more likely than terbinafine to cause liver injury. The rate of liver injury with terbinafine is only about 2 out of 100,000. It’s five times more likely with itraconazole and 21 times more likely with Augmentin.”
She recommends obtaining a baseline LFT in patients starting terbinafine therapy “to make sure their liver is normal from the start.” In addition, she advised, “let them know that there is a TB seroconversion risk of about 1 in 50,000 people, and that if it happens there would be symptomatic changes. They would maybe notice pruritus and have a darkening in their urine, and they’d have some flu-like symptoms, which would mean stop the drug and get some care.”
Dr. Kirby emphasized that a patient’s propensity for developing drug-induced liver injury from terbinafine use is not predictable from LFT monitoring. “What you’re more likely to find is an asymptomatic LFT rise in about 1% of people,” she said.
She disclosed that she has received honoraria from AbbVie, ChemoCentryx, Incyte, Janssen, Novartis, and UCB Pharma.
From time to time, Joslyn Kirby, MD, asks other physicians about their experience with certain medications used in dermatology, especially when something new hits the market.
“Sometimes I get an answer like, ‘The last time I used that medicine, my patient needed a liver transplant,’ ” Dr. Kirby, associate professor of dermatology, Penn State University, Hershey, said during the Orlando Dermatology Aesthetic and Clinical Conference. “It’s typically a story of something rare, uncommon, and awful. The challenge with an anecdote is that for all its power, it has a lower level of evidence. But it sticks with us and influences us more than a better level of evidence because it’s a situation and a story that we might relate to.”
Dr. Kirby said that when she thinks about managing side effects from drugs used in dermatology, it usually relates to something common and low-risk such as sore, dry skin with isotretinoin use. In contrast, if there is an uncommon but serious side effect, then mitigation rather than management is key. “I want to mitigate the risk – meaning warn my patient about it or be careful about how I select my patients when it is a serious side effect that happens infrequently,” she said. “The worst combination is a frequent and severe side effect. That is something we should avoid, for sure.”
Isotretinoin
But another aspect of prescribing a new drug for patients can be less clear-cut, Dr. Kirby continued, such as the rationale for routine lab monitoring. She began by discussing one of her male patients with moderate to severe acne. After he failed oral antibiotics and topical retinoids, she recommended isotretinoin, which carries a risk of hypertriglyceridemia-associated pancreatitis. “Early in my career, I was getting a lot of monthly labs in patients on this drug that were totally normal and not influencing my practice,” Dr. Kirby recalled. “We’ve seen studies coming out on isotretinoin lab monitoring, showing us that we can keep our patients safe and that we really don’t need to be checking labs as often, because lab changes are infrequent.”
In one of those studies, researchers evaluated 1,863 patients treated with isotretinoin for acne between Jan. 1, 2008, and June 30, 2017 (J Am Acad Dermatol. 2020 Jan;82[1]:72-9).Over time, fewer than 1% of patients screened developed grade 3 or greater triglyceride testing abnormalities, while fewer than 0.5% developed liver function testing (LFT) abnormalities. Authors of a separate systematic review concluded that for patients on isotretinoin therapy without elevated baseline triglycerides, or risk thereof, monitoring triglycerides is of little value (Br J Dermatol. 2017 Oct;177[4]:960-6). Of the 25 patients in the analysis who developed pancreatitis on isotretinoin, only 3 had elevated triglycerides at baseline.
“I was taught that I need to check triglycerides frequently due to the risk of pancreatitis developing with isotretinoin use,” Dr. Kirby said. “Lipid changes on therapy are expected, but they tend to peak early, meaning the first 3 months of treatment when we’re ramping up from a starting dose to a maintenance dose. It’s rare for somebody to be a late bloomer, meaning that they have totally normal labs in the first 3 months and then suddenly develop an abnormality. People are either going to demonstrate an abnormality early or not have one at all.”
When Dr. Kirby starts patients on isotretinoin, she orders baseline LFTs and a lipid panel and repeats them 60 days later. “If everything is fine or only mildly high, we don’t do more testing, only a review of systems,” she said. “This is valuable to our patients because fear of needles and fainting peak during adolescence.”
Spironolactone
The clinical use of regularly monitoring potassium levels in young women taking spironolactone for acne has also been questioned. The drug has been linked to an increased risk for hyperkalemia, but the prevalence is unclear. “I got a lot of normal potassium levels in these patients [when] I was in training and I really questioned, ‘Why am I doing this? What is the rationale?’ ” Dr. Kirby said.
In a study that informed her own practice, researchers reviewed the rate of hyperkalemia in 974 healthy young women taking spironolactone for acne or for an endocrine disorder with associated acne between Dec. 1, 2000, and March 31, 2014 (JAMA Dermatol. 2015 Sep;151[9]:941-4). Of the total of 1,802 serum potassium measurements taken during treatment, 13 (0.72%) were mildly elevated levels and none of the patients had a potassium level above 5.5 mEq/L. Retesting within 1 to 3 weeks in 6 of 13 patients with elevated levels found that potassium levels were normal. “The recommendation for spironolactone in healthy women is not to check the potassium level,” Dr. Kirby said, adding that she does counsel patients about the risk of breast tenderness (which can occur 5% to 40% of the time) and spotting (which can occur in 10% to 20% of patients). Gynecomastia can occur in 10% to 30% of men, which is one of the reasons she does not use spironolactone in male patients.
TB testing and biologics
Whether or not to test for TB in patients with psoriasis taking biologic therapies represents another conundrum, she continued. Patients taking biologics are at risk of reactivation of latent TB infection, but in her experience, package inserts contain language like “perform TB testing at baseline, then periodically,” or “use at baseline, then with active TB symptoms,” and “after treatment is discontinued.”
“What the inserts didn’t recommend was to perform TB testing every year, which is what my routine had been,” Dr. Kirby said. “In the United States, thankfully we don’t have a lot of TB.” In a study that informed her own practice, researchers at a single academic medical center retrospectively reviewed the TB seroconversion rate among 316 patients treated with second-generation biologics (J Am Acad Dermatol. 2020 Oct 1;S0190-9622[20]32676-1. doi: 10.1016/j.jaad.2020.09.075). It found that only six patients (2%) converted and had a positive TB test later during treatment with the biologic. “Of these six people, all had grown up outside the U.S., had traveled outside of the U.S., or were in a group living situation,” said Dr. Kirby, who was not affiliated with the study.
“This informs our rationale for how we can do this testing. If insurance requires it every year, fine. But if they don’t, I ask patients about travel, about their living situation, and how they’re feeling. If everything’s going great, I don’t order TB testing. I do favor the interferon-gamma release assays because they’re a lot more effective than PPDs [purified protein derivative skin tests]. Also, PPDs are difficult for patients who have a low rate of returning to have that test read.”
Terbinafine for onychomycosis
Dr. Kirby also discussed the rationale for ordering regular LFTs in patients taking terbinafine for onychomycosis. “There is a risk of drug-induced liver injury from taking terbinafine, but it’s rare,” she said. “Can we be thoughtful about which patients we expose?”
Evidence suggests that patients with hyperkeratosis greater than 2 mm, with nail matrix involvement, with 50% or more of the nail involved, or having concomitant peripheral vascular disease and diabetes are recalcitrant to treatment with terbinafine
(J Am Acad Dermatol. 2019 Apr;80[4]:853-67). “If we can frame this risk, then we can frame it for our patients,” she said. “We’re more likely to cause liver injury with an antibiotic. When it comes to an oral antifungal, itraconazole is more likely than terbinafine to cause liver injury. The rate of liver injury with terbinafine is only about 2 out of 100,000. It’s five times more likely with itraconazole and 21 times more likely with Augmentin.”
She recommends obtaining a baseline LFT in patients starting terbinafine therapy “to make sure their liver is normal from the start.” In addition, she advised, “let them know that there is a TB seroconversion risk of about 1 in 50,000 people, and that if it happens there would be symptomatic changes. They would maybe notice pruritus and have a darkening in their urine, and they’d have some flu-like symptoms, which would mean stop the drug and get some care.”
Dr. Kirby emphasized that a patient’s propensity for developing drug-induced liver injury from terbinafine use is not predictable from LFT monitoring. “What you’re more likely to find is an asymptomatic LFT rise in about 1% of people,” she said.
She disclosed that she has received honoraria from AbbVie, ChemoCentryx, Incyte, Janssen, Novartis, and UCB Pharma.
FROM ODAC 2021
Treatment of horizontal neck lines
The interplay of the neck subunits, as outlined in the recent article by Friedman and colleagues, requires multiple combination treatments, including fat removal, augmentation of deficient bony prominences, relaxation of hyperkinetic muscles, tissue tightening, suture anchoring, skin resurfacing, and treatment of dyschromia.
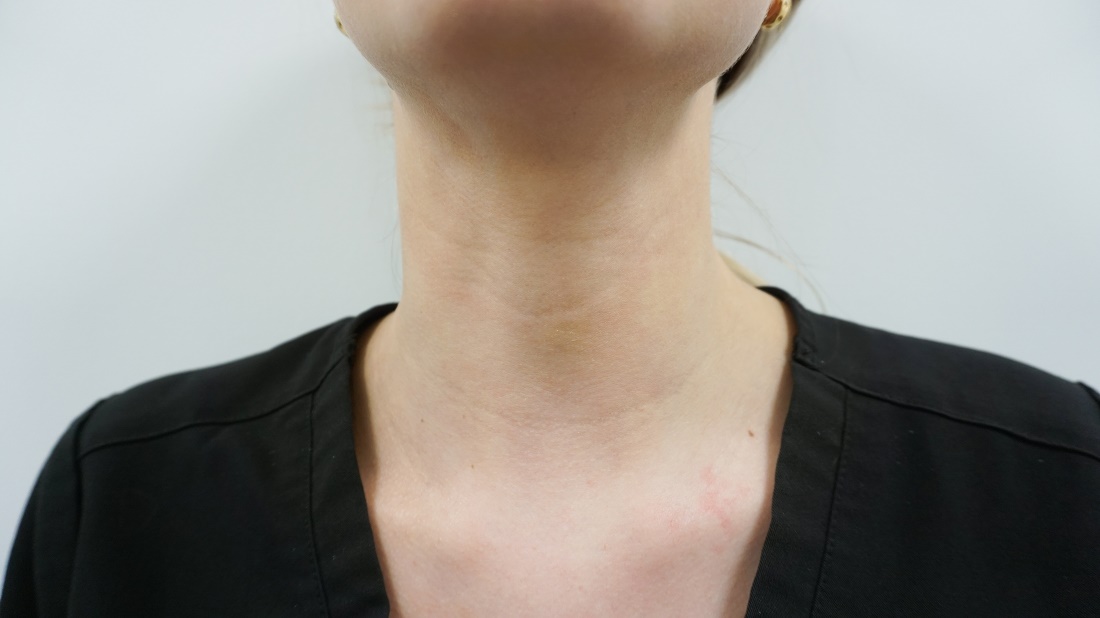
Horizontal neck lines are linear etched lines or furrows that commonly appear at a young age and are not caused by the aging process. The anatomy of the neck and the manner in which it bends contributes to their development at an early age. It is hypothesized that variable adipose tissue thickness and fibromuscular bands contribute to deepening of these lines in overweight patients. The widespread use of cell phones, laptops, and tablets has increased their prevalence and this has become one of the most common concerns of patients aged under 30 years in my clinic.
Various treatments have been recommended for neck rejuvenation, including hyaluronic acid and dilute calcium hydroxylapatite. In my experience, neither of these treatments adequately resolves the horizontal neck lines, and more importantly, prevents them from reoccurring. In addition, given the variability in skin and adipose thickness in the anterior neck, side effects including lumps, irregular correction, and the Tyndall effect, are common, particularly with incorrect choice of filler and injection depth.
The fibromuscular bands along the transverse neck lines pose one of the complexities in treatment with injectable filler. I have had significant improvement in the aesthetic outcome of my patients by using subcision along the transverse bands extensively prior to injection with hyaluronic acid fillers. The subcision is done with a 27-gauge needle to release the fibrous bands that tether the tissue down. If a patient has excess adipose tissue on either side of the bands, injectable fillers often do not improve the appearance of the lines and can make the neck appear heavier. The use of subcision followed by one to six treatments of deoxycholic acid in the adjacent adipose tissue prior to injection with a filler will help even out the contour of the neck, decrease adipose tissue bulges, release the fibrous bands, and fill the lines properly.
Working from home and on handheld devices has increased the appearance of neck lines in young populations. Despite the vast array of treatments in the aging neck, none have been very successful for this particular problem in the young. We need an improved understanding of these lines and better studies to investigate treatment options and long-term correction.
References:
Friedman O et al. J Cosmet Dermatol. 2021 Feb;20(2):569-76.
Brandt FS and Boker A. Dermatol Clin. 2004 Apr;22(2):159-66.
Tseng F and Yu H. Plast Reconstr Surg Glob Open. 2019 Aug 19;7(8):e2366.
Dibernardo BE. J Cosmet Laser Ther. 2013 Apr;15(2):56-64.
Jones D et al. Dermatol Surg. 2016 Oct;4 Suppl 1(Suppl 1):S235-42.
Lee SK and Kim HS. J Cosmet Dermatol. 2018 Aug;17(4):590-5.
Chao YY et al. Dermatol Surg. 2011 Oct;37(10):1542-5.
Han TY et al. Dermatol Surg. 2011 Sep;37(9):1291-6.
Dr. Wesley and Dr. Talakoub are cocontributors to this column. Dr. Wesley practices dermatology in Beverly Hills, Calif. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Talakoub. Write to them at dermnews@mdedge.com. They had no relevant disclosures.
The interplay of the neck subunits, as outlined in the recent article by Friedman and colleagues, requires multiple combination treatments, including fat removal, augmentation of deficient bony prominences, relaxation of hyperkinetic muscles, tissue tightening, suture anchoring, skin resurfacing, and treatment of dyschromia.
Horizontal neck lines are linear etched lines or furrows that commonly appear at a young age and are not caused by the aging process. The anatomy of the neck and the manner in which it bends contributes to their development at an early age. It is hypothesized that variable adipose tissue thickness and fibromuscular bands contribute to deepening of these lines in overweight patients. The widespread use of cell phones, laptops, and tablets has increased their prevalence and this has become one of the most common concerns of patients aged under 30 years in my clinic.
Various treatments have been recommended for neck rejuvenation, including hyaluronic acid and dilute calcium hydroxylapatite. In my experience, neither of these treatments adequately resolves the horizontal neck lines, and more importantly, prevents them from reoccurring. In addition, given the variability in skin and adipose thickness in the anterior neck, side effects including lumps, irregular correction, and the Tyndall effect, are common, particularly with incorrect choice of filler and injection depth.
The fibromuscular bands along the transverse neck lines pose one of the complexities in treatment with injectable filler. I have had significant improvement in the aesthetic outcome of my patients by using subcision along the transverse bands extensively prior to injection with hyaluronic acid fillers. The subcision is done with a 27-gauge needle to release the fibrous bands that tether the tissue down. If a patient has excess adipose tissue on either side of the bands, injectable fillers often do not improve the appearance of the lines and can make the neck appear heavier. The use of subcision followed by one to six treatments of deoxycholic acid in the adjacent adipose tissue prior to injection with a filler will help even out the contour of the neck, decrease adipose tissue bulges, release the fibrous bands, and fill the lines properly.
Working from home and on handheld devices has increased the appearance of neck lines in young populations. Despite the vast array of treatments in the aging neck, none have been very successful for this particular problem in the young. We need an improved understanding of these lines and better studies to investigate treatment options and long-term correction.
References:
Friedman O et al. J Cosmet Dermatol. 2021 Feb;20(2):569-76.
Brandt FS and Boker A. Dermatol Clin. 2004 Apr;22(2):159-66.
Tseng F and Yu H. Plast Reconstr Surg Glob Open. 2019 Aug 19;7(8):e2366.
Dibernardo BE. J Cosmet Laser Ther. 2013 Apr;15(2):56-64.
Jones D et al. Dermatol Surg. 2016 Oct;4 Suppl 1(Suppl 1):S235-42.
Lee SK and Kim HS. J Cosmet Dermatol. 2018 Aug;17(4):590-5.
Chao YY et al. Dermatol Surg. 2011 Oct;37(10):1542-5.
Han TY et al. Dermatol Surg. 2011 Sep;37(9):1291-6.
Dr. Wesley and Dr. Talakoub are cocontributors to this column. Dr. Wesley practices dermatology in Beverly Hills, Calif. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Talakoub. Write to them at dermnews@mdedge.com. They had no relevant disclosures.
The interplay of the neck subunits, as outlined in the recent article by Friedman and colleagues, requires multiple combination treatments, including fat removal, augmentation of deficient bony prominences, relaxation of hyperkinetic muscles, tissue tightening, suture anchoring, skin resurfacing, and treatment of dyschromia.
Horizontal neck lines are linear etched lines or furrows that commonly appear at a young age and are not caused by the aging process. The anatomy of the neck and the manner in which it bends contributes to their development at an early age. It is hypothesized that variable adipose tissue thickness and fibromuscular bands contribute to deepening of these lines in overweight patients. The widespread use of cell phones, laptops, and tablets has increased their prevalence and this has become one of the most common concerns of patients aged under 30 years in my clinic.
Various treatments have been recommended for neck rejuvenation, including hyaluronic acid and dilute calcium hydroxylapatite. In my experience, neither of these treatments adequately resolves the horizontal neck lines, and more importantly, prevents them from reoccurring. In addition, given the variability in skin and adipose thickness in the anterior neck, side effects including lumps, irregular correction, and the Tyndall effect, are common, particularly with incorrect choice of filler and injection depth.
The fibromuscular bands along the transverse neck lines pose one of the complexities in treatment with injectable filler. I have had significant improvement in the aesthetic outcome of my patients by using subcision along the transverse bands extensively prior to injection with hyaluronic acid fillers. The subcision is done with a 27-gauge needle to release the fibrous bands that tether the tissue down. If a patient has excess adipose tissue on either side of the bands, injectable fillers often do not improve the appearance of the lines and can make the neck appear heavier. The use of subcision followed by one to six treatments of deoxycholic acid in the adjacent adipose tissue prior to injection with a filler will help even out the contour of the neck, decrease adipose tissue bulges, release the fibrous bands, and fill the lines properly.
Working from home and on handheld devices has increased the appearance of neck lines in young populations. Despite the vast array of treatments in the aging neck, none have been very successful for this particular problem in the young. We need an improved understanding of these lines and better studies to investigate treatment options and long-term correction.
References:
Friedman O et al. J Cosmet Dermatol. 2021 Feb;20(2):569-76.
Brandt FS and Boker A. Dermatol Clin. 2004 Apr;22(2):159-66.
Tseng F and Yu H. Plast Reconstr Surg Glob Open. 2019 Aug 19;7(8):e2366.
Dibernardo BE. J Cosmet Laser Ther. 2013 Apr;15(2):56-64.
Jones D et al. Dermatol Surg. 2016 Oct;4 Suppl 1(Suppl 1):S235-42.
Lee SK and Kim HS. J Cosmet Dermatol. 2018 Aug;17(4):590-5.
Chao YY et al. Dermatol Surg. 2011 Oct;37(10):1542-5.
Han TY et al. Dermatol Surg. 2011 Sep;37(9):1291-6.
Dr. Wesley and Dr. Talakoub are cocontributors to this column. Dr. Wesley practices dermatology in Beverly Hills, Calif. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Talakoub. Write to them at dermnews@mdedge.com. They had no relevant disclosures.
Molecular insights suggest novel therapies for hidradenitis suppurativa
at the virtual annual congress of the European Academy of Dermatology and Venereology.
He presented highlights of a multicenter translational study, which utilized whole transcriptome analysis of lesional and nonlesional skin from patients with HS and normal controls along with quantitative real-time PCR and immunohistochemistry. The purpose was to further define the molecular taxonomy of this inflammatory disease. And while this objective was achieved, the results also underscored a truism regarding the painful and scarring disease: “HS is characterized by an ever-growing complexity, which translates into multiple potential mechanistic drivers,” observed Dr. da Costa, head of immunology precision medicine at AstraZeneca in Gothenburg, Sweden.
Indeed, the study identified a panel of immune-related drivers in HS that influence innate immunity and cell differentiation in follicular and epidermal keratinocytes. The research by Dr. da Costa and coinvestigators identified a broad array of promising novel therapeutic targets in HS.
“Our findings provide evidence of an inflammatory process coupled with impaired barrier function, altered epidermal cell differentiation, and possibly abnormal microbiome activity which can be seen at the follicular and epidermal keratinocytes and also to a minor degree at the level of the skin glands,” Dr. da Costa said.
There is a huge unmet need for new therapies for HS, since at present adalimumab (Humira) is the only approved medication for this debilitating inflammatory disease. Some good news that emerged from this translational study is that some of the novel molecular mediators implicated in HS are targeted by multiple Food and Drug Administration–approved therapies that have other indications. From a drug development standpoint, repurposing a commercially available drug for a novel indication is a much more efficient and less costly endeavor than is necessary to establish the safety and efficacy of an unproven new agent.
The translational work demonstrated that the proteins calgranulin-A and -B and serpin-B4 were strongly expressed in the hair root sheaths of patients with HS. Connexin-32 and koebnerisin were present in stratum granulosum, matrix metallopeptidase-9 was strongly expressed in resident monocytes, small prolin-rich protein 3 in apocrine sweat glands and ducts as well as in sebaceous glands and ducts, and transcobalamin-1 was prominent in stratum spinosum.
Of the 19 key molecular mediators of HS identified in the study, FDA-approved agents are already available that target 12 of them. For example, apremilast (Otezla) targets interferon-gamma and tumor necrosis factor–alpha. Gentamicin targets growth arrest-specific 6 (GAS6) and interleukin-17 (IL-17). Secukinumab (Cosentyx) and ixekizumab (Taltz) target IL-17A, and brodalumab (Siliq) more broadly targets IL-17A as well as all the other IL-17 receptors. Thalidomide targets hepatocyte growth factor (HGF) and TNF-alpha. Spironolactone targets androgen receptor (AR) and TNF-alpha. Colchicine targets tubulin. Anakinra (Kineret) homes in on the IL-1 receptor. And prednisone targets NFxB.
Other key molecular mediators of HS, which are targeted by commercially available drugs, include epidermal growth factor (EGF), macrophage colony-stimulating factor (MCSF), epiregulin (EREG), fibroblast growth factor 1 (FGF1), FGF2, insulin-like growth factor 2 (IGF2), and IL-6, according to Dr. da Costa.
In addition, clinical trials are underway in HS involving totally investigational agents, including several Janus kinase inhibitors and tyrosine kinase 2 inhibitors.
The work described by Dr. da Costa had multiple funding sources, including the European Hidradenitis Suppurativa Foundation, the University of Copenhagen, the Icahn School of Medicine at Mount Sinai, AstraZeneca, and the German Federal Ministry of Education and Research. Dr. da Costa is an employee of AstraZeneca, Gothenburg, Sweden.
at the virtual annual congress of the European Academy of Dermatology and Venereology.
He presented highlights of a multicenter translational study, which utilized whole transcriptome analysis of lesional and nonlesional skin from patients with HS and normal controls along with quantitative real-time PCR and immunohistochemistry. The purpose was to further define the molecular taxonomy of this inflammatory disease. And while this objective was achieved, the results also underscored a truism regarding the painful and scarring disease: “HS is characterized by an ever-growing complexity, which translates into multiple potential mechanistic drivers,” observed Dr. da Costa, head of immunology precision medicine at AstraZeneca in Gothenburg, Sweden.
Indeed, the study identified a panel of immune-related drivers in HS that influence innate immunity and cell differentiation in follicular and epidermal keratinocytes. The research by Dr. da Costa and coinvestigators identified a broad array of promising novel therapeutic targets in HS.
“Our findings provide evidence of an inflammatory process coupled with impaired barrier function, altered epidermal cell differentiation, and possibly abnormal microbiome activity which can be seen at the follicular and epidermal keratinocytes and also to a minor degree at the level of the skin glands,” Dr. da Costa said.
There is a huge unmet need for new therapies for HS, since at present adalimumab (Humira) is the only approved medication for this debilitating inflammatory disease. Some good news that emerged from this translational study is that some of the novel molecular mediators implicated in HS are targeted by multiple Food and Drug Administration–approved therapies that have other indications. From a drug development standpoint, repurposing a commercially available drug for a novel indication is a much more efficient and less costly endeavor than is necessary to establish the safety and efficacy of an unproven new agent.
The translational work demonstrated that the proteins calgranulin-A and -B and serpin-B4 were strongly expressed in the hair root sheaths of patients with HS. Connexin-32 and koebnerisin were present in stratum granulosum, matrix metallopeptidase-9 was strongly expressed in resident monocytes, small prolin-rich protein 3 in apocrine sweat glands and ducts as well as in sebaceous glands and ducts, and transcobalamin-1 was prominent in stratum spinosum.
Of the 19 key molecular mediators of HS identified in the study, FDA-approved agents are already available that target 12 of them. For example, apremilast (Otezla) targets interferon-gamma and tumor necrosis factor–alpha. Gentamicin targets growth arrest-specific 6 (GAS6) and interleukin-17 (IL-17). Secukinumab (Cosentyx) and ixekizumab (Taltz) target IL-17A, and brodalumab (Siliq) more broadly targets IL-17A as well as all the other IL-17 receptors. Thalidomide targets hepatocyte growth factor (HGF) and TNF-alpha. Spironolactone targets androgen receptor (AR) and TNF-alpha. Colchicine targets tubulin. Anakinra (Kineret) homes in on the IL-1 receptor. And prednisone targets NFxB.
Other key molecular mediators of HS, which are targeted by commercially available drugs, include epidermal growth factor (EGF), macrophage colony-stimulating factor (MCSF), epiregulin (EREG), fibroblast growth factor 1 (FGF1), FGF2, insulin-like growth factor 2 (IGF2), and IL-6, according to Dr. da Costa.
In addition, clinical trials are underway in HS involving totally investigational agents, including several Janus kinase inhibitors and tyrosine kinase 2 inhibitors.
The work described by Dr. da Costa had multiple funding sources, including the European Hidradenitis Suppurativa Foundation, the University of Copenhagen, the Icahn School of Medicine at Mount Sinai, AstraZeneca, and the German Federal Ministry of Education and Research. Dr. da Costa is an employee of AstraZeneca, Gothenburg, Sweden.
at the virtual annual congress of the European Academy of Dermatology and Venereology.
He presented highlights of a multicenter translational study, which utilized whole transcriptome analysis of lesional and nonlesional skin from patients with HS and normal controls along with quantitative real-time PCR and immunohistochemistry. The purpose was to further define the molecular taxonomy of this inflammatory disease. And while this objective was achieved, the results also underscored a truism regarding the painful and scarring disease: “HS is characterized by an ever-growing complexity, which translates into multiple potential mechanistic drivers,” observed Dr. da Costa, head of immunology precision medicine at AstraZeneca in Gothenburg, Sweden.
Indeed, the study identified a panel of immune-related drivers in HS that influence innate immunity and cell differentiation in follicular and epidermal keratinocytes. The research by Dr. da Costa and coinvestigators identified a broad array of promising novel therapeutic targets in HS.
“Our findings provide evidence of an inflammatory process coupled with impaired barrier function, altered epidermal cell differentiation, and possibly abnormal microbiome activity which can be seen at the follicular and epidermal keratinocytes and also to a minor degree at the level of the skin glands,” Dr. da Costa said.
There is a huge unmet need for new therapies for HS, since at present adalimumab (Humira) is the only approved medication for this debilitating inflammatory disease. Some good news that emerged from this translational study is that some of the novel molecular mediators implicated in HS are targeted by multiple Food and Drug Administration–approved therapies that have other indications. From a drug development standpoint, repurposing a commercially available drug for a novel indication is a much more efficient and less costly endeavor than is necessary to establish the safety and efficacy of an unproven new agent.
The translational work demonstrated that the proteins calgranulin-A and -B and serpin-B4 were strongly expressed in the hair root sheaths of patients with HS. Connexin-32 and koebnerisin were present in stratum granulosum, matrix metallopeptidase-9 was strongly expressed in resident monocytes, small prolin-rich protein 3 in apocrine sweat glands and ducts as well as in sebaceous glands and ducts, and transcobalamin-1 was prominent in stratum spinosum.
Of the 19 key molecular mediators of HS identified in the study, FDA-approved agents are already available that target 12 of them. For example, apremilast (Otezla) targets interferon-gamma and tumor necrosis factor–alpha. Gentamicin targets growth arrest-specific 6 (GAS6) and interleukin-17 (IL-17). Secukinumab (Cosentyx) and ixekizumab (Taltz) target IL-17A, and brodalumab (Siliq) more broadly targets IL-17A as well as all the other IL-17 receptors. Thalidomide targets hepatocyte growth factor (HGF) and TNF-alpha. Spironolactone targets androgen receptor (AR) and TNF-alpha. Colchicine targets tubulin. Anakinra (Kineret) homes in on the IL-1 receptor. And prednisone targets NFxB.
Other key molecular mediators of HS, which are targeted by commercially available drugs, include epidermal growth factor (EGF), macrophage colony-stimulating factor (MCSF), epiregulin (EREG), fibroblast growth factor 1 (FGF1), FGF2, insulin-like growth factor 2 (IGF2), and IL-6, according to Dr. da Costa.
In addition, clinical trials are underway in HS involving totally investigational agents, including several Janus kinase inhibitors and tyrosine kinase 2 inhibitors.
The work described by Dr. da Costa had multiple funding sources, including the European Hidradenitis Suppurativa Foundation, the University of Copenhagen, the Icahn School of Medicine at Mount Sinai, AstraZeneca, and the German Federal Ministry of Education and Research. Dr. da Costa is an employee of AstraZeneca, Gothenburg, Sweden.
FROM THE EADV CONGRESS
Anybody for a nanobody? Novel psoriasis therapy impresses in phase 2b
in a phase 2b randomized trial, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A nanobody is a tiny antibody fragment with a much smaller molecular weight than the monoclonal antibodies utilized today in treating psoriasis or atopic dermatitis. The sonelokinab nanobody, derived from animals in the camel family, is a recombinant sequence-optimized nanobody specific for human IL-17F, IL-17A, the heterodimer IL-17A/F, and serum albumin. The binding to serum albumin give sonelokinab a lengthy half-life of 10-12 hours, which may be therapeutically relevant, explained Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
He presented the 24-week results of a multicenter, double-blind, double-dummy randomized trial including 313 North American and European adults with an average 18-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of about 21. They were randomized to one of six treatment arms for the first 12 weeks: subcutaneous injection of sonelokinab at 30, 60, or 120 mg at weeks 0, 2, 4, and 8; enhanced–loading-dose sonelokinab at 120 mg every 2 weeks through week 10; the IL-17A inhibitor secukinumab (Cosentyx) at its standard dosing as an active comparator; or placebo. Data analysis was by rigorous nonresponder imputation, meaning anyone who didn’t complete the study was scored as a nonresponder.
“This yields a conservative data analysis somewhat biased against sonelokinab,” the dermatologist pointed out.
The primary outcome in the trial was the week-12 rate of an Investigator’s Global Assessment score of 0 or 1, indicative of clear or almost clear skin. This was achieved in 88.2% of patients in the highest-dose arm of sonelokinab. That group also had a week-12 PASI 90 response rate of 76.5% and a PASI 100 response rate of 33.3%. By comparison, patients on standard-dose secukinumab had a less robust week-12 IGA 0/1 rate of 77.4%, a PASI 90 of 64.2%, and a PASI 100 of 28.3%. Of note, however, this secukinumab performance was better than seen in the 30-mg sonelokinab group, and comparable to outcomes with 60 mg of sonelokinab.
Dose escalation was performed from weeks 12-24. Patients with a week-12 IGA score greater than 1 after being on sonelokinab at 30 or 60 mg were upgraded to 120 mg at week 12 and again every 4 weeks thereafter. Placebo-treated controls were switched to 120 mg at weeks 12, 14, 16, and every 4 weeks thereafter. The group on the enhanced–loading-dose sonelokinab moved to 120 mg every 4 weeks, while those who had gotten four doses of sonelokinab at 120 mg during the first 12 weeks were switched to 120 mg every 8 weeks. The secukinumab group remained on the approved dosing through week 24.
At week 24, superior outcomes were seen in the enhanced–loading-dose sonelokinab group, with an IGA 0/1 response rate of 94.2%, a PASI 90 of 90.4%, and a PASI 100 of 56.9%. The corresponding week-24 rates in patients on 120 mg of sonelokinab every 8 weeks from week 12 on were 80.4%, 79.2%, and 40.4%, outcomes similar to those seen with secukinumab.
The rapidity of response to sonelokinab at 120 mg was striking, with approximately one-third of treated patients achieving a PASI 90 response by week 4.
“This could reflect the smaller molecular profile. There is possibly rapid increased absorption or bioavailability, quicker time to achieving serum half-life, better penetration into target tissue, and perhaps more effective engagement at the target. All of those things are possibilities. These are things that are yet to be explored, but it’s very enticing to see that uncharacteristically rapid initial response. It’s all very gratifying – and tantalizing,” Dr. Papp said in response to an audience question.
The safety profile of sonelokinab was reassuring. The most common adverse events were nasopharyngitis in 13.5% of patients and pruritus in 6.7%, with most cases being mild or moderate. As with other IL-17 blockers, there was an increase in oral candidiasis. This side effect appeared to occur in dose-dependent fashion: The incidence was zero in the 30-mg group, 1.9% with 60 mg, 3.8% with sonelokinab at 120 mg without an enhanced loading dose, and 5.9% with the enhanced loading dose.
The study was conducted by Avillion in partnership with Merck. Dr. Papp reported receiving research funding from and serving as a consultant to those and numerous other pharmaceutical companies.
in a phase 2b randomized trial, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A nanobody is a tiny antibody fragment with a much smaller molecular weight than the monoclonal antibodies utilized today in treating psoriasis or atopic dermatitis. The sonelokinab nanobody, derived from animals in the camel family, is a recombinant sequence-optimized nanobody specific for human IL-17F, IL-17A, the heterodimer IL-17A/F, and serum albumin. The binding to serum albumin give sonelokinab a lengthy half-life of 10-12 hours, which may be therapeutically relevant, explained Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
He presented the 24-week results of a multicenter, double-blind, double-dummy randomized trial including 313 North American and European adults with an average 18-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of about 21. They were randomized to one of six treatment arms for the first 12 weeks: subcutaneous injection of sonelokinab at 30, 60, or 120 mg at weeks 0, 2, 4, and 8; enhanced–loading-dose sonelokinab at 120 mg every 2 weeks through week 10; the IL-17A inhibitor secukinumab (Cosentyx) at its standard dosing as an active comparator; or placebo. Data analysis was by rigorous nonresponder imputation, meaning anyone who didn’t complete the study was scored as a nonresponder.
“This yields a conservative data analysis somewhat biased against sonelokinab,” the dermatologist pointed out.
The primary outcome in the trial was the week-12 rate of an Investigator’s Global Assessment score of 0 or 1, indicative of clear or almost clear skin. This was achieved in 88.2% of patients in the highest-dose arm of sonelokinab. That group also had a week-12 PASI 90 response rate of 76.5% and a PASI 100 response rate of 33.3%. By comparison, patients on standard-dose secukinumab had a less robust week-12 IGA 0/1 rate of 77.4%, a PASI 90 of 64.2%, and a PASI 100 of 28.3%. Of note, however, this secukinumab performance was better than seen in the 30-mg sonelokinab group, and comparable to outcomes with 60 mg of sonelokinab.
Dose escalation was performed from weeks 12-24. Patients with a week-12 IGA score greater than 1 after being on sonelokinab at 30 or 60 mg were upgraded to 120 mg at week 12 and again every 4 weeks thereafter. Placebo-treated controls were switched to 120 mg at weeks 12, 14, 16, and every 4 weeks thereafter. The group on the enhanced–loading-dose sonelokinab moved to 120 mg every 4 weeks, while those who had gotten four doses of sonelokinab at 120 mg during the first 12 weeks were switched to 120 mg every 8 weeks. The secukinumab group remained on the approved dosing through week 24.
At week 24, superior outcomes were seen in the enhanced–loading-dose sonelokinab group, with an IGA 0/1 response rate of 94.2%, a PASI 90 of 90.4%, and a PASI 100 of 56.9%. The corresponding week-24 rates in patients on 120 mg of sonelokinab every 8 weeks from week 12 on were 80.4%, 79.2%, and 40.4%, outcomes similar to those seen with secukinumab.
The rapidity of response to sonelokinab at 120 mg was striking, with approximately one-third of treated patients achieving a PASI 90 response by week 4.
“This could reflect the smaller molecular profile. There is possibly rapid increased absorption or bioavailability, quicker time to achieving serum half-life, better penetration into target tissue, and perhaps more effective engagement at the target. All of those things are possibilities. These are things that are yet to be explored, but it’s very enticing to see that uncharacteristically rapid initial response. It’s all very gratifying – and tantalizing,” Dr. Papp said in response to an audience question.
The safety profile of sonelokinab was reassuring. The most common adverse events were nasopharyngitis in 13.5% of patients and pruritus in 6.7%, with most cases being mild or moderate. As with other IL-17 blockers, there was an increase in oral candidiasis. This side effect appeared to occur in dose-dependent fashion: The incidence was zero in the 30-mg group, 1.9% with 60 mg, 3.8% with sonelokinab at 120 mg without an enhanced loading dose, and 5.9% with the enhanced loading dose.
The study was conducted by Avillion in partnership with Merck. Dr. Papp reported receiving research funding from and serving as a consultant to those and numerous other pharmaceutical companies.
in a phase 2b randomized trial, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A nanobody is a tiny antibody fragment with a much smaller molecular weight than the monoclonal antibodies utilized today in treating psoriasis or atopic dermatitis. The sonelokinab nanobody, derived from animals in the camel family, is a recombinant sequence-optimized nanobody specific for human IL-17F, IL-17A, the heterodimer IL-17A/F, and serum albumin. The binding to serum albumin give sonelokinab a lengthy half-life of 10-12 hours, which may be therapeutically relevant, explained Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
He presented the 24-week results of a multicenter, double-blind, double-dummy randomized trial including 313 North American and European adults with an average 18-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of about 21. They were randomized to one of six treatment arms for the first 12 weeks: subcutaneous injection of sonelokinab at 30, 60, or 120 mg at weeks 0, 2, 4, and 8; enhanced–loading-dose sonelokinab at 120 mg every 2 weeks through week 10; the IL-17A inhibitor secukinumab (Cosentyx) at its standard dosing as an active comparator; or placebo. Data analysis was by rigorous nonresponder imputation, meaning anyone who didn’t complete the study was scored as a nonresponder.
“This yields a conservative data analysis somewhat biased against sonelokinab,” the dermatologist pointed out.
The primary outcome in the trial was the week-12 rate of an Investigator’s Global Assessment score of 0 or 1, indicative of clear or almost clear skin. This was achieved in 88.2% of patients in the highest-dose arm of sonelokinab. That group also had a week-12 PASI 90 response rate of 76.5% and a PASI 100 response rate of 33.3%. By comparison, patients on standard-dose secukinumab had a less robust week-12 IGA 0/1 rate of 77.4%, a PASI 90 of 64.2%, and a PASI 100 of 28.3%. Of note, however, this secukinumab performance was better than seen in the 30-mg sonelokinab group, and comparable to outcomes with 60 mg of sonelokinab.
Dose escalation was performed from weeks 12-24. Patients with a week-12 IGA score greater than 1 after being on sonelokinab at 30 or 60 mg were upgraded to 120 mg at week 12 and again every 4 weeks thereafter. Placebo-treated controls were switched to 120 mg at weeks 12, 14, 16, and every 4 weeks thereafter. The group on the enhanced–loading-dose sonelokinab moved to 120 mg every 4 weeks, while those who had gotten four doses of sonelokinab at 120 mg during the first 12 weeks were switched to 120 mg every 8 weeks. The secukinumab group remained on the approved dosing through week 24.
At week 24, superior outcomes were seen in the enhanced–loading-dose sonelokinab group, with an IGA 0/1 response rate of 94.2%, a PASI 90 of 90.4%, and a PASI 100 of 56.9%. The corresponding week-24 rates in patients on 120 mg of sonelokinab every 8 weeks from week 12 on were 80.4%, 79.2%, and 40.4%, outcomes similar to those seen with secukinumab.
The rapidity of response to sonelokinab at 120 mg was striking, with approximately one-third of treated patients achieving a PASI 90 response by week 4.
“This could reflect the smaller molecular profile. There is possibly rapid increased absorption or bioavailability, quicker time to achieving serum half-life, better penetration into target tissue, and perhaps more effective engagement at the target. All of those things are possibilities. These are things that are yet to be explored, but it’s very enticing to see that uncharacteristically rapid initial response. It’s all very gratifying – and tantalizing,” Dr. Papp said in response to an audience question.
The safety profile of sonelokinab was reassuring. The most common adverse events were nasopharyngitis in 13.5% of patients and pruritus in 6.7%, with most cases being mild or moderate. As with other IL-17 blockers, there was an increase in oral candidiasis. This side effect appeared to occur in dose-dependent fashion: The incidence was zero in the 30-mg group, 1.9% with 60 mg, 3.8% with sonelokinab at 120 mg without an enhanced loading dose, and 5.9% with the enhanced loading dose.
The study was conducted by Avillion in partnership with Merck. Dr. Papp reported receiving research funding from and serving as a consultant to those and numerous other pharmaceutical companies.
FROM the eadv congress
Neoadjuvant immunotherapy shows promise in stage III melanoma
The next dramatic , John M. Kirkwood, MD, predicted at a virtual forum on cutaneous malignancies jointly presented by the Postgraduate Institute for Medicine and Global Academy for Medical Education.

These agents have already demonstrated profound efficacy, first in stage IV metastatic disease and more recently as adjuvant therapy for resected stage III melanoma. Now, there is a great interest in learning whether by prescribing them preoperatively, patients might reduce their risk of advancing to metastatic disease. And neoadjuvant therapy offers an extremely attractive feature: It yields results in an accelerated fashion.
“The major problem with postoperative adjuvant trials in melanoma since 1984 is the long time to maturity. Many of us don’t want to wait the full 9 or 10 years for a full-bore, phase 3 adjuvant trial in stage III melanoma to mature,” explained Dr. Kirkwood, professor of medicine, dermatology, and translational science and coleader of the melanoma and skin cancer program at the University of Pittsburgh. “The opportunity to treat a patient who presents with a bulky lymph node, has a biopsy, and then can be treated for 3 or 6 weeks or sometimes even longer periods with a therapy that’s promising allows us to ask what’s going on in the tumor tissue, what’s going on in the clinical response at 3 or 6 weeks, and if there’s pathological complete or near-complete response under the microscope.”
Because pathological complete response is a strong predictor of relapse-free survival, this neoadjuvant-forward therapeutic strategy has the potential to provide patients and their physicians with an early forecast of likely clinical outcome only 4-6 weeks into treatment. Also, there is both preclinical and clinical evidence that neoadjuvant therapy may offer a survival advantage over adjuvant therapy, perhaps as a result of early treatment of micrometastatic disease. Another benefit of neoadjuvant therapy for melanoma is the resultant tumor shrinkage, which can permit less extensive surgery.
Dr. Kirkwood highlighted a phase 2 clinical trial conducted at the University of Pittsburgh to illustrate the potential of neoadjuvant therapy in melanoma. The ongoing single-arm study includes 32 patients with stage IIIB or IIIC resectable melanoma along with accessible tumor for biopsy and intratumoral injections of CMP-001, a toll-like receptor 9 agonist. According to the Eighth Edition of the American Joint Committee on Cancer staging manual, stage IIIB melanoma has a 10-year mortality of 23%, and stage IIIC disease has 40%.
CMP-001 triggers type 1 interferon production through activation of plasmacytoid dendritic cells. The resultant inflammatory response draws T cells into the tumor to enhance the response to immunotherapy, which in this study was nivolumab (Opdivo), a human programmed death ligand 1 (PD-L1)–blocking antibody. The neoadjuvant regimen consisted of seven once-weekly intratumoral injections of CMP-001, plus three 240-mg doses of nivolumab given at 2-week intervals. This was followed by resection, then 1 year of adjuvant therapy with nivolumab at 480 mg every 4 weeks and intratumoral CMP-001 every 4 weeks.
In an interim analysis, a major pathologic response occurred in an impressive 15 of 21 patients (71%) after 6 weeks of neoadjuvant therapy. Thirteen of the 15 had a pathologic complete response. Encouragingly, no one with a pathologic complete or near-complete response has relapsed to date.
“A pathologic complete response or near-complete response with neoadjuvant therapy appears to be a biomarker of durable disease control and is associated with excellent outcomes,” Dr. Kirkwood observed, adding that the Pittsburgh experience has been mirrored in reports from the Netherlands, Australia, and University of Texas M.D. Anderson Cancer Center, Houston, involving other neoadjuvant agents.
Other potential early biomarkers of favorable outcome with neoadjuvant therapy include CD8+ T cells in the tumor at baseline, tumor mutational burden, T-cell clonality, and a T-cell–inflamed gene-expression profile.
There were no dose-limiting toxicities or delays in surgery related to the neoadjuvant treatment.
Of note, imaging often inaccurately showed only a partial response in patients who actually had a pathologic complete response, meaning totally devoid of tumor, Dr. Kirkwood said.
Corroboration of these findings is planned in the national multicenter ECOG-ACRIN neoadjuvant trial EA6194.
“Consider referring to this trial any patients who present with bulky nodal disease for whom a treatment assessment at 4-6 weeks is desired in order to predict what the outcome may be,” he suggested.
Dr. Kirkwood reported receiving research grants from Amgen, BMS, Castle Biosciences, Checkmate, Immunocore, Iovance, and Novartis and serving as a consultant to a handful of companies.
Global Academy for Medical Education and this news organization are owned by the same company.
The next dramatic , John M. Kirkwood, MD, predicted at a virtual forum on cutaneous malignancies jointly presented by the Postgraduate Institute for Medicine and Global Academy for Medical Education.
These agents have already demonstrated profound efficacy, first in stage IV metastatic disease and more recently as adjuvant therapy for resected stage III melanoma. Now, there is a great interest in learning whether by prescribing them preoperatively, patients might reduce their risk of advancing to metastatic disease. And neoadjuvant therapy offers an extremely attractive feature: It yields results in an accelerated fashion.
“The major problem with postoperative adjuvant trials in melanoma since 1984 is the long time to maturity. Many of us don’t want to wait the full 9 or 10 years for a full-bore, phase 3 adjuvant trial in stage III melanoma to mature,” explained Dr. Kirkwood, professor of medicine, dermatology, and translational science and coleader of the melanoma and skin cancer program at the University of Pittsburgh. “The opportunity to treat a patient who presents with a bulky lymph node, has a biopsy, and then can be treated for 3 or 6 weeks or sometimes even longer periods with a therapy that’s promising allows us to ask what’s going on in the tumor tissue, what’s going on in the clinical response at 3 or 6 weeks, and if there’s pathological complete or near-complete response under the microscope.”
Because pathological complete response is a strong predictor of relapse-free survival, this neoadjuvant-forward therapeutic strategy has the potential to provide patients and their physicians with an early forecast of likely clinical outcome only 4-6 weeks into treatment. Also, there is both preclinical and clinical evidence that neoadjuvant therapy may offer a survival advantage over adjuvant therapy, perhaps as a result of early treatment of micrometastatic disease. Another benefit of neoadjuvant therapy for melanoma is the resultant tumor shrinkage, which can permit less extensive surgery.
Dr. Kirkwood highlighted a phase 2 clinical trial conducted at the University of Pittsburgh to illustrate the potential of neoadjuvant therapy in melanoma. The ongoing single-arm study includes 32 patients with stage IIIB or IIIC resectable melanoma along with accessible tumor for biopsy and intratumoral injections of CMP-001, a toll-like receptor 9 agonist. According to the Eighth Edition of the American Joint Committee on Cancer staging manual, stage IIIB melanoma has a 10-year mortality of 23%, and stage IIIC disease has 40%.
CMP-001 triggers type 1 interferon production through activation of plasmacytoid dendritic cells. The resultant inflammatory response draws T cells into the tumor to enhance the response to immunotherapy, which in this study was nivolumab (Opdivo), a human programmed death ligand 1 (PD-L1)–blocking antibody. The neoadjuvant regimen consisted of seven once-weekly intratumoral injections of CMP-001, plus three 240-mg doses of nivolumab given at 2-week intervals. This was followed by resection, then 1 year of adjuvant therapy with nivolumab at 480 mg every 4 weeks and intratumoral CMP-001 every 4 weeks.
In an interim analysis, a major pathologic response occurred in an impressive 15 of 21 patients (71%) after 6 weeks of neoadjuvant therapy. Thirteen of the 15 had a pathologic complete response. Encouragingly, no one with a pathologic complete or near-complete response has relapsed to date.
“A pathologic complete response or near-complete response with neoadjuvant therapy appears to be a biomarker of durable disease control and is associated with excellent outcomes,” Dr. Kirkwood observed, adding that the Pittsburgh experience has been mirrored in reports from the Netherlands, Australia, and University of Texas M.D. Anderson Cancer Center, Houston, involving other neoadjuvant agents.
Other potential early biomarkers of favorable outcome with neoadjuvant therapy include CD8+ T cells in the tumor at baseline, tumor mutational burden, T-cell clonality, and a T-cell–inflamed gene-expression profile.
There were no dose-limiting toxicities or delays in surgery related to the neoadjuvant treatment.
Of note, imaging often inaccurately showed only a partial response in patients who actually had a pathologic complete response, meaning totally devoid of tumor, Dr. Kirkwood said.
Corroboration of these findings is planned in the national multicenter ECOG-ACRIN neoadjuvant trial EA6194.
“Consider referring to this trial any patients who present with bulky nodal disease for whom a treatment assessment at 4-6 weeks is desired in order to predict what the outcome may be,” he suggested.
Dr. Kirkwood reported receiving research grants from Amgen, BMS, Castle Biosciences, Checkmate, Immunocore, Iovance, and Novartis and serving as a consultant to a handful of companies.
Global Academy for Medical Education and this news organization are owned by the same company.
The next dramatic , John M. Kirkwood, MD, predicted at a virtual forum on cutaneous malignancies jointly presented by the Postgraduate Institute for Medicine and Global Academy for Medical Education.
These agents have already demonstrated profound efficacy, first in stage IV metastatic disease and more recently as adjuvant therapy for resected stage III melanoma. Now, there is a great interest in learning whether by prescribing them preoperatively, patients might reduce their risk of advancing to metastatic disease. And neoadjuvant therapy offers an extremely attractive feature: It yields results in an accelerated fashion.
“The major problem with postoperative adjuvant trials in melanoma since 1984 is the long time to maturity. Many of us don’t want to wait the full 9 or 10 years for a full-bore, phase 3 adjuvant trial in stage III melanoma to mature,” explained Dr. Kirkwood, professor of medicine, dermatology, and translational science and coleader of the melanoma and skin cancer program at the University of Pittsburgh. “The opportunity to treat a patient who presents with a bulky lymph node, has a biopsy, and then can be treated for 3 or 6 weeks or sometimes even longer periods with a therapy that’s promising allows us to ask what’s going on in the tumor tissue, what’s going on in the clinical response at 3 or 6 weeks, and if there’s pathological complete or near-complete response under the microscope.”
Because pathological complete response is a strong predictor of relapse-free survival, this neoadjuvant-forward therapeutic strategy has the potential to provide patients and their physicians with an early forecast of likely clinical outcome only 4-6 weeks into treatment. Also, there is both preclinical and clinical evidence that neoadjuvant therapy may offer a survival advantage over adjuvant therapy, perhaps as a result of early treatment of micrometastatic disease. Another benefit of neoadjuvant therapy for melanoma is the resultant tumor shrinkage, which can permit less extensive surgery.
Dr. Kirkwood highlighted a phase 2 clinical trial conducted at the University of Pittsburgh to illustrate the potential of neoadjuvant therapy in melanoma. The ongoing single-arm study includes 32 patients with stage IIIB or IIIC resectable melanoma along with accessible tumor for biopsy and intratumoral injections of CMP-001, a toll-like receptor 9 agonist. According to the Eighth Edition of the American Joint Committee on Cancer staging manual, stage IIIB melanoma has a 10-year mortality of 23%, and stage IIIC disease has 40%.
CMP-001 triggers type 1 interferon production through activation of plasmacytoid dendritic cells. The resultant inflammatory response draws T cells into the tumor to enhance the response to immunotherapy, which in this study was nivolumab (Opdivo), a human programmed death ligand 1 (PD-L1)–blocking antibody. The neoadjuvant regimen consisted of seven once-weekly intratumoral injections of CMP-001, plus three 240-mg doses of nivolumab given at 2-week intervals. This was followed by resection, then 1 year of adjuvant therapy with nivolumab at 480 mg every 4 weeks and intratumoral CMP-001 every 4 weeks.
In an interim analysis, a major pathologic response occurred in an impressive 15 of 21 patients (71%) after 6 weeks of neoadjuvant therapy. Thirteen of the 15 had a pathologic complete response. Encouragingly, no one with a pathologic complete or near-complete response has relapsed to date.
“A pathologic complete response or near-complete response with neoadjuvant therapy appears to be a biomarker of durable disease control and is associated with excellent outcomes,” Dr. Kirkwood observed, adding that the Pittsburgh experience has been mirrored in reports from the Netherlands, Australia, and University of Texas M.D. Anderson Cancer Center, Houston, involving other neoadjuvant agents.
Other potential early biomarkers of favorable outcome with neoadjuvant therapy include CD8+ T cells in the tumor at baseline, tumor mutational burden, T-cell clonality, and a T-cell–inflamed gene-expression profile.
There were no dose-limiting toxicities or delays in surgery related to the neoadjuvant treatment.
Of note, imaging often inaccurately showed only a partial response in patients who actually had a pathologic complete response, meaning totally devoid of tumor, Dr. Kirkwood said.
Corroboration of these findings is planned in the national multicenter ECOG-ACRIN neoadjuvant trial EA6194.
“Consider referring to this trial any patients who present with bulky nodal disease for whom a treatment assessment at 4-6 weeks is desired in order to predict what the outcome may be,” he suggested.
Dr. Kirkwood reported receiving research grants from Amgen, BMS, Castle Biosciences, Checkmate, Immunocore, Iovance, and Novartis and serving as a consultant to a handful of companies.
Global Academy for Medical Education and this news organization are owned by the same company.
FROM THE CUTANEOUS MALIGNANCIES FORUM
Study: COVID cases have been ‘severely undercounted’
Large numbers of COVID-19 cases have been undetected and unreported, which has resulted in severe undercounting of the total number of people who have been infected during the pandemic, according to a new study published Monday in the journal PLOS ONE.
In the United States, the number of COVID-19 cases is likely three times that of reported cases. According to the study, more than 71 million Americans have contracted the virus during the pandemic, and 7 million were infected or potentially contagious last week.
Public health officials rely on case counts to guide decisions, so the undercounting should be considered while trying to end the pandemic.
“The estimates of actual infections reveal for the first time the true severity of COVID-19 across the U.S. and in countries worldwide,” Jungsik Noh, PhD, a bioinformatics professor at the University of Texas Southwestern Medical Center, said in a statement.
Dr. Noh and colleague Gaudenz Danuser created a computational model that uses machine-learning strategies to estimate the actual number of daily cases in the United States and the 50 most-infected countries.
The model pulls data from the Johns Hopkins University database and the COVID Tracking Project, as well as large-scale surveys conducted by the CDC and several states. The algorithm uses the number of reported deaths, which is thought to be more accurate than the number of lab-confirmed cases, as the basis for calculations.
In 25 of the 50 countries, the “actual” cumulative cases were estimated to be 5-20 times greater than the confirmed cases. In the United States, Belgium, and Brazil, about 10% of the population has contracted the coronavirus, according to the model. At the beginning of February, about 11% of the population in Pennsylvania had current infections, which was the highest rate of any state. About 0.15% of residents in Minnesota had infections, and about 2.5% of residents in New York and Texas had infections.
“Knowing the true severity in different regions will help us effectively fight against the virus spreading,” Dr. Noh said. “The currently infected population is the cause of future infections and deaths. Its actual size in a region is a crucial variable required when determining the severity of COVID-19 and building strategies against regional outbreaks.”
A version of this article first appeared on WebMD.com.
Large numbers of COVID-19 cases have been undetected and unreported, which has resulted in severe undercounting of the total number of people who have been infected during the pandemic, according to a new study published Monday in the journal PLOS ONE.
In the United States, the number of COVID-19 cases is likely three times that of reported cases. According to the study, more than 71 million Americans have contracted the virus during the pandemic, and 7 million were infected or potentially contagious last week.
Public health officials rely on case counts to guide decisions, so the undercounting should be considered while trying to end the pandemic.
“The estimates of actual infections reveal for the first time the true severity of COVID-19 across the U.S. and in countries worldwide,” Jungsik Noh, PhD, a bioinformatics professor at the University of Texas Southwestern Medical Center, said in a statement.
Dr. Noh and colleague Gaudenz Danuser created a computational model that uses machine-learning strategies to estimate the actual number of daily cases in the United States and the 50 most-infected countries.
The model pulls data from the Johns Hopkins University database and the COVID Tracking Project, as well as large-scale surveys conducted by the CDC and several states. The algorithm uses the number of reported deaths, which is thought to be more accurate than the number of lab-confirmed cases, as the basis for calculations.
In 25 of the 50 countries, the “actual” cumulative cases were estimated to be 5-20 times greater than the confirmed cases. In the United States, Belgium, and Brazil, about 10% of the population has contracted the coronavirus, according to the model. At the beginning of February, about 11% of the population in Pennsylvania had current infections, which was the highest rate of any state. About 0.15% of residents in Minnesota had infections, and about 2.5% of residents in New York and Texas had infections.
“Knowing the true severity in different regions will help us effectively fight against the virus spreading,” Dr. Noh said. “The currently infected population is the cause of future infections and deaths. Its actual size in a region is a crucial variable required when determining the severity of COVID-19 and building strategies against regional outbreaks.”
A version of this article first appeared on WebMD.com.
Large numbers of COVID-19 cases have been undetected and unreported, which has resulted in severe undercounting of the total number of people who have been infected during the pandemic, according to a new study published Monday in the journal PLOS ONE.
In the United States, the number of COVID-19 cases is likely three times that of reported cases. According to the study, more than 71 million Americans have contracted the virus during the pandemic, and 7 million were infected or potentially contagious last week.
Public health officials rely on case counts to guide decisions, so the undercounting should be considered while trying to end the pandemic.
“The estimates of actual infections reveal for the first time the true severity of COVID-19 across the U.S. and in countries worldwide,” Jungsik Noh, PhD, a bioinformatics professor at the University of Texas Southwestern Medical Center, said in a statement.
Dr. Noh and colleague Gaudenz Danuser created a computational model that uses machine-learning strategies to estimate the actual number of daily cases in the United States and the 50 most-infected countries.
The model pulls data from the Johns Hopkins University database and the COVID Tracking Project, as well as large-scale surveys conducted by the CDC and several states. The algorithm uses the number of reported deaths, which is thought to be more accurate than the number of lab-confirmed cases, as the basis for calculations.
In 25 of the 50 countries, the “actual” cumulative cases were estimated to be 5-20 times greater than the confirmed cases. In the United States, Belgium, and Brazil, about 10% of the population has contracted the coronavirus, according to the model. At the beginning of February, about 11% of the population in Pennsylvania had current infections, which was the highest rate of any state. About 0.15% of residents in Minnesota had infections, and about 2.5% of residents in New York and Texas had infections.
“Knowing the true severity in different regions will help us effectively fight against the virus spreading,” Dr. Noh said. “The currently infected population is the cause of future infections and deaths. Its actual size in a region is a crucial variable required when determining the severity of COVID-19 and building strategies against regional outbreaks.”
A version of this article first appeared on WebMD.com.
COVID-19: Peginterferon lambda may prevent clinical deterioration, shorten viral shedding
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.

Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.
Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.
Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
FROM THE LANCET RESPIRATORY MEDICINE
Teenagers get in the queue for COVID-19 vaccines
The vaccinations can’t come soon enough for parents like Stacy Hillenburg, a developmental therapist in Aurora, Ill., whose 9-year-old son takes immunosuppressants because he had a heart transplant when he was 7 weeks old. Although school-age children aren’t yet included in clinical trials, if her 12- and 13-year-old daughters could get vaccinated, along with both parents, then the family could relax some of the protocols they currently follow to prevent infection.
Whenever they are around other people, even masked and socially distanced, they come home and immediately shower and change their clothes. So far, no one in the family has been infected with COVID, but the anxiety is ever-present. “I can’t wait for it to come out,” Ms. Hillenburg said of a pediatric COVID vaccine. “It will ease my mind so much.”
She isn’t alone in that anticipation. In the fall, the American Academy of Pediatrics and other pediatric vaccine experts urged faster action on pediatric vaccine trials and worried that children would be left behind as adults gained protection from COVID. But recent developments have eased those concerns.
“Over the next couple of months, we will be doing trials in an age-deescalation manner,” with studies moving gradually to younger children, Anthony S. Fauci, MD, chief medical adviser on COVID-19 to the president, said in a coronavirus response team briefing on Jan. 29. “So that hopefully, as we get to the late spring and summer, we will have children being able to be vaccinated.”
Pfizer completed enrollment of 2,259 teens aged 12-15 years in late January and expects to move forward with a separate pediatric trial of children aged 5-11 years by this spring, Keanna Ghazvini, senior associate for global media relations at Pfizer, said in an interview.
Enrollment in Moderna’s TeenCove study of adolescents ages 12-17 years began slowly in late December, but the pace has since picked up, said company spokesperson Colleen Hussey. “We continue to bring clinical trial sites online, and we are on track to provide updated data around mid-year 2021.” A trial extension in children 11 years and younger is expected to begin later in 2021.
Johnson & Johnson and AstraZeneca said they expect to begin adolescent trials in early 2021, according to data shared by the Advisory Committee on Immunization Practices. An interim analysis of J&J’s Janssen COVID-19 vaccine trial data, released on Jan. 29, showed it was 72% effective in US participants aged 18 years or older. AstraZeneca’s U.S. trial in adults is ongoing.
Easing the burden
Vaccination could lessen children’s risk of severe disease as well as the social and emotional burdens of the pandemic, says James Campbell, MD, a pediatric infectious disease specialist at the University of Maryland’s Center for Vaccine Development in Baltimore, which was involved in the Moderna and early-phase Pfizer trials. He coauthored a September 2020 article in Clinical Infectious Diseases titled: “Warp Speed for COVID-19 vaccines: Why are children stuck in neutral?”
The adolescent trials are a vital step to ensure timely vaccine access for teens and younger children. “It is reasonable, when you have limited vaccine, that your rollout goes to the highest priority and then moves to lower and lower priorities. In adults, we’re just saying: ‘Wait your turn,’ ” he said of the current vaccination effort. “If we didn’t have the [vaccine trial] data in children, we’d be saying: ‘You don’t have a turn.’ ”
As the pandemic has worn on, the burden on children has grown. As of Tuesday, 269 children had died of COVID-19. That is well above the highest annual death toll recorded during a regular flu season – 188 flu deaths among children and adolescents under 18 in the 2019-2020 and 2017-2018 flu seasons.
Children are less likely to transmit COVID-19 in their household than adults, according to a meta-analysis of 54 studies published in JAMA Network Open. But that does not necessarily mean children are less infectious, the authors said, noting that unmeasured factors could have affected the spread of infection among adults.
Moreover, children and adolescents need protection from COVID infection – and from the potential for severe disease or lingering effects – and, given that there are 74 million children and teens in the United States, their vaccination is an important part of stopping the pandemic, said Grace Lee, MD, professor of pediatrics at Stanford (Calif.) University, and cochair of ACIP’s COVID-19 Vaccine Safety Technical Subgroup.
“In order to interrupt transmission, I don’t see how we’re going to do that without vaccinating children and adolescents,” she said.
Dr. Lee said her 16-year-old daughter misses the normal teenage social life and is excited about getting the vaccine when she is eligible. (Adolescents without high-risk conditions are in the lowest vaccination tier, according to ACIP recommendations.) “There is truly individual protection to be gained,” Dr. Lee said.
She noted that researchers continue to assess the immune responses to the adult vaccines – even looking at immune characteristics of the small percentage of people who aren’t protected from infection – and that information helps in the evaluation of the pediatric immune responses. As the trials expand to younger children and infants, dosing will be a major focus. “How many doses do they need they need to receive the same immunity? Safety considerations will be critically important,” she said.
Teen trials underway
Pfizer/BioNTech extended its adult trial to 16- and 17-year-olds in October, which enabled older teens to be included in its emergency-use authorization. They and younger teens, ages 12-15, receive the same dose as adults.
The ongoing trials with Pfizer and Moderna vaccines are immunobridging trials, designed to study safety and immunogenicity. Investigators will compare the teens’ immune response with the findings from the larger adult trials. When the trials expand to school-age children (6-12 years), protocols call for testing the safety and immunogenicity of a half-dose vaccine as well as the full dose.
Children ages 2-5 years and infants and toddlers will be enrolled in future trials, studying safety and immunogenicity of full, half, or even quarter dosages. The Pediatric Research Equity Act of 2003 requires licensed vaccines to be tested for safety and efficacy in children, unless they are not appropriate for a pediatric population.
Demand for the teen trials has been strong. At Cincinnati Children’s Hospital Medical Center, 259 teenagers joined the Pfizer/BioNTech trial, but some teenagers were turned away when the trial’s national enrollment closed in late January.
“Many of the children are having no side effects, and if they are, they’re having the same [effects] as the young adults – local redness or pain, fatigue, and headaches,” said Robert Frenck, MD, director of the Cincinnati Children’s Gamble Program for Clinical Studies.
Parents may share some of the vaccine hesitancy that has affected adult vaccination. But that is balanced by the hope that vaccines will end the pandemic and usher in a new normal. “If it looks like [vaccines] will increase the likelihood of children returning to school safely, that may be a motivating factor,” Dr. Frenck said.
Cody Meissner, MD, chief of the pediatric infectious disease service at Tufts Medical Center, Boston, was initially cautious about the extension of vaccination to adolescents. A member of the Vaccine and Related Biological Products Advisory Committee, which evaluates data and makes recommendations to the Food and Drug Administration, Dr. Meissner initially abstained in the vote on the Pfizer/BioNTech emergency-use authorization for people 16 and older.
He noted that, at the time the committee reviewed the Pfizer vaccine, the company had data available for just 134 teenagers, half of whom received a placebo. But the vaccination of 34 million adults has provided robust data about the vaccine’s safety, and the trial expansion into adolescents is important.
“I’m comfortable with the way these trials are going now,” he said. “This is the way I was hoping they would go.”
Ms. Hillenburg is on the parent advisory board of Voices for Vaccines, an organization of parents supporting vaccination that is affiliated with the Task Force for Global Health, an Atlanta-based independent public health organization. Dr. Campbell’s institution has received funds to conduct clinical trials from the National Institutes of Health and several companies, including Merck, GlaxoSmithKline, Sanofi, Pfizer, and Moderna. He has served pro bono on many safety and data monitoring committees. Dr. Frenck’s institution is receiving funds to conduct the Pfizer trial. In the past 5 years, he has also participated in clinical trials for GlaxoSmithKline, Merck, and Meissa vaccines. Dr. Lee and Dr. Meissner disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The vaccinations can’t come soon enough for parents like Stacy Hillenburg, a developmental therapist in Aurora, Ill., whose 9-year-old son takes immunosuppressants because he had a heart transplant when he was 7 weeks old. Although school-age children aren’t yet included in clinical trials, if her 12- and 13-year-old daughters could get vaccinated, along with both parents, then the family could relax some of the protocols they currently follow to prevent infection.
Whenever they are around other people, even masked and socially distanced, they come home and immediately shower and change their clothes. So far, no one in the family has been infected with COVID, but the anxiety is ever-present. “I can’t wait for it to come out,” Ms. Hillenburg said of a pediatric COVID vaccine. “It will ease my mind so much.”
She isn’t alone in that anticipation. In the fall, the American Academy of Pediatrics and other pediatric vaccine experts urged faster action on pediatric vaccine trials and worried that children would be left behind as adults gained protection from COVID. But recent developments have eased those concerns.
“Over the next couple of months, we will be doing trials in an age-deescalation manner,” with studies moving gradually to younger children, Anthony S. Fauci, MD, chief medical adviser on COVID-19 to the president, said in a coronavirus response team briefing on Jan. 29. “So that hopefully, as we get to the late spring and summer, we will have children being able to be vaccinated.”
Pfizer completed enrollment of 2,259 teens aged 12-15 years in late January and expects to move forward with a separate pediatric trial of children aged 5-11 years by this spring, Keanna Ghazvini, senior associate for global media relations at Pfizer, said in an interview.
Enrollment in Moderna’s TeenCove study of adolescents ages 12-17 years began slowly in late December, but the pace has since picked up, said company spokesperson Colleen Hussey. “We continue to bring clinical trial sites online, and we are on track to provide updated data around mid-year 2021.” A trial extension in children 11 years and younger is expected to begin later in 2021.
Johnson & Johnson and AstraZeneca said they expect to begin adolescent trials in early 2021, according to data shared by the Advisory Committee on Immunization Practices. An interim analysis of J&J’s Janssen COVID-19 vaccine trial data, released on Jan. 29, showed it was 72% effective in US participants aged 18 years or older. AstraZeneca’s U.S. trial in adults is ongoing.
Easing the burden
Vaccination could lessen children’s risk of severe disease as well as the social and emotional burdens of the pandemic, says James Campbell, MD, a pediatric infectious disease specialist at the University of Maryland’s Center for Vaccine Development in Baltimore, which was involved in the Moderna and early-phase Pfizer trials. He coauthored a September 2020 article in Clinical Infectious Diseases titled: “Warp Speed for COVID-19 vaccines: Why are children stuck in neutral?”
The adolescent trials are a vital step to ensure timely vaccine access for teens and younger children. “It is reasonable, when you have limited vaccine, that your rollout goes to the highest priority and then moves to lower and lower priorities. In adults, we’re just saying: ‘Wait your turn,’ ” he said of the current vaccination effort. “If we didn’t have the [vaccine trial] data in children, we’d be saying: ‘You don’t have a turn.’ ”
As the pandemic has worn on, the burden on children has grown. As of Tuesday, 269 children had died of COVID-19. That is well above the highest annual death toll recorded during a regular flu season – 188 flu deaths among children and adolescents under 18 in the 2019-2020 and 2017-2018 flu seasons.
Children are less likely to transmit COVID-19 in their household than adults, according to a meta-analysis of 54 studies published in JAMA Network Open. But that does not necessarily mean children are less infectious, the authors said, noting that unmeasured factors could have affected the spread of infection among adults.
Moreover, children and adolescents need protection from COVID infection – and from the potential for severe disease or lingering effects – and, given that there are 74 million children and teens in the United States, their vaccination is an important part of stopping the pandemic, said Grace Lee, MD, professor of pediatrics at Stanford (Calif.) University, and cochair of ACIP’s COVID-19 Vaccine Safety Technical Subgroup.
“In order to interrupt transmission, I don’t see how we’re going to do that without vaccinating children and adolescents,” she said.
Dr. Lee said her 16-year-old daughter misses the normal teenage social life and is excited about getting the vaccine when she is eligible. (Adolescents without high-risk conditions are in the lowest vaccination tier, according to ACIP recommendations.) “There is truly individual protection to be gained,” Dr. Lee said.
She noted that researchers continue to assess the immune responses to the adult vaccines – even looking at immune characteristics of the small percentage of people who aren’t protected from infection – and that information helps in the evaluation of the pediatric immune responses. As the trials expand to younger children and infants, dosing will be a major focus. “How many doses do they need they need to receive the same immunity? Safety considerations will be critically important,” she said.
Teen trials underway
Pfizer/BioNTech extended its adult trial to 16- and 17-year-olds in October, which enabled older teens to be included in its emergency-use authorization. They and younger teens, ages 12-15, receive the same dose as adults.
The ongoing trials with Pfizer and Moderna vaccines are immunobridging trials, designed to study safety and immunogenicity. Investigators will compare the teens’ immune response with the findings from the larger adult trials. When the trials expand to school-age children (6-12 years), protocols call for testing the safety and immunogenicity of a half-dose vaccine as well as the full dose.
Children ages 2-5 years and infants and toddlers will be enrolled in future trials, studying safety and immunogenicity of full, half, or even quarter dosages. The Pediatric Research Equity Act of 2003 requires licensed vaccines to be tested for safety and efficacy in children, unless they are not appropriate for a pediatric population.
Demand for the teen trials has been strong. At Cincinnati Children’s Hospital Medical Center, 259 teenagers joined the Pfizer/BioNTech trial, but some teenagers were turned away when the trial’s national enrollment closed in late January.
“Many of the children are having no side effects, and if they are, they’re having the same [effects] as the young adults – local redness or pain, fatigue, and headaches,” said Robert Frenck, MD, director of the Cincinnati Children’s Gamble Program for Clinical Studies.
Parents may share some of the vaccine hesitancy that has affected adult vaccination. But that is balanced by the hope that vaccines will end the pandemic and usher in a new normal. “If it looks like [vaccines] will increase the likelihood of children returning to school safely, that may be a motivating factor,” Dr. Frenck said.
Cody Meissner, MD, chief of the pediatric infectious disease service at Tufts Medical Center, Boston, was initially cautious about the extension of vaccination to adolescents. A member of the Vaccine and Related Biological Products Advisory Committee, which evaluates data and makes recommendations to the Food and Drug Administration, Dr. Meissner initially abstained in the vote on the Pfizer/BioNTech emergency-use authorization for people 16 and older.
He noted that, at the time the committee reviewed the Pfizer vaccine, the company had data available for just 134 teenagers, half of whom received a placebo. But the vaccination of 34 million adults has provided robust data about the vaccine’s safety, and the trial expansion into adolescents is important.
“I’m comfortable with the way these trials are going now,” he said. “This is the way I was hoping they would go.”
Ms. Hillenburg is on the parent advisory board of Voices for Vaccines, an organization of parents supporting vaccination that is affiliated with the Task Force for Global Health, an Atlanta-based independent public health organization. Dr. Campbell’s institution has received funds to conduct clinical trials from the National Institutes of Health and several companies, including Merck, GlaxoSmithKline, Sanofi, Pfizer, and Moderna. He has served pro bono on many safety and data monitoring committees. Dr. Frenck’s institution is receiving funds to conduct the Pfizer trial. In the past 5 years, he has also participated in clinical trials for GlaxoSmithKline, Merck, and Meissa vaccines. Dr. Lee and Dr. Meissner disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The vaccinations can’t come soon enough for parents like Stacy Hillenburg, a developmental therapist in Aurora, Ill., whose 9-year-old son takes immunosuppressants because he had a heart transplant when he was 7 weeks old. Although school-age children aren’t yet included in clinical trials, if her 12- and 13-year-old daughters could get vaccinated, along with both parents, then the family could relax some of the protocols they currently follow to prevent infection.
Whenever they are around other people, even masked and socially distanced, they come home and immediately shower and change their clothes. So far, no one in the family has been infected with COVID, but the anxiety is ever-present. “I can’t wait for it to come out,” Ms. Hillenburg said of a pediatric COVID vaccine. “It will ease my mind so much.”
She isn’t alone in that anticipation. In the fall, the American Academy of Pediatrics and other pediatric vaccine experts urged faster action on pediatric vaccine trials and worried that children would be left behind as adults gained protection from COVID. But recent developments have eased those concerns.
“Over the next couple of months, we will be doing trials in an age-deescalation manner,” with studies moving gradually to younger children, Anthony S. Fauci, MD, chief medical adviser on COVID-19 to the president, said in a coronavirus response team briefing on Jan. 29. “So that hopefully, as we get to the late spring and summer, we will have children being able to be vaccinated.”
Pfizer completed enrollment of 2,259 teens aged 12-15 years in late January and expects to move forward with a separate pediatric trial of children aged 5-11 years by this spring, Keanna Ghazvini, senior associate for global media relations at Pfizer, said in an interview.
Enrollment in Moderna’s TeenCove study of adolescents ages 12-17 years began slowly in late December, but the pace has since picked up, said company spokesperson Colleen Hussey. “We continue to bring clinical trial sites online, and we are on track to provide updated data around mid-year 2021.” A trial extension in children 11 years and younger is expected to begin later in 2021.
Johnson & Johnson and AstraZeneca said they expect to begin adolescent trials in early 2021, according to data shared by the Advisory Committee on Immunization Practices. An interim analysis of J&J’s Janssen COVID-19 vaccine trial data, released on Jan. 29, showed it was 72% effective in US participants aged 18 years or older. AstraZeneca’s U.S. trial in adults is ongoing.
Easing the burden
Vaccination could lessen children’s risk of severe disease as well as the social and emotional burdens of the pandemic, says James Campbell, MD, a pediatric infectious disease specialist at the University of Maryland’s Center for Vaccine Development in Baltimore, which was involved in the Moderna and early-phase Pfizer trials. He coauthored a September 2020 article in Clinical Infectious Diseases titled: “Warp Speed for COVID-19 vaccines: Why are children stuck in neutral?”
The adolescent trials are a vital step to ensure timely vaccine access for teens and younger children. “It is reasonable, when you have limited vaccine, that your rollout goes to the highest priority and then moves to lower and lower priorities. In adults, we’re just saying: ‘Wait your turn,’ ” he said of the current vaccination effort. “If we didn’t have the [vaccine trial] data in children, we’d be saying: ‘You don’t have a turn.’ ”
As the pandemic has worn on, the burden on children has grown. As of Tuesday, 269 children had died of COVID-19. That is well above the highest annual death toll recorded during a regular flu season – 188 flu deaths among children and adolescents under 18 in the 2019-2020 and 2017-2018 flu seasons.
Children are less likely to transmit COVID-19 in their household than adults, according to a meta-analysis of 54 studies published in JAMA Network Open. But that does not necessarily mean children are less infectious, the authors said, noting that unmeasured factors could have affected the spread of infection among adults.
Moreover, children and adolescents need protection from COVID infection – and from the potential for severe disease or lingering effects – and, given that there are 74 million children and teens in the United States, their vaccination is an important part of stopping the pandemic, said Grace Lee, MD, professor of pediatrics at Stanford (Calif.) University, and cochair of ACIP’s COVID-19 Vaccine Safety Technical Subgroup.
“In order to interrupt transmission, I don’t see how we’re going to do that without vaccinating children and adolescents,” she said.
Dr. Lee said her 16-year-old daughter misses the normal teenage social life and is excited about getting the vaccine when she is eligible. (Adolescents without high-risk conditions are in the lowest vaccination tier, according to ACIP recommendations.) “There is truly individual protection to be gained,” Dr. Lee said.
She noted that researchers continue to assess the immune responses to the adult vaccines – even looking at immune characteristics of the small percentage of people who aren’t protected from infection – and that information helps in the evaluation of the pediatric immune responses. As the trials expand to younger children and infants, dosing will be a major focus. “How many doses do they need they need to receive the same immunity? Safety considerations will be critically important,” she said.
Teen trials underway
Pfizer/BioNTech extended its adult trial to 16- and 17-year-olds in October, which enabled older teens to be included in its emergency-use authorization. They and younger teens, ages 12-15, receive the same dose as adults.
The ongoing trials with Pfizer and Moderna vaccines are immunobridging trials, designed to study safety and immunogenicity. Investigators will compare the teens’ immune response with the findings from the larger adult trials. When the trials expand to school-age children (6-12 years), protocols call for testing the safety and immunogenicity of a half-dose vaccine as well as the full dose.
Children ages 2-5 years and infants and toddlers will be enrolled in future trials, studying safety and immunogenicity of full, half, or even quarter dosages. The Pediatric Research Equity Act of 2003 requires licensed vaccines to be tested for safety and efficacy in children, unless they are not appropriate for a pediatric population.
Demand for the teen trials has been strong. At Cincinnati Children’s Hospital Medical Center, 259 teenagers joined the Pfizer/BioNTech trial, but some teenagers were turned away when the trial’s national enrollment closed in late January.
“Many of the children are having no side effects, and if they are, they’re having the same [effects] as the young adults – local redness or pain, fatigue, and headaches,” said Robert Frenck, MD, director of the Cincinnati Children’s Gamble Program for Clinical Studies.
Parents may share some of the vaccine hesitancy that has affected adult vaccination. But that is balanced by the hope that vaccines will end the pandemic and usher in a new normal. “If it looks like [vaccines] will increase the likelihood of children returning to school safely, that may be a motivating factor,” Dr. Frenck said.
Cody Meissner, MD, chief of the pediatric infectious disease service at Tufts Medical Center, Boston, was initially cautious about the extension of vaccination to adolescents. A member of the Vaccine and Related Biological Products Advisory Committee, which evaluates data and makes recommendations to the Food and Drug Administration, Dr. Meissner initially abstained in the vote on the Pfizer/BioNTech emergency-use authorization for people 16 and older.
He noted that, at the time the committee reviewed the Pfizer vaccine, the company had data available for just 134 teenagers, half of whom received a placebo. But the vaccination of 34 million adults has provided robust data about the vaccine’s safety, and the trial expansion into adolescents is important.
“I’m comfortable with the way these trials are going now,” he said. “This is the way I was hoping they would go.”
Ms. Hillenburg is on the parent advisory board of Voices for Vaccines, an organization of parents supporting vaccination that is affiliated with the Task Force for Global Health, an Atlanta-based independent public health organization. Dr. Campbell’s institution has received funds to conduct clinical trials from the National Institutes of Health and several companies, including Merck, GlaxoSmithKline, Sanofi, Pfizer, and Moderna. He has served pro bono on many safety and data monitoring committees. Dr. Frenck’s institution is receiving funds to conduct the Pfizer trial. In the past 5 years, he has also participated in clinical trials for GlaxoSmithKline, Merck, and Meissa vaccines. Dr. Lee and Dr. Meissner disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The cutaneous benefits of bee venom, Part I: Atopic dermatitis and acne
Honeybees, Apis mellifera, play an important role in the web of life. We rely on bees for pollinating approximately one-third of our crops, including multiple fruits, vegetables, nuts, and seeds.1,2 Bees are also instrumental in the propagation of other plants, flower nectar, and flower pollen. A. mellifera, the European honeybee, is the main pollinator in Europe and North America, but other species, including A. cerana, A. dorsata, A. floria, A. andreniformis, A. koschevnikov, and A. laboriosa, yield honey.3 Honey, propolis, and royal jelly, along with beeswax and bee pollen, are among some of the celebrated bee products that have been found to confer health benefits to human beings.4,5 Bee venom, a toxin bees use for protection, is a convoluted combination of peptides and toxic proteins such as phospholipase A2 (PLA2) and melittin that has garnered significant scientific attention of late and is used to treat various inflammatory conditions.6-8 This column will focus on the investigation of the use of bee venom to treat atopic dermatitis (AD) and acne.

Atopic dermatitis
In 2013, Kim et al. assessed the impact of bee venom on AD-related symptoms in mice, finding that it attenuated the effects of AD-simulating compounds in 48 of 80 patients injected subcutaneously. They concluded that bee venom acted by suppressing mast cell degranulation and proinflammatory cytokine expression.9 Three years later, You et al. conducted a double-blind, randomized, base-controlled multicenter study of 136 patients with AD to ascertain the effects of a bee venom emollient. For 4 weeks, patients applied an emollient with bee venom and silk protein or a vehicle lacking bee venom twice daily. Eczema area and severity index (EASI) scores were significantly lower in the bee venom group, as were the visual analogue scale (VAS) scores. The investigators concluded that bee venom is an effective and safe therapeutic choice for treating patients with AD.10 Further, in 2018, Shin et al. demonstrated that PLA2 derived from bee venom mitigates atopic skin inflammation via the CD206 mannose receptor. They had previously shown in a mouse model that PLA2 from bee venom exerts such activity against AD-like lesions induced by 2,4-dinitrochlorobenzene (DNCB) and house dust mite (Dermatophagoides farinae) extract.11 Gu et al. observed later that year that intraperitoneal administration of bee venom eased the symptoms of ovalbumin-induced AD-like skin lesions in an experimental mouse model. Bee venom also lowered serum immunoglobulin E levels and suppressed infiltration of eosinophils and mast cells. They concluded that bee venom is a viable alternative for attenuating the allergic skin inflammation characteristic of AD.12 At the end of 2018, An et al. reported on the use of an in vivo female Balb/c mouse AD model in which 1-chloro-DNCB acted as inducer in cultures of human keratinocytes, stimulated by TNF-alpha/IFN-gamma. The investigators found that bee venom and melittin displayed robust antiatopic effects as evidenced by reduced lesions. The bee products were also found to have hindered elevated expression of various chemokines and proinflammatory cytokines. The authors suggested that bee venom and melittin appear to warrant consideration as a topical treatment for AD.13 In 2019, Kim et al. demonstrated in mice that bee venom eases the symptoms of AD by inactivating the complement system, particularly through CD55 induction, which might account for its effectiveness in AD treatment in humans, they suggested.6 Early in 2020, Lee et al. demonstrated in a Balb/c mouse model that bee venom appears to be a possible therapeutic macromolecule for treating phthalic anhydride-induced AD.7
Acne
In 2013, in vitro experiments by Han et al. showed that purified bee venom exhibited antimicrobial activity, in a concentration-dependent manner, against Cutibacterium acnes (or Propionibacterium acnes). They followed up with a small randomized, double-blind, controlled trial with 12 subjects who were treated with cosmetics with pure bee venom or cosmetics without it for two weeks. The group receiving bee venom experienced significantly fewer inflammatory and noninflammatory lesions, and a significant decline in adenosine triphosphate levels (a 57.5% reduction) was noted in subjects in the bee venom group, with a nonsignificant decrease of 4.7% observed in the control group. The investigators concluded the purified bee venom may be suitable as an antiacne agent.14 Using a mouse model, An et al. studied the therapeutic effects of bee venom against C. acnes–induced skin inflammation. They found that bee venom significantly diminished the volume of infiltrated inflammatory cells in the treated mice, compared with untreated mice. Bee venom also decreased expression levels of tumor necrosis factor (TNF)-α, and interleukin (IL)-1beta and suppressed Toll-like receptor (TLR)2 and CD14 expression in C. acnes–injected tissue. The investigators concluded that bee venom imparts notable anti-inflammatory activity and has potential for use in treating acne and as an anti-inflammatory agent in skin care.15

In 2015, Kim et al. studied the influence of bee venom against C. acnes–induced inflammation in human keratinocytes (HaCaT) and monocytes (THP-1). They found that bee venom successfully suppressed the secretion of interferon-gamma, IL-1beta, IL-8, and TNF-alpha. It also galvanized the expression of IL-8 and TLR2 in HaCaT cells but hampered their expression in heat-killed C. acnes. The researchers concluded that bee venom displays considerable anti-inflammatory activity against C. acnes and warrants consideration as an alternative to antibiotic acne treatment.16 It is worth noting that early that year, in a comprehensive database review to evaluate the effects and safety of a wide range of complementary treatments for acne, Cao et al. found, among 35 studies including parallel-group randomized controlled trials, that one trial indicated bee venom was superior to control in lowering the number of acne lesions.17
Conclusion
More research, in the form of randomized, controlled trials, is required before bee venom can be incorporated into the dermatologic armamentarium as a first-line therapy for common and vexing cutaneous conditions. Nevertheless, .
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann has written two textbooks and a New York Times Best Sellers book for consumers. Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Galderma, Revance, Evolus, and Burt’s Bees. She is the CEO of Skin Type Solutions Inc., a company that independently tests skin care products and makes recommendations to physicians on which skin care technologies are best. Write to her at dermnews@mdedge.com.
References
1. Walsh B. The plight of the honeybee: Mass deaths in bee colonies may mean disaster for farmers – and your favorite Foods. Time Magazine, 2013 Aug 19.
2. Klein AM et al. Proc Biol Sci. 2007 Feb 7;274(1608):303-13. doi: 10.1098/rspb.2006.3721.
3. Ediriweera ER and Premarathna NY. AYU. 2012 Apr;33(2):178-82. doi: 10.4103/0974-8520.105233.
4. Baumann, L. Honey/Propolis/Royal Jelly. In Cosmeceuticals and Cosmetic Ingredients. New York:McGraw-Hill; 2014:203-212.
5. Cornara L et al. Front Pharmacol. 2017 Jun 28;8:412. doi: 10.3389/fphar.2017.00412.
6. Kim Y et al. Toxins (Basel). 2019 Apr 26;11(5):239. doi: 10.3390/toxins11050239.
7. Lee YJ et al. Inflammopharmacology. 2020 Feb;28(1):253-63. doi: 10.1007/s10787-019-00646-w.
8. Lee G and Bae H. Molecules. 2016 May 11;21(5):616. doi: 10.3390/molecules21050616.
9. Kim KH et al. Int J Clin Exp Pathol. 2013 Nov 15;6(12):2896-903.
10. You CE et al. Ann Dermatol. 2016 Oct;28(5):593-9. doi: 10.5021/ad.2016.28.5.593.
11. Shin D et al. Toxins (Basel). 2018 Apr 2;10(4):146. doi: 10.3390/toxins10040146.
12. Gu H et al. Mol Med Rep. 2018 Oct;18(4):3711-8. doi: 10.3892/mmr.2018.9398.
13. An HJ et al. Br J Pharmacol. 2018 Dec;175(23):4310-24. doi: 10.1111/bph.14487.
14. Han SM et al. J Integr Med. 2013 Sep;11(5):320-6. doi: 10.3736/jintegrmed2013043.
15. An HJ et al. Int J Mol Med. 2014 Nov;34(5):1341-8. doi: 10.3892/ijmm.2014.1933.
16. Kim JY et al. Int J Mol Med. 2015 Jun;35(6):1651-6. doi: 10.3892/ijmm.2015.2180.
17. Cao H et al. Cochrane Database Syst Rev. 2015 Jan 19;1:CD009436. doi: 10.1002/14651858.CD009436.pub2.
Honeybees, Apis mellifera, play an important role in the web of life. We rely on bees for pollinating approximately one-third of our crops, including multiple fruits, vegetables, nuts, and seeds.1,2 Bees are also instrumental in the propagation of other plants, flower nectar, and flower pollen. A. mellifera, the European honeybee, is the main pollinator in Europe and North America, but other species, including A. cerana, A. dorsata, A. floria, A. andreniformis, A. koschevnikov, and A. laboriosa, yield honey.3 Honey, propolis, and royal jelly, along with beeswax and bee pollen, are among some of the celebrated bee products that have been found to confer health benefits to human beings.4,5 Bee venom, a toxin bees use for protection, is a convoluted combination of peptides and toxic proteins such as phospholipase A2 (PLA2) and melittin that has garnered significant scientific attention of late and is used to treat various inflammatory conditions.6-8 This column will focus on the investigation of the use of bee venom to treat atopic dermatitis (AD) and acne.
Atopic dermatitis
In 2013, Kim et al. assessed the impact of bee venom on AD-related symptoms in mice, finding that it attenuated the effects of AD-simulating compounds in 48 of 80 patients injected subcutaneously. They concluded that bee venom acted by suppressing mast cell degranulation and proinflammatory cytokine expression.9 Three years later, You et al. conducted a double-blind, randomized, base-controlled multicenter study of 136 patients with AD to ascertain the effects of a bee venom emollient. For 4 weeks, patients applied an emollient with bee venom and silk protein or a vehicle lacking bee venom twice daily. Eczema area and severity index (EASI) scores were significantly lower in the bee venom group, as were the visual analogue scale (VAS) scores. The investigators concluded that bee venom is an effective and safe therapeutic choice for treating patients with AD.10 Further, in 2018, Shin et al. demonstrated that PLA2 derived from bee venom mitigates atopic skin inflammation via the CD206 mannose receptor. They had previously shown in a mouse model that PLA2 from bee venom exerts such activity against AD-like lesions induced by 2,4-dinitrochlorobenzene (DNCB) and house dust mite (Dermatophagoides farinae) extract.11 Gu et al. observed later that year that intraperitoneal administration of bee venom eased the symptoms of ovalbumin-induced AD-like skin lesions in an experimental mouse model. Bee venom also lowered serum immunoglobulin E levels and suppressed infiltration of eosinophils and mast cells. They concluded that bee venom is a viable alternative for attenuating the allergic skin inflammation characteristic of AD.12 At the end of 2018, An et al. reported on the use of an in vivo female Balb/c mouse AD model in which 1-chloro-DNCB acted as inducer in cultures of human keratinocytes, stimulated by TNF-alpha/IFN-gamma. The investigators found that bee venom and melittin displayed robust antiatopic effects as evidenced by reduced lesions. The bee products were also found to have hindered elevated expression of various chemokines and proinflammatory cytokines. The authors suggested that bee venom and melittin appear to warrant consideration as a topical treatment for AD.13 In 2019, Kim et al. demonstrated in mice that bee venom eases the symptoms of AD by inactivating the complement system, particularly through CD55 induction, which might account for its effectiveness in AD treatment in humans, they suggested.6 Early in 2020, Lee et al. demonstrated in a Balb/c mouse model that bee venom appears to be a possible therapeutic macromolecule for treating phthalic anhydride-induced AD.7
Acne
In 2013, in vitro experiments by Han et al. showed that purified bee venom exhibited antimicrobial activity, in a concentration-dependent manner, against Cutibacterium acnes (or Propionibacterium acnes). They followed up with a small randomized, double-blind, controlled trial with 12 subjects who were treated with cosmetics with pure bee venom or cosmetics without it for two weeks. The group receiving bee venom experienced significantly fewer inflammatory and noninflammatory lesions, and a significant decline in adenosine triphosphate levels (a 57.5% reduction) was noted in subjects in the bee venom group, with a nonsignificant decrease of 4.7% observed in the control group. The investigators concluded the purified bee venom may be suitable as an antiacne agent.14 Using a mouse model, An et al. studied the therapeutic effects of bee venom against C. acnes–induced skin inflammation. They found that bee venom significantly diminished the volume of infiltrated inflammatory cells in the treated mice, compared with untreated mice. Bee venom also decreased expression levels of tumor necrosis factor (TNF)-α, and interleukin (IL)-1beta and suppressed Toll-like receptor (TLR)2 and CD14 expression in C. acnes–injected tissue. The investigators concluded that bee venom imparts notable anti-inflammatory activity and has potential for use in treating acne and as an anti-inflammatory agent in skin care.15
In 2015, Kim et al. studied the influence of bee venom against C. acnes–induced inflammation in human keratinocytes (HaCaT) and monocytes (THP-1). They found that bee venom successfully suppressed the secretion of interferon-gamma, IL-1beta, IL-8, and TNF-alpha. It also galvanized the expression of IL-8 and TLR2 in HaCaT cells but hampered their expression in heat-killed C. acnes. The researchers concluded that bee venom displays considerable anti-inflammatory activity against C. acnes and warrants consideration as an alternative to antibiotic acne treatment.16 It is worth noting that early that year, in a comprehensive database review to evaluate the effects and safety of a wide range of complementary treatments for acne, Cao et al. found, among 35 studies including parallel-group randomized controlled trials, that one trial indicated bee venom was superior to control in lowering the number of acne lesions.17
Conclusion
More research, in the form of randomized, controlled trials, is required before bee venom can be incorporated into the dermatologic armamentarium as a first-line therapy for common and vexing cutaneous conditions. Nevertheless, .
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann has written two textbooks and a New York Times Best Sellers book for consumers. Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Galderma, Revance, Evolus, and Burt’s Bees. She is the CEO of Skin Type Solutions Inc., a company that independently tests skin care products and makes recommendations to physicians on which skin care technologies are best. Write to her at dermnews@mdedge.com.
References
1. Walsh B. The plight of the honeybee: Mass deaths in bee colonies may mean disaster for farmers – and your favorite Foods. Time Magazine, 2013 Aug 19.
2. Klein AM et al. Proc Biol Sci. 2007 Feb 7;274(1608):303-13. doi: 10.1098/rspb.2006.3721.
3. Ediriweera ER and Premarathna NY. AYU. 2012 Apr;33(2):178-82. doi: 10.4103/0974-8520.105233.
4. Baumann, L. Honey/Propolis/Royal Jelly. In Cosmeceuticals and Cosmetic Ingredients. New York:McGraw-Hill; 2014:203-212.
5. Cornara L et al. Front Pharmacol. 2017 Jun 28;8:412. doi: 10.3389/fphar.2017.00412.
6. Kim Y et al. Toxins (Basel). 2019 Apr 26;11(5):239. doi: 10.3390/toxins11050239.
7. Lee YJ et al. Inflammopharmacology. 2020 Feb;28(1):253-63. doi: 10.1007/s10787-019-00646-w.
8. Lee G and Bae H. Molecules. 2016 May 11;21(5):616. doi: 10.3390/molecules21050616.
9. Kim KH et al. Int J Clin Exp Pathol. 2013 Nov 15;6(12):2896-903.
10. You CE et al. Ann Dermatol. 2016 Oct;28(5):593-9. doi: 10.5021/ad.2016.28.5.593.
11. Shin D et al. Toxins (Basel). 2018 Apr 2;10(4):146. doi: 10.3390/toxins10040146.
12. Gu H et al. Mol Med Rep. 2018 Oct;18(4):3711-8. doi: 10.3892/mmr.2018.9398.
13. An HJ et al. Br J Pharmacol. 2018 Dec;175(23):4310-24. doi: 10.1111/bph.14487.
14. Han SM et al. J Integr Med. 2013 Sep;11(5):320-6. doi: 10.3736/jintegrmed2013043.
15. An HJ et al. Int J Mol Med. 2014 Nov;34(5):1341-8. doi: 10.3892/ijmm.2014.1933.
16. Kim JY et al. Int J Mol Med. 2015 Jun;35(6):1651-6. doi: 10.3892/ijmm.2015.2180.
17. Cao H et al. Cochrane Database Syst Rev. 2015 Jan 19;1:CD009436. doi: 10.1002/14651858.CD009436.pub2.
Honeybees, Apis mellifera, play an important role in the web of life. We rely on bees for pollinating approximately one-third of our crops, including multiple fruits, vegetables, nuts, and seeds.1,2 Bees are also instrumental in the propagation of other plants, flower nectar, and flower pollen. A. mellifera, the European honeybee, is the main pollinator in Europe and North America, but other species, including A. cerana, A. dorsata, A. floria, A. andreniformis, A. koschevnikov, and A. laboriosa, yield honey.3 Honey, propolis, and royal jelly, along with beeswax and bee pollen, are among some of the celebrated bee products that have been found to confer health benefits to human beings.4,5 Bee venom, a toxin bees use for protection, is a convoluted combination of peptides and toxic proteins such as phospholipase A2 (PLA2) and melittin that has garnered significant scientific attention of late and is used to treat various inflammatory conditions.6-8 This column will focus on the investigation of the use of bee venom to treat atopic dermatitis (AD) and acne.
Atopic dermatitis
In 2013, Kim et al. assessed the impact of bee venom on AD-related symptoms in mice, finding that it attenuated the effects of AD-simulating compounds in 48 of 80 patients injected subcutaneously. They concluded that bee venom acted by suppressing mast cell degranulation and proinflammatory cytokine expression.9 Three years later, You et al. conducted a double-blind, randomized, base-controlled multicenter study of 136 patients with AD to ascertain the effects of a bee venom emollient. For 4 weeks, patients applied an emollient with bee venom and silk protein or a vehicle lacking bee venom twice daily. Eczema area and severity index (EASI) scores were significantly lower in the bee venom group, as were the visual analogue scale (VAS) scores. The investigators concluded that bee venom is an effective and safe therapeutic choice for treating patients with AD.10 Further, in 2018, Shin et al. demonstrated that PLA2 derived from bee venom mitigates atopic skin inflammation via the CD206 mannose receptor. They had previously shown in a mouse model that PLA2 from bee venom exerts such activity against AD-like lesions induced by 2,4-dinitrochlorobenzene (DNCB) and house dust mite (Dermatophagoides farinae) extract.11 Gu et al. observed later that year that intraperitoneal administration of bee venom eased the symptoms of ovalbumin-induced AD-like skin lesions in an experimental mouse model. Bee venom also lowered serum immunoglobulin E levels and suppressed infiltration of eosinophils and mast cells. They concluded that bee venom is a viable alternative for attenuating the allergic skin inflammation characteristic of AD.12 At the end of 2018, An et al. reported on the use of an in vivo female Balb/c mouse AD model in which 1-chloro-DNCB acted as inducer in cultures of human keratinocytes, stimulated by TNF-alpha/IFN-gamma. The investigators found that bee venom and melittin displayed robust antiatopic effects as evidenced by reduced lesions. The bee products were also found to have hindered elevated expression of various chemokines and proinflammatory cytokines. The authors suggested that bee venom and melittin appear to warrant consideration as a topical treatment for AD.13 In 2019, Kim et al. demonstrated in mice that bee venom eases the symptoms of AD by inactivating the complement system, particularly through CD55 induction, which might account for its effectiveness in AD treatment in humans, they suggested.6 Early in 2020, Lee et al. demonstrated in a Balb/c mouse model that bee venom appears to be a possible therapeutic macromolecule for treating phthalic anhydride-induced AD.7
Acne
In 2013, in vitro experiments by Han et al. showed that purified bee venom exhibited antimicrobial activity, in a concentration-dependent manner, against Cutibacterium acnes (or Propionibacterium acnes). They followed up with a small randomized, double-blind, controlled trial with 12 subjects who were treated with cosmetics with pure bee venom or cosmetics without it for two weeks. The group receiving bee venom experienced significantly fewer inflammatory and noninflammatory lesions, and a significant decline in adenosine triphosphate levels (a 57.5% reduction) was noted in subjects in the bee venom group, with a nonsignificant decrease of 4.7% observed in the control group. The investigators concluded the purified bee venom may be suitable as an antiacne agent.14 Using a mouse model, An et al. studied the therapeutic effects of bee venom against C. acnes–induced skin inflammation. They found that bee venom significantly diminished the volume of infiltrated inflammatory cells in the treated mice, compared with untreated mice. Bee venom also decreased expression levels of tumor necrosis factor (TNF)-α, and interleukin (IL)-1beta and suppressed Toll-like receptor (TLR)2 and CD14 expression in C. acnes–injected tissue. The investigators concluded that bee venom imparts notable anti-inflammatory activity and has potential for use in treating acne and as an anti-inflammatory agent in skin care.15
In 2015, Kim et al. studied the influence of bee venom against C. acnes–induced inflammation in human keratinocytes (HaCaT) and monocytes (THP-1). They found that bee venom successfully suppressed the secretion of interferon-gamma, IL-1beta, IL-8, and TNF-alpha. It also galvanized the expression of IL-8 and TLR2 in HaCaT cells but hampered their expression in heat-killed C. acnes. The researchers concluded that bee venom displays considerable anti-inflammatory activity against C. acnes and warrants consideration as an alternative to antibiotic acne treatment.16 It is worth noting that early that year, in a comprehensive database review to evaluate the effects and safety of a wide range of complementary treatments for acne, Cao et al. found, among 35 studies including parallel-group randomized controlled trials, that one trial indicated bee venom was superior to control in lowering the number of acne lesions.17
Conclusion
More research, in the form of randomized, controlled trials, is required before bee venom can be incorporated into the dermatologic armamentarium as a first-line therapy for common and vexing cutaneous conditions. Nevertheless, .
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann has written two textbooks and a New York Times Best Sellers book for consumers. Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Galderma, Revance, Evolus, and Burt’s Bees. She is the CEO of Skin Type Solutions Inc., a company that independently tests skin care products and makes recommendations to physicians on which skin care technologies are best. Write to her at dermnews@mdedge.com.
References
1. Walsh B. The plight of the honeybee: Mass deaths in bee colonies may mean disaster for farmers – and your favorite Foods. Time Magazine, 2013 Aug 19.
2. Klein AM et al. Proc Biol Sci. 2007 Feb 7;274(1608):303-13. doi: 10.1098/rspb.2006.3721.
3. Ediriweera ER and Premarathna NY. AYU. 2012 Apr;33(2):178-82. doi: 10.4103/0974-8520.105233.
4. Baumann, L. Honey/Propolis/Royal Jelly. In Cosmeceuticals and Cosmetic Ingredients. New York:McGraw-Hill; 2014:203-212.
5. Cornara L et al. Front Pharmacol. 2017 Jun 28;8:412. doi: 10.3389/fphar.2017.00412.
6. Kim Y et al. Toxins (Basel). 2019 Apr 26;11(5):239. doi: 10.3390/toxins11050239.
7. Lee YJ et al. Inflammopharmacology. 2020 Feb;28(1):253-63. doi: 10.1007/s10787-019-00646-w.
8. Lee G and Bae H. Molecules. 2016 May 11;21(5):616. doi: 10.3390/molecules21050616.
9. Kim KH et al. Int J Clin Exp Pathol. 2013 Nov 15;6(12):2896-903.
10. You CE et al. Ann Dermatol. 2016 Oct;28(5):593-9. doi: 10.5021/ad.2016.28.5.593.
11. Shin D et al. Toxins (Basel). 2018 Apr 2;10(4):146. doi: 10.3390/toxins10040146.
12. Gu H et al. Mol Med Rep. 2018 Oct;18(4):3711-8. doi: 10.3892/mmr.2018.9398.
13. An HJ et al. Br J Pharmacol. 2018 Dec;175(23):4310-24. doi: 10.1111/bph.14487.
14. Han SM et al. J Integr Med. 2013 Sep;11(5):320-6. doi: 10.3736/jintegrmed2013043.
15. An HJ et al. Int J Mol Med. 2014 Nov;34(5):1341-8. doi: 10.3892/ijmm.2014.1933.
16. Kim JY et al. Int J Mol Med. 2015 Jun;35(6):1651-6. doi: 10.3892/ijmm.2015.2180.
17. Cao H et al. Cochrane Database Syst Rev. 2015 Jan 19;1:CD009436. doi: 10.1002/14651858.CD009436.pub2.
Some COVID-19 vaccine reactions could be pseudoallergic, experts say
On Jan. 13, 2 days after a drive-through vaccination “superstation” opened in San Diego, six people were treated for anaphylaxis after they received the Moderna vaccine, leading the California state epidemiologist to recommend pausing the administration of that particular lot.
A group of allergy and immunology experts and public health officials reviewed the cases, as well as an incident that occurred the day before, and concluded that at least some of the responses were angioedema, or swelling — a serious allergic reaction — but none were actually anaphylaxis. No similar clusters had occurred with the same vaccine lot in other states, and California resumed using the doses.
Yet questions remain about the reactions and the mechanisms for them. Some might have been triggered by an allergy to a vaccine component, most likely the polyethylene glycol (PEG) that stabilizes the lipid surrounding the mRNA, the key vaccine component in both the Moderna and Pfizer vaccines. Another possible explanation is that some could be pseudoallergic reactions to a blood protein known as complement, a little-understood process that resembles an antigen-based reaction but doesn’t leave an immune memory and might not recur.
Cases of complement-activation-related pseudoallergy look like a severe allergic reaction but occur through a different mechanism and don’t require previous exposure to an allergen.
“It has the same signs and symptoms and is treated the same way, but it occurs through a different pathway,” explained Neal Halsey, MD, director emeritus of the Institute for Vaccine Safety and emeritus professor at the Johns Hopkins Bloomberg School of Public Health in Baltimore.
Pseudoallergies are not well understood, but they have been associated with reactions to the contrast media used in imaging, such as with MRI. “If people have had an anaphylaxis-type reaction following the injection of contrast-dye material, that is a strong signal that it might be a complement-activation-related pseudoallergy,” said Dr. Halsey, a member of the Clinical Immunization Safety Assessment Network. “Those are the people who definitely need to consider seeing an allergist before getting the COVID vaccines.”
When Aleena Banerji, MD, clinical director of the allergy and clinical immunology unit at Massachusetts General Hospital in Boston, talks to patients about vaccine reactions, she addresses the risk for COVID-19 infection. All of the people who developed allergies after the Pfizer and Moderna vaccines recovered, but more than 445,000 Americans have died from COVID-19.
Most people with common allergies, such as to food or oral medications, don’t need to worry about reactions, said Dr. Banerji, lead author of a review that assessed the risk for allergic reactions to the Pfizer and Moderna vaccines.
Investigating reactions
As investigators search for the answers to what causes reactions, transparency is crucial to trust, said Kathryn Edwards, MD, principal investigator of the Clinical Immunization Safety Assessment Project, a vaccine safety network funded by the Centers for Disease Control and Prevention.
“Unless the public knows that we’re really investigating and we’re taking this seriously, then I think the vaccine hesitancy is going to increase,” said Dr. Edwards, professor of pediatrics at Vanderbilt University Medical Center and scientific director of the Vanderbilt Vaccine Research Program in Nashville, Tenn.
First reports of anaphylaxis came quickly after COVID-19 vaccinations began. In the 2 weeks before the holidays, almost 2 million health care workers received the Pfizer vaccine, and 21 of them developed anaphylaxis, according to CDC researchers who reviewed case reports from the Vaccine Adverse Event Reporting System (VAERS). That rate of about 1 in 100,000 is 10 times higher than the occurrence with other vaccines. No deaths from anaphylaxis were reported.
As the vaccinations ramped up, the rate declined. As of Jan. 18, 50 cases of anaphylaxis were reported to VAERS after the administration of 9,943,247 Pfizer doses, for a rate of 5.0 per million, according to data presented at the Jan. 27 meeting of the CDC Advisory Committee on Immunization Practices. And 21 cases of anaphylaxis were reported to VAERS after the administration of 7,581,429 Moderna doses, for a rate of 2.8 per million.
The anaphylaxis occurred almost exclusively in women; only three of the VAERS anaphylaxis reports were from men. Only 24% had a history of anaphylaxis.
The earlier CDC report explored the potential link to allergies. One person with anaphylaxis had a history of allergy to iodinated contrast media, and others had allergies to various medications, vaccines, foods, and animals. The researchers reported 86 nonanaphylaxis allergic reactions and 61 nonallergic adverse events among the 175 case reports they reviewed as possible cases of severe allergic reaction.
Of 1,266 reports that VAERS received from Dec. 21 to Jan. 10, the CDC identified 108 possible cases of severe allergic reaction after the Moderna vaccine. Only 10 met the case definition of anaphylaxis put forward by the Brighton Collaboration, a vaccine safety organization. All but one case involved a history of allergies or allergic reactions; only five had a previously experienced anaphylaxis.
There were 47 nonanaphylaxis allergic reactions.
The San Diego cluster also met the Brighton case definition for anaphylaxis, Dr. Edwards reported. This discrepancy highlights the difficulties in characterizing vaccine reactions.
Measuring a pseudoallergic reaction is a challenge. It requires that a blood sample be drawn soon after the incident and then frozen to protect heat-sensitive blood markers, Dr. Edwards explained.
And as vaccinations rise, so do adverse-event reports. But unlike in clinical trials, there is no control group for comparison. That is why vaccine safety experts urge caution when evaluating events and, where possible, advise looking at background rates.
“A major way to determine whether the adverse event is causally related is to assess the incidence of the adverse event in vaccines versus nonvaccines,” said Walter Orenstein, MD, who directed the U.S. Immunization Program from 1988 to 2004 and is now associate director of the Emory Vaccine Center and professor of infectious diseases at Emory University in Atlanta. Public health officials could then identify vaccine risk factors, he said.
When a reaction occurs almost immediately after vaccination, vaccine safety investigators look for probable triggers. If allergy to PEG is the culprit in anaphylactic reactions, then the individuals would have had a previous exposure, perhaps from injectable medications, Dr. Edwards said.
It might be feasible to perform a skin test for allergy to PEG. “If the skin testing is negative, that doesn’t completely rule out allergy, but it can be used in the decision-making about giving the first or second vaccine dose,” Dr. Banerji said.
Other vaccines, such as childhood vaccines, contain polysorbate as a stabilizer, which has a similar chemical structure, and it’s not clear why someone would react to PEG but not to polysorbate, Dr. Edwards said.
Meanwhile, other illnesses and even deaths sometimes occur in the days after vaccination, but that doesn’t mean the vaccine caused them, cautioned Steve Black, MD, emeritus professor of pediatrics at Cincinnati Children’s Hospital and cofounder of the Global Vaccine Data Network, an international vaccine safety collaboration.
“Different events and clusters of events will occur by chance alone, as these events can occur without vaccines. We need to not immediately assume that they’re due to the vaccine,” he said. “You don’t want to undermine the whole vaccine program every time something comes up and assume that it’s associated with the vaccine.”
The CDC only has three contraindications for the vaccines:
- Severe allergic reaction (such as anaphylaxis) after a previous dose of an mRNA COVID-19 vaccine or any of its components.
- Immediate allergic reaction of any severity to a previous dose of an mRNA COVID-19 vaccine or any of its components (including PEG).
- Immediate allergic reaction of any severity to polysorbate (due to potential cross-reactive hypersensitivity with PEG).
People who have had an immediate allergic reaction to other vaccines or injectable therapies should consider consulting with an allergist or immunologist before getting the Pfizer or Moderna vaccines, the CDC advises.
The CDC also says that people with a history of anaphylaxis from any cause should be observed for 30 minutes after vaccination. Vaccination protocol calls for everyone else to wait on site for 15 minutes after vaccination.
A version of this article first appeared on Medscape.com.
On Jan. 13, 2 days after a drive-through vaccination “superstation” opened in San Diego, six people were treated for anaphylaxis after they received the Moderna vaccine, leading the California state epidemiologist to recommend pausing the administration of that particular lot.
A group of allergy and immunology experts and public health officials reviewed the cases, as well as an incident that occurred the day before, and concluded that at least some of the responses were angioedema, or swelling — a serious allergic reaction — but none were actually anaphylaxis. No similar clusters had occurred with the same vaccine lot in other states, and California resumed using the doses.
Yet questions remain about the reactions and the mechanisms for them. Some might have been triggered by an allergy to a vaccine component, most likely the polyethylene glycol (PEG) that stabilizes the lipid surrounding the mRNA, the key vaccine component in both the Moderna and Pfizer vaccines. Another possible explanation is that some could be pseudoallergic reactions to a blood protein known as complement, a little-understood process that resembles an antigen-based reaction but doesn’t leave an immune memory and might not recur.
Cases of complement-activation-related pseudoallergy look like a severe allergic reaction but occur through a different mechanism and don’t require previous exposure to an allergen.
“It has the same signs and symptoms and is treated the same way, but it occurs through a different pathway,” explained Neal Halsey, MD, director emeritus of the Institute for Vaccine Safety and emeritus professor at the Johns Hopkins Bloomberg School of Public Health in Baltimore.
Pseudoallergies are not well understood, but they have been associated with reactions to the contrast media used in imaging, such as with MRI. “If people have had an anaphylaxis-type reaction following the injection of contrast-dye material, that is a strong signal that it might be a complement-activation-related pseudoallergy,” said Dr. Halsey, a member of the Clinical Immunization Safety Assessment Network. “Those are the people who definitely need to consider seeing an allergist before getting the COVID vaccines.”
When Aleena Banerji, MD, clinical director of the allergy and clinical immunology unit at Massachusetts General Hospital in Boston, talks to patients about vaccine reactions, she addresses the risk for COVID-19 infection. All of the people who developed allergies after the Pfizer and Moderna vaccines recovered, but more than 445,000 Americans have died from COVID-19.
Most people with common allergies, such as to food or oral medications, don’t need to worry about reactions, said Dr. Banerji, lead author of a review that assessed the risk for allergic reactions to the Pfizer and Moderna vaccines.
Investigating reactions
As investigators search for the answers to what causes reactions, transparency is crucial to trust, said Kathryn Edwards, MD, principal investigator of the Clinical Immunization Safety Assessment Project, a vaccine safety network funded by the Centers for Disease Control and Prevention.
“Unless the public knows that we’re really investigating and we’re taking this seriously, then I think the vaccine hesitancy is going to increase,” said Dr. Edwards, professor of pediatrics at Vanderbilt University Medical Center and scientific director of the Vanderbilt Vaccine Research Program in Nashville, Tenn.
First reports of anaphylaxis came quickly after COVID-19 vaccinations began. In the 2 weeks before the holidays, almost 2 million health care workers received the Pfizer vaccine, and 21 of them developed anaphylaxis, according to CDC researchers who reviewed case reports from the Vaccine Adverse Event Reporting System (VAERS). That rate of about 1 in 100,000 is 10 times higher than the occurrence with other vaccines. No deaths from anaphylaxis were reported.
As the vaccinations ramped up, the rate declined. As of Jan. 18, 50 cases of anaphylaxis were reported to VAERS after the administration of 9,943,247 Pfizer doses, for a rate of 5.0 per million, according to data presented at the Jan. 27 meeting of the CDC Advisory Committee on Immunization Practices. And 21 cases of anaphylaxis were reported to VAERS after the administration of 7,581,429 Moderna doses, for a rate of 2.8 per million.
The anaphylaxis occurred almost exclusively in women; only three of the VAERS anaphylaxis reports were from men. Only 24% had a history of anaphylaxis.
The earlier CDC report explored the potential link to allergies. One person with anaphylaxis had a history of allergy to iodinated contrast media, and others had allergies to various medications, vaccines, foods, and animals. The researchers reported 86 nonanaphylaxis allergic reactions and 61 nonallergic adverse events among the 175 case reports they reviewed as possible cases of severe allergic reaction.
Of 1,266 reports that VAERS received from Dec. 21 to Jan. 10, the CDC identified 108 possible cases of severe allergic reaction after the Moderna vaccine. Only 10 met the case definition of anaphylaxis put forward by the Brighton Collaboration, a vaccine safety organization. All but one case involved a history of allergies or allergic reactions; only five had a previously experienced anaphylaxis.
There were 47 nonanaphylaxis allergic reactions.
The San Diego cluster also met the Brighton case definition for anaphylaxis, Dr. Edwards reported. This discrepancy highlights the difficulties in characterizing vaccine reactions.
Measuring a pseudoallergic reaction is a challenge. It requires that a blood sample be drawn soon after the incident and then frozen to protect heat-sensitive blood markers, Dr. Edwards explained.
And as vaccinations rise, so do adverse-event reports. But unlike in clinical trials, there is no control group for comparison. That is why vaccine safety experts urge caution when evaluating events and, where possible, advise looking at background rates.
“A major way to determine whether the adverse event is causally related is to assess the incidence of the adverse event in vaccines versus nonvaccines,” said Walter Orenstein, MD, who directed the U.S. Immunization Program from 1988 to 2004 and is now associate director of the Emory Vaccine Center and professor of infectious diseases at Emory University in Atlanta. Public health officials could then identify vaccine risk factors, he said.
When a reaction occurs almost immediately after vaccination, vaccine safety investigators look for probable triggers. If allergy to PEG is the culprit in anaphylactic reactions, then the individuals would have had a previous exposure, perhaps from injectable medications, Dr. Edwards said.
It might be feasible to perform a skin test for allergy to PEG. “If the skin testing is negative, that doesn’t completely rule out allergy, but it can be used in the decision-making about giving the first or second vaccine dose,” Dr. Banerji said.
Other vaccines, such as childhood vaccines, contain polysorbate as a stabilizer, which has a similar chemical structure, and it’s not clear why someone would react to PEG but not to polysorbate, Dr. Edwards said.
Meanwhile, other illnesses and even deaths sometimes occur in the days after vaccination, but that doesn’t mean the vaccine caused them, cautioned Steve Black, MD, emeritus professor of pediatrics at Cincinnati Children’s Hospital and cofounder of the Global Vaccine Data Network, an international vaccine safety collaboration.
“Different events and clusters of events will occur by chance alone, as these events can occur without vaccines. We need to not immediately assume that they’re due to the vaccine,” he said. “You don’t want to undermine the whole vaccine program every time something comes up and assume that it’s associated with the vaccine.”
The CDC only has three contraindications for the vaccines:
- Severe allergic reaction (such as anaphylaxis) after a previous dose of an mRNA COVID-19 vaccine or any of its components.
- Immediate allergic reaction of any severity to a previous dose of an mRNA COVID-19 vaccine or any of its components (including PEG).
- Immediate allergic reaction of any severity to polysorbate (due to potential cross-reactive hypersensitivity with PEG).
People who have had an immediate allergic reaction to other vaccines or injectable therapies should consider consulting with an allergist or immunologist before getting the Pfizer or Moderna vaccines, the CDC advises.
The CDC also says that people with a history of anaphylaxis from any cause should be observed for 30 minutes after vaccination. Vaccination protocol calls for everyone else to wait on site for 15 minutes after vaccination.
A version of this article first appeared on Medscape.com.
On Jan. 13, 2 days after a drive-through vaccination “superstation” opened in San Diego, six people were treated for anaphylaxis after they received the Moderna vaccine, leading the California state epidemiologist to recommend pausing the administration of that particular lot.
A group of allergy and immunology experts and public health officials reviewed the cases, as well as an incident that occurred the day before, and concluded that at least some of the responses were angioedema, or swelling — a serious allergic reaction — but none were actually anaphylaxis. No similar clusters had occurred with the same vaccine lot in other states, and California resumed using the doses.
Yet questions remain about the reactions and the mechanisms for them. Some might have been triggered by an allergy to a vaccine component, most likely the polyethylene glycol (PEG) that stabilizes the lipid surrounding the mRNA, the key vaccine component in both the Moderna and Pfizer vaccines. Another possible explanation is that some could be pseudoallergic reactions to a blood protein known as complement, a little-understood process that resembles an antigen-based reaction but doesn’t leave an immune memory and might not recur.
Cases of complement-activation-related pseudoallergy look like a severe allergic reaction but occur through a different mechanism and don’t require previous exposure to an allergen.
“It has the same signs and symptoms and is treated the same way, but it occurs through a different pathway,” explained Neal Halsey, MD, director emeritus of the Institute for Vaccine Safety and emeritus professor at the Johns Hopkins Bloomberg School of Public Health in Baltimore.
Pseudoallergies are not well understood, but they have been associated with reactions to the contrast media used in imaging, such as with MRI. “If people have had an anaphylaxis-type reaction following the injection of contrast-dye material, that is a strong signal that it might be a complement-activation-related pseudoallergy,” said Dr. Halsey, a member of the Clinical Immunization Safety Assessment Network. “Those are the people who definitely need to consider seeing an allergist before getting the COVID vaccines.”
When Aleena Banerji, MD, clinical director of the allergy and clinical immunology unit at Massachusetts General Hospital in Boston, talks to patients about vaccine reactions, she addresses the risk for COVID-19 infection. All of the people who developed allergies after the Pfizer and Moderna vaccines recovered, but more than 445,000 Americans have died from COVID-19.
Most people with common allergies, such as to food or oral medications, don’t need to worry about reactions, said Dr. Banerji, lead author of a review that assessed the risk for allergic reactions to the Pfizer and Moderna vaccines.
Investigating reactions
As investigators search for the answers to what causes reactions, transparency is crucial to trust, said Kathryn Edwards, MD, principal investigator of the Clinical Immunization Safety Assessment Project, a vaccine safety network funded by the Centers for Disease Control and Prevention.
“Unless the public knows that we’re really investigating and we’re taking this seriously, then I think the vaccine hesitancy is going to increase,” said Dr. Edwards, professor of pediatrics at Vanderbilt University Medical Center and scientific director of the Vanderbilt Vaccine Research Program in Nashville, Tenn.
First reports of anaphylaxis came quickly after COVID-19 vaccinations began. In the 2 weeks before the holidays, almost 2 million health care workers received the Pfizer vaccine, and 21 of them developed anaphylaxis, according to CDC researchers who reviewed case reports from the Vaccine Adverse Event Reporting System (VAERS). That rate of about 1 in 100,000 is 10 times higher than the occurrence with other vaccines. No deaths from anaphylaxis were reported.
As the vaccinations ramped up, the rate declined. As of Jan. 18, 50 cases of anaphylaxis were reported to VAERS after the administration of 9,943,247 Pfizer doses, for a rate of 5.0 per million, according to data presented at the Jan. 27 meeting of the CDC Advisory Committee on Immunization Practices. And 21 cases of anaphylaxis were reported to VAERS after the administration of 7,581,429 Moderna doses, for a rate of 2.8 per million.
The anaphylaxis occurred almost exclusively in women; only three of the VAERS anaphylaxis reports were from men. Only 24% had a history of anaphylaxis.
The earlier CDC report explored the potential link to allergies. One person with anaphylaxis had a history of allergy to iodinated contrast media, and others had allergies to various medications, vaccines, foods, and animals. The researchers reported 86 nonanaphylaxis allergic reactions and 61 nonallergic adverse events among the 175 case reports they reviewed as possible cases of severe allergic reaction.
Of 1,266 reports that VAERS received from Dec. 21 to Jan. 10, the CDC identified 108 possible cases of severe allergic reaction after the Moderna vaccine. Only 10 met the case definition of anaphylaxis put forward by the Brighton Collaboration, a vaccine safety organization. All but one case involved a history of allergies or allergic reactions; only five had a previously experienced anaphylaxis.
There were 47 nonanaphylaxis allergic reactions.
The San Diego cluster also met the Brighton case definition for anaphylaxis, Dr. Edwards reported. This discrepancy highlights the difficulties in characterizing vaccine reactions.
Measuring a pseudoallergic reaction is a challenge. It requires that a blood sample be drawn soon after the incident and then frozen to protect heat-sensitive blood markers, Dr. Edwards explained.
And as vaccinations rise, so do adverse-event reports. But unlike in clinical trials, there is no control group for comparison. That is why vaccine safety experts urge caution when evaluating events and, where possible, advise looking at background rates.
“A major way to determine whether the adverse event is causally related is to assess the incidence of the adverse event in vaccines versus nonvaccines,” said Walter Orenstein, MD, who directed the U.S. Immunization Program from 1988 to 2004 and is now associate director of the Emory Vaccine Center and professor of infectious diseases at Emory University in Atlanta. Public health officials could then identify vaccine risk factors, he said.
When a reaction occurs almost immediately after vaccination, vaccine safety investigators look for probable triggers. If allergy to PEG is the culprit in anaphylactic reactions, then the individuals would have had a previous exposure, perhaps from injectable medications, Dr. Edwards said.
It might be feasible to perform a skin test for allergy to PEG. “If the skin testing is negative, that doesn’t completely rule out allergy, but it can be used in the decision-making about giving the first or second vaccine dose,” Dr. Banerji said.
Other vaccines, such as childhood vaccines, contain polysorbate as a stabilizer, which has a similar chemical structure, and it’s not clear why someone would react to PEG but not to polysorbate, Dr. Edwards said.
Meanwhile, other illnesses and even deaths sometimes occur in the days after vaccination, but that doesn’t mean the vaccine caused them, cautioned Steve Black, MD, emeritus professor of pediatrics at Cincinnati Children’s Hospital and cofounder of the Global Vaccine Data Network, an international vaccine safety collaboration.
“Different events and clusters of events will occur by chance alone, as these events can occur without vaccines. We need to not immediately assume that they’re due to the vaccine,” he said. “You don’t want to undermine the whole vaccine program every time something comes up and assume that it’s associated with the vaccine.”
The CDC only has three contraindications for the vaccines:
- Severe allergic reaction (such as anaphylaxis) after a previous dose of an mRNA COVID-19 vaccine or any of its components.
- Immediate allergic reaction of any severity to a previous dose of an mRNA COVID-19 vaccine or any of its components (including PEG).
- Immediate allergic reaction of any severity to polysorbate (due to potential cross-reactive hypersensitivity with PEG).
People who have had an immediate allergic reaction to other vaccines or injectable therapies should consider consulting with an allergist or immunologist before getting the Pfizer or Moderna vaccines, the CDC advises.
The CDC also says that people with a history of anaphylaxis from any cause should be observed for 30 minutes after vaccination. Vaccination protocol calls for everyone else to wait on site for 15 minutes after vaccination.
A version of this article first appeared on Medscape.com.