User login
An MD’s nightmare began with reporting her manic episode to the medical board
Susan Haney, MD, a board-certified emergency physician in Coos Bay, Ore., was 2 years into her career when she had her first manic episode, likely a side effect of the steroid prednisone, which she had been prescribed for an asthma flare-up. Her boss at Bay Area Hospital told her that if she wanted to return to work, she would need to have written clearance from the medical board.
In retrospect, Dr. Haney says, “I don’t think they had any idea of what they would set in motion.”
Dr. Haney says the Oregon Medical Board posted her name and the nondisciplinary action on their website and in their newsletter. Her local newspaper read it and ran a story about her. “They effectively announced my mental illness to the general public despite my objections,” she says.
During the next decade, she had two more manic episodes, and more board investigations and actions followed. Despite being cleared for work each time, Dr. Haney says the board actions decimated her career in emergency medicine and her income, which is about half of what she would have earned by now. She is frustrated, sad, and angry about what happened but considers herself lucky to be practicing medicine in urgent care.
Being investigated is scary
After her first manic episode in 2006, Dr. Haney contacted the board’s medical director, a retired general surgeon, who told her the only way the board would authorize her return to work was if she agreed to open a board investigation.
She gave them the green light because she thought she had nothing to fear – she was cooperating fully and wasn’t impaired. Now Dr. Haney says she was naive. “The board is not your friend,” she says.
Dr. Haney was also anxious to return to work. She worked in a seven-person emergency department, and two colleagues were on maternity leave or medical leave.
“My colleagues kept calling asking me when I was going to return to work, and I kept saying, ‘I don’t know because the board won’t tell me,’ “ she says.
She was also feeling a lot of financial pressure. She was 2 years out of residency, owed $100,000 in student loans, and had just bought a house.
“I was really scared – I didn’t know how long this would last or if they would let me return to work. Early on, I even got a fitness for duty evaluation from the state’s consulting psychiatrist, who cleared me for work, and the board still wouldn’t let me return. They told me I had to go through their bureaucracy and a board meeting, which didn’t make sense to me.”
Dr. Haney consented to give the board’s investigative staff access to her medical records because she feared that if she challenged them, they would suspend or revoke her license immediately.
After investigating her for 4 months, the board cleared Dr. Haney to return to work at Bay Area Hospital. She agreed to the board’s “corrective action” terms: She would continue to receive psychiatric care, maintain a physician-patient relationship with a primary care physician, and enroll in the Health Physicians Program (HPP) for substance abuse monitoring.
Dr. Haney suspects that the board investigation damaged her reputation at work. “Before this, my work evaluations were consistently excellent. Afterwards, they were all adequate. I don’t think that was a coincidence.”
Worst time of her life
Five years later, after taking prednisone for another asthma flare-up, Dr. Haney had a more severe manic episode and was hospitalized.
The consulting psychiatrist who evaluated her reported her case to the medical board, stating she had bipolar disorder, was mentally incompetent, and shouldn’t be practicing medicine. The board opened a second investigation of her in 2012, which lasted 4 months.
Dr. Haney had quit her job at Bay Area Hospital in 2011 because she was pregnant and was planning to take a year off to care for the baby at home.
“That was the worst time of my life. I lost the baby at 4 months, I wasn’t working, and now I was under investigation by the board again,” she says.
The board issued an “interim stipulated order” that required that she be monitored regularly for mental illness and substance abuse by the Health Professionals Services Program (HPSP) for 2 years. “The board accused me of abusing prednisone, which I wasn’t. I was using it as prescribed and medically indicated,” she said.
The board order was reported to the National Practitioner Databank and is now permanently in her record. Although the board cleared her to work, she could not find a permanent job in a hospital emergency department.
“The repeated ‘nondisciplinary’ public board orders have had the same net impact on my career as if I had been disciplined for killing or harming my patients. For all intents and purposes, people treat it as a disciplinary action for the rest of your career,” she said.
To keep afloat financially, she found locum tenens work in local emergency departments until 2019.
Mental health toll
Dr. Haney feels that the stress of repeated board investigations has affected her mental health. “Both times this happened, it made my mental health worse, made the mania worse, and subsequent depression worse.”
Particularly distressing to her was the fact that the administrative staff who investigated her were attorneys and persons in law enforcement, rather than medical professionals with mental health training.
“I was required to disclose intimate personal details of my psychological and psychiatric history to anybody at the board who requested them. These investigators were asking me about my childhood history. That was traumatic and none of their business!”
Dr. Haney had quietly managed episodes of major depression since she was in her early 20s with the help of a psychiatrist. Her third episode of mania, which occurred in 2014, triggered a more severe depression, which she says deepened when she learned that the HPSP had notified the board about her manic symptoms and that she would not be released from the 2-year monitoring contract. When the board notified her 2 weeks later that they were opening another investigation, Dr. Haney says she had an emotional crisis, attempted suicide, and was briefly hospitalized. Several weeks later, she decided to take a mood stabilizer, which she continues to take.
The board’s 2015 corrective action agreement required Dr. Haney to practice medicine only in settings that the board’s medical director preapproved and to obtain a preapproved monitoring health care provider who would send quarterly reports to the medical director. Dr. Haney says the “nondisciplinary” action agreement was also reported to the National Practitioner Data Bank.
She also agreed to ongoing monitoring by the HPSP for mental illness and substance abuse, which involved random drug testing. When she didn’t call in one day in 2019 and missed a scheduled test, the board opened another investigation on her that lasted 7 months until July 2020. Dr. Haney said this was despite three subsequent negative tests.
Dr. Haney believes that the “open investigation” doomed a job offer from a hospital emergency department in the Virgin Islands. “I had passed all the required credentialing and explained previous board orders. They pulled the rug from under me 1 week before I was supposed to move there,” says Dr. Haney.
Her license was inactivated again because she hadn’t practiced medicine for a year, which she says was a new board policy. Although Dr. Haney says the medical director reactivated her license after talking with her, “By the time I was able to apply emergency medicine jobs, no one was interested in me anymore.”
Financial toll
Dr. Haney started her medical career when she was 42 as a second career. She says the board investigations and actions have resulted in a significant loss of work and income. “I have only worked 14 of the past 17 years as a doctor. I live cheaply because I never know how much longer my career will last,” says Dr. Haney.
The ordeal has devastated her finances. She has shelled out at least $200,000 in legal fees – she hired an attorney in 2007 and filed a lawsuit against the board in Oregon district court alleging that members had violated several of her rights. The district judge sided with the state medical board, and it was upheld on appeal in 2012, referring to state laws that gave the board absolute immunity from civil lawsuits. “I had no legal recourse to contest their decisions, no matter how injurious or unjust,” says Dr. Haney.
She has also shelled out at least $100,000 to be evaluated and monitored by the health physician program (now HPSP) for several years. Physicians who agree to be monitored by these health programs have to pay their fees. The board finally agreed last July to end her HPSP participation.
Dr. Haney also filed a complaint in 2007 with the federal Department of Health & Human Services Office for Civil Rights, alleging that the board violated her civil rights under the Americans with Disabilities Act. She says that her lawsuit and the OCR investigation of the board enabled her to withdraw from the HPP in good standing in 2008..
What would she have done differently?
She regrets not hiring an attorney earlier because “most likely the board action would not have been made public. It snowballed after that -- any mistake I made in my career was viewed in the lens of potential impairment.”
She also regrets telling her employer about the nature of her illness and reporting it to the board. A psychiatrist she saw later shared advice he gives to other patients who want to remain anonymous: get help but go out of town, use a false name, and pay cash.
“I wish I had that advice when all this started. That was the best way to protect my career,” says Dr. Haney.
Protecting the public?
The Oregon Medical Board declined to comment on Dr. Haney’s experience because investigations are confidential, but the executive director, Nicole Krishnaswami, JD, answered questions in an email about how the current board operates.
She says the board has 11 medical professionals and employs a medical director and expert consultants in specialty-specific fields. MDs with mental health training are involved in investigating/reviewing cases involving doctors with mental illnesses.
“State medical boards have a responsibility to protect and inform the public. State laws further require state agencies to provide access and transparency regarding the board’s official actions. If the board receives a complaint that a licensee is impaired and thus unable to safely practice, the board has a responsibility to investigate and ensure the licensee is practicing medicine safely,” Ms. Krishnaswami said.
The HPSP is the monitoring program established by state law to provide oversight in order to ensure that licensees are not practicing while impaired. HPSP is separate from the board and the board adopted a statement outlining its perspective on the program in support of doctors with substance abuse and mental health disorder.
The board also founded the Oregon Wellness Program, which provides free, confidential counseling to all Oregon-licensed physicians and physician assistants.
Stigma continues
Dr. Haney feels there is huge stigma associated with mental illness in the medical profession. “If I had cancer twice, I wouldn’t have been put in this position and would be at the peak of my career,” she says.
Nearly half of the 862 emergency medicine physicians surveyed last October said they were reluctant to seek mental health treatment. The reasons included fear of professional repercussions and stigma in the workplace. Several physicians said they were concerned about potentially having to report the treatment on medical license applications in the future, according to a survey by the American College of Emergency Physicians.
In addition, 26% of the more than 12,000 physicians who responded to a Medscape survey last year said they didn’t want to risk disclosure (20%) or that they distrusted mental health professionals (6%).
Another physician fights back
Steven Miles, MD, an award-winning professor emeritus of medicine and bioethics at the Center for Bioethics at the University of Minnesota, in Minneapolis, understands their reluctance. In 1996, he disclosed on his license renewal application that he had recently been diagnosed with a mainly depressive type of bipolar disorder and was in treatment. He had already told his employer, who was supportive.
That set off a 14-month investigation of him by the Minnesota Board of Medical Practice. Dr. Miles and his psychiatrist refused to release his confidential records to a panel of physicians, most of whom had no expertise in mental health care. He also filed a federal claim that the board’s requests violated the ADA, and he won the case.
“Had the board given me evidence of impaired ability to practice with ordinary skill and safety, I would have cooperated. Instead, they proposed a course of action, which would have degraded the privacy of my relationship with my psychiatrist and arguably increased the barrier to getting proper care and the risk of impairment,” he said.
The board kept renewing his license, and Dr. Miles continued to work full time. “I was empowered and protected by my stature in the field at the time my mental illness was diagnosed. Early-career physicians do not yet have that protection and should be very careful of disclosing, given the still widespread stigma of mental illnesses,” he said.
His advocacy led to changes
Dr. Miles went public to mobilize support for his ADA claim. He wrote editorials that were published in JAMA and Minnesota Medicine that refer to the American Psychiatric Association’s 1984 position paper, which says that the mandatory disclosure of the physician’s confidential medical record is without merit. Dr. Miles adds that major newspapers ran stories based on his editorials.
The board backed down after Dr. Miles won his ADA case, and it met with him. “I said this is not good stewardship of the medical profession; you are injuring doctors by keeping them from psychiatric care, which is out of line with the medical view of the treatability of depression and that needs to change,” he says.
Dr. Miles says he won a victory because his practice continued. “I also won a victory in the way the board was handling these questions, which was an opening salvo in a process that continues to this day.”
The original form asked whether he had ever been diagnosed with or treated for manic depression, schizophrenia, compulsive gambling, or other psychiatric conditions.
The revised form asks, “Do you have a physical or mental condition that would affect your ability, with or without reasonable accommodation, to provide appropriate care to patients and otherwise perform the essential functions of a practitioner in your area of practice without posing a health or safety risk to your patients? If yes, what accommodations would help you provide appropriate care to patients and perform other essential functions?”
Dr. Miles says that the final wording wasn’t ideal and that it was confusing to physicians. He says this prompted additional changes in wording by the board. Starting in January, applicants will be asked, “Do you currently have any condition that is not being appropriately treated that is likely to impair or adversely affect your ability to practice medicine with reasonable skill and safety in a competent, ethical, and professional manner?” the medical board’s executive director, Ruth M. Martinez, said in an email.
When asked whether the board still investigates physicians who reveal mental illnesses on licensing applications, Ms. Martinez responded, “All disclosures are evaluated to assure that the practitioner is qualified and safe to practice.”
This article was updated 11/4/21.
A version of this article first appeared on Medscape.com.
Susan Haney, MD, a board-certified emergency physician in Coos Bay, Ore., was 2 years into her career when she had her first manic episode, likely a side effect of the steroid prednisone, which she had been prescribed for an asthma flare-up. Her boss at Bay Area Hospital told her that if she wanted to return to work, she would need to have written clearance from the medical board.
In retrospect, Dr. Haney says, “I don’t think they had any idea of what they would set in motion.”
Dr. Haney says the Oregon Medical Board posted her name and the nondisciplinary action on their website and in their newsletter. Her local newspaper read it and ran a story about her. “They effectively announced my mental illness to the general public despite my objections,” she says.
During the next decade, she had two more manic episodes, and more board investigations and actions followed. Despite being cleared for work each time, Dr. Haney says the board actions decimated her career in emergency medicine and her income, which is about half of what she would have earned by now. She is frustrated, sad, and angry about what happened but considers herself lucky to be practicing medicine in urgent care.
Being investigated is scary
After her first manic episode in 2006, Dr. Haney contacted the board’s medical director, a retired general surgeon, who told her the only way the board would authorize her return to work was if she agreed to open a board investigation.
She gave them the green light because she thought she had nothing to fear – she was cooperating fully and wasn’t impaired. Now Dr. Haney says she was naive. “The board is not your friend,” she says.
Dr. Haney was also anxious to return to work. She worked in a seven-person emergency department, and two colleagues were on maternity leave or medical leave.
“My colleagues kept calling asking me when I was going to return to work, and I kept saying, ‘I don’t know because the board won’t tell me,’ “ she says.
She was also feeling a lot of financial pressure. She was 2 years out of residency, owed $100,000 in student loans, and had just bought a house.
“I was really scared – I didn’t know how long this would last or if they would let me return to work. Early on, I even got a fitness for duty evaluation from the state’s consulting psychiatrist, who cleared me for work, and the board still wouldn’t let me return. They told me I had to go through their bureaucracy and a board meeting, which didn’t make sense to me.”
Dr. Haney consented to give the board’s investigative staff access to her medical records because she feared that if she challenged them, they would suspend or revoke her license immediately.
After investigating her for 4 months, the board cleared Dr. Haney to return to work at Bay Area Hospital. She agreed to the board’s “corrective action” terms: She would continue to receive psychiatric care, maintain a physician-patient relationship with a primary care physician, and enroll in the Health Physicians Program (HPP) for substance abuse monitoring.
Dr. Haney suspects that the board investigation damaged her reputation at work. “Before this, my work evaluations were consistently excellent. Afterwards, they were all adequate. I don’t think that was a coincidence.”
Worst time of her life
Five years later, after taking prednisone for another asthma flare-up, Dr. Haney had a more severe manic episode and was hospitalized.
The consulting psychiatrist who evaluated her reported her case to the medical board, stating she had bipolar disorder, was mentally incompetent, and shouldn’t be practicing medicine. The board opened a second investigation of her in 2012, which lasted 4 months.
Dr. Haney had quit her job at Bay Area Hospital in 2011 because she was pregnant and was planning to take a year off to care for the baby at home.
“That was the worst time of my life. I lost the baby at 4 months, I wasn’t working, and now I was under investigation by the board again,” she says.
The board issued an “interim stipulated order” that required that she be monitored regularly for mental illness and substance abuse by the Health Professionals Services Program (HPSP) for 2 years. “The board accused me of abusing prednisone, which I wasn’t. I was using it as prescribed and medically indicated,” she said.
The board order was reported to the National Practitioner Databank and is now permanently in her record. Although the board cleared her to work, she could not find a permanent job in a hospital emergency department.
“The repeated ‘nondisciplinary’ public board orders have had the same net impact on my career as if I had been disciplined for killing or harming my patients. For all intents and purposes, people treat it as a disciplinary action for the rest of your career,” she said.
To keep afloat financially, she found locum tenens work in local emergency departments until 2019.
Mental health toll
Dr. Haney feels that the stress of repeated board investigations has affected her mental health. “Both times this happened, it made my mental health worse, made the mania worse, and subsequent depression worse.”
Particularly distressing to her was the fact that the administrative staff who investigated her were attorneys and persons in law enforcement, rather than medical professionals with mental health training.
“I was required to disclose intimate personal details of my psychological and psychiatric history to anybody at the board who requested them. These investigators were asking me about my childhood history. That was traumatic and none of their business!”
Dr. Haney had quietly managed episodes of major depression since she was in her early 20s with the help of a psychiatrist. Her third episode of mania, which occurred in 2014, triggered a more severe depression, which she says deepened when she learned that the HPSP had notified the board about her manic symptoms and that she would not be released from the 2-year monitoring contract. When the board notified her 2 weeks later that they were opening another investigation, Dr. Haney says she had an emotional crisis, attempted suicide, and was briefly hospitalized. Several weeks later, she decided to take a mood stabilizer, which she continues to take.
The board’s 2015 corrective action agreement required Dr. Haney to practice medicine only in settings that the board’s medical director preapproved and to obtain a preapproved monitoring health care provider who would send quarterly reports to the medical director. Dr. Haney says the “nondisciplinary” action agreement was also reported to the National Practitioner Data Bank.
She also agreed to ongoing monitoring by the HPSP for mental illness and substance abuse, which involved random drug testing. When she didn’t call in one day in 2019 and missed a scheduled test, the board opened another investigation on her that lasted 7 months until July 2020. Dr. Haney said this was despite three subsequent negative tests.
Dr. Haney believes that the “open investigation” doomed a job offer from a hospital emergency department in the Virgin Islands. “I had passed all the required credentialing and explained previous board orders. They pulled the rug from under me 1 week before I was supposed to move there,” says Dr. Haney.
Her license was inactivated again because she hadn’t practiced medicine for a year, which she says was a new board policy. Although Dr. Haney says the medical director reactivated her license after talking with her, “By the time I was able to apply emergency medicine jobs, no one was interested in me anymore.”
Financial toll
Dr. Haney started her medical career when she was 42 as a second career. She says the board investigations and actions have resulted in a significant loss of work and income. “I have only worked 14 of the past 17 years as a doctor. I live cheaply because I never know how much longer my career will last,” says Dr. Haney.
The ordeal has devastated her finances. She has shelled out at least $200,000 in legal fees – she hired an attorney in 2007 and filed a lawsuit against the board in Oregon district court alleging that members had violated several of her rights. The district judge sided with the state medical board, and it was upheld on appeal in 2012, referring to state laws that gave the board absolute immunity from civil lawsuits. “I had no legal recourse to contest their decisions, no matter how injurious or unjust,” says Dr. Haney.
She has also shelled out at least $100,000 to be evaluated and monitored by the health physician program (now HPSP) for several years. Physicians who agree to be monitored by these health programs have to pay their fees. The board finally agreed last July to end her HPSP participation.
Dr. Haney also filed a complaint in 2007 with the federal Department of Health & Human Services Office for Civil Rights, alleging that the board violated her civil rights under the Americans with Disabilities Act. She says that her lawsuit and the OCR investigation of the board enabled her to withdraw from the HPP in good standing in 2008..
What would she have done differently?
She regrets not hiring an attorney earlier because “most likely the board action would not have been made public. It snowballed after that -- any mistake I made in my career was viewed in the lens of potential impairment.”
She also regrets telling her employer about the nature of her illness and reporting it to the board. A psychiatrist she saw later shared advice he gives to other patients who want to remain anonymous: get help but go out of town, use a false name, and pay cash.
“I wish I had that advice when all this started. That was the best way to protect my career,” says Dr. Haney.
Protecting the public?
The Oregon Medical Board declined to comment on Dr. Haney’s experience because investigations are confidential, but the executive director, Nicole Krishnaswami, JD, answered questions in an email about how the current board operates.
She says the board has 11 medical professionals and employs a medical director and expert consultants in specialty-specific fields. MDs with mental health training are involved in investigating/reviewing cases involving doctors with mental illnesses.
“State medical boards have a responsibility to protect and inform the public. State laws further require state agencies to provide access and transparency regarding the board’s official actions. If the board receives a complaint that a licensee is impaired and thus unable to safely practice, the board has a responsibility to investigate and ensure the licensee is practicing medicine safely,” Ms. Krishnaswami said.
The HPSP is the monitoring program established by state law to provide oversight in order to ensure that licensees are not practicing while impaired. HPSP is separate from the board and the board adopted a statement outlining its perspective on the program in support of doctors with substance abuse and mental health disorder.
The board also founded the Oregon Wellness Program, which provides free, confidential counseling to all Oregon-licensed physicians and physician assistants.
Stigma continues
Dr. Haney feels there is huge stigma associated with mental illness in the medical profession. “If I had cancer twice, I wouldn’t have been put in this position and would be at the peak of my career,” she says.
Nearly half of the 862 emergency medicine physicians surveyed last October said they were reluctant to seek mental health treatment. The reasons included fear of professional repercussions and stigma in the workplace. Several physicians said they were concerned about potentially having to report the treatment on medical license applications in the future, according to a survey by the American College of Emergency Physicians.
In addition, 26% of the more than 12,000 physicians who responded to a Medscape survey last year said they didn’t want to risk disclosure (20%) or that they distrusted mental health professionals (6%).
Another physician fights back
Steven Miles, MD, an award-winning professor emeritus of medicine and bioethics at the Center for Bioethics at the University of Minnesota, in Minneapolis, understands their reluctance. In 1996, he disclosed on his license renewal application that he had recently been diagnosed with a mainly depressive type of bipolar disorder and was in treatment. He had already told his employer, who was supportive.
That set off a 14-month investigation of him by the Minnesota Board of Medical Practice. Dr. Miles and his psychiatrist refused to release his confidential records to a panel of physicians, most of whom had no expertise in mental health care. He also filed a federal claim that the board’s requests violated the ADA, and he won the case.
“Had the board given me evidence of impaired ability to practice with ordinary skill and safety, I would have cooperated. Instead, they proposed a course of action, which would have degraded the privacy of my relationship with my psychiatrist and arguably increased the barrier to getting proper care and the risk of impairment,” he said.
The board kept renewing his license, and Dr. Miles continued to work full time. “I was empowered and protected by my stature in the field at the time my mental illness was diagnosed. Early-career physicians do not yet have that protection and should be very careful of disclosing, given the still widespread stigma of mental illnesses,” he said.
His advocacy led to changes
Dr. Miles went public to mobilize support for his ADA claim. He wrote editorials that were published in JAMA and Minnesota Medicine that refer to the American Psychiatric Association’s 1984 position paper, which says that the mandatory disclosure of the physician’s confidential medical record is without merit. Dr. Miles adds that major newspapers ran stories based on his editorials.
The board backed down after Dr. Miles won his ADA case, and it met with him. “I said this is not good stewardship of the medical profession; you are injuring doctors by keeping them from psychiatric care, which is out of line with the medical view of the treatability of depression and that needs to change,” he says.
Dr. Miles says he won a victory because his practice continued. “I also won a victory in the way the board was handling these questions, which was an opening salvo in a process that continues to this day.”
The original form asked whether he had ever been diagnosed with or treated for manic depression, schizophrenia, compulsive gambling, or other psychiatric conditions.
The revised form asks, “Do you have a physical or mental condition that would affect your ability, with or without reasonable accommodation, to provide appropriate care to patients and otherwise perform the essential functions of a practitioner in your area of practice without posing a health or safety risk to your patients? If yes, what accommodations would help you provide appropriate care to patients and perform other essential functions?”
Dr. Miles says that the final wording wasn’t ideal and that it was confusing to physicians. He says this prompted additional changes in wording by the board. Starting in January, applicants will be asked, “Do you currently have any condition that is not being appropriately treated that is likely to impair or adversely affect your ability to practice medicine with reasonable skill and safety in a competent, ethical, and professional manner?” the medical board’s executive director, Ruth M. Martinez, said in an email.
When asked whether the board still investigates physicians who reveal mental illnesses on licensing applications, Ms. Martinez responded, “All disclosures are evaluated to assure that the practitioner is qualified and safe to practice.”
This article was updated 11/4/21.
A version of this article first appeared on Medscape.com.
Susan Haney, MD, a board-certified emergency physician in Coos Bay, Ore., was 2 years into her career when she had her first manic episode, likely a side effect of the steroid prednisone, which she had been prescribed for an asthma flare-up. Her boss at Bay Area Hospital told her that if she wanted to return to work, she would need to have written clearance from the medical board.
In retrospect, Dr. Haney says, “I don’t think they had any idea of what they would set in motion.”
Dr. Haney says the Oregon Medical Board posted her name and the nondisciplinary action on their website and in their newsletter. Her local newspaper read it and ran a story about her. “They effectively announced my mental illness to the general public despite my objections,” she says.
During the next decade, she had two more manic episodes, and more board investigations and actions followed. Despite being cleared for work each time, Dr. Haney says the board actions decimated her career in emergency medicine and her income, which is about half of what she would have earned by now. She is frustrated, sad, and angry about what happened but considers herself lucky to be practicing medicine in urgent care.
Being investigated is scary
After her first manic episode in 2006, Dr. Haney contacted the board’s medical director, a retired general surgeon, who told her the only way the board would authorize her return to work was if she agreed to open a board investigation.
She gave them the green light because she thought she had nothing to fear – she was cooperating fully and wasn’t impaired. Now Dr. Haney says she was naive. “The board is not your friend,” she says.
Dr. Haney was also anxious to return to work. She worked in a seven-person emergency department, and two colleagues were on maternity leave or medical leave.
“My colleagues kept calling asking me when I was going to return to work, and I kept saying, ‘I don’t know because the board won’t tell me,’ “ she says.
She was also feeling a lot of financial pressure. She was 2 years out of residency, owed $100,000 in student loans, and had just bought a house.
“I was really scared – I didn’t know how long this would last or if they would let me return to work. Early on, I even got a fitness for duty evaluation from the state’s consulting psychiatrist, who cleared me for work, and the board still wouldn’t let me return. They told me I had to go through their bureaucracy and a board meeting, which didn’t make sense to me.”
Dr. Haney consented to give the board’s investigative staff access to her medical records because she feared that if she challenged them, they would suspend or revoke her license immediately.
After investigating her for 4 months, the board cleared Dr. Haney to return to work at Bay Area Hospital. She agreed to the board’s “corrective action” terms: She would continue to receive psychiatric care, maintain a physician-patient relationship with a primary care physician, and enroll in the Health Physicians Program (HPP) for substance abuse monitoring.
Dr. Haney suspects that the board investigation damaged her reputation at work. “Before this, my work evaluations were consistently excellent. Afterwards, they were all adequate. I don’t think that was a coincidence.”
Worst time of her life
Five years later, after taking prednisone for another asthma flare-up, Dr. Haney had a more severe manic episode and was hospitalized.
The consulting psychiatrist who evaluated her reported her case to the medical board, stating she had bipolar disorder, was mentally incompetent, and shouldn’t be practicing medicine. The board opened a second investigation of her in 2012, which lasted 4 months.
Dr. Haney had quit her job at Bay Area Hospital in 2011 because she was pregnant and was planning to take a year off to care for the baby at home.
“That was the worst time of my life. I lost the baby at 4 months, I wasn’t working, and now I was under investigation by the board again,” she says.
The board issued an “interim stipulated order” that required that she be monitored regularly for mental illness and substance abuse by the Health Professionals Services Program (HPSP) for 2 years. “The board accused me of abusing prednisone, which I wasn’t. I was using it as prescribed and medically indicated,” she said.
The board order was reported to the National Practitioner Databank and is now permanently in her record. Although the board cleared her to work, she could not find a permanent job in a hospital emergency department.
“The repeated ‘nondisciplinary’ public board orders have had the same net impact on my career as if I had been disciplined for killing or harming my patients. For all intents and purposes, people treat it as a disciplinary action for the rest of your career,” she said.
To keep afloat financially, she found locum tenens work in local emergency departments until 2019.
Mental health toll
Dr. Haney feels that the stress of repeated board investigations has affected her mental health. “Both times this happened, it made my mental health worse, made the mania worse, and subsequent depression worse.”
Particularly distressing to her was the fact that the administrative staff who investigated her were attorneys and persons in law enforcement, rather than medical professionals with mental health training.
“I was required to disclose intimate personal details of my psychological and psychiatric history to anybody at the board who requested them. These investigators were asking me about my childhood history. That was traumatic and none of their business!”
Dr. Haney had quietly managed episodes of major depression since she was in her early 20s with the help of a psychiatrist. Her third episode of mania, which occurred in 2014, triggered a more severe depression, which she says deepened when she learned that the HPSP had notified the board about her manic symptoms and that she would not be released from the 2-year monitoring contract. When the board notified her 2 weeks later that they were opening another investigation, Dr. Haney says she had an emotional crisis, attempted suicide, and was briefly hospitalized. Several weeks later, she decided to take a mood stabilizer, which she continues to take.
The board’s 2015 corrective action agreement required Dr. Haney to practice medicine only in settings that the board’s medical director preapproved and to obtain a preapproved monitoring health care provider who would send quarterly reports to the medical director. Dr. Haney says the “nondisciplinary” action agreement was also reported to the National Practitioner Data Bank.
She also agreed to ongoing monitoring by the HPSP for mental illness and substance abuse, which involved random drug testing. When she didn’t call in one day in 2019 and missed a scheduled test, the board opened another investigation on her that lasted 7 months until July 2020. Dr. Haney said this was despite three subsequent negative tests.
Dr. Haney believes that the “open investigation” doomed a job offer from a hospital emergency department in the Virgin Islands. “I had passed all the required credentialing and explained previous board orders. They pulled the rug from under me 1 week before I was supposed to move there,” says Dr. Haney.
Her license was inactivated again because she hadn’t practiced medicine for a year, which she says was a new board policy. Although Dr. Haney says the medical director reactivated her license after talking with her, “By the time I was able to apply emergency medicine jobs, no one was interested in me anymore.”
Financial toll
Dr. Haney started her medical career when she was 42 as a second career. She says the board investigations and actions have resulted in a significant loss of work and income. “I have only worked 14 of the past 17 years as a doctor. I live cheaply because I never know how much longer my career will last,” says Dr. Haney.
The ordeal has devastated her finances. She has shelled out at least $200,000 in legal fees – she hired an attorney in 2007 and filed a lawsuit against the board in Oregon district court alleging that members had violated several of her rights. The district judge sided with the state medical board, and it was upheld on appeal in 2012, referring to state laws that gave the board absolute immunity from civil lawsuits. “I had no legal recourse to contest their decisions, no matter how injurious or unjust,” says Dr. Haney.
She has also shelled out at least $100,000 to be evaluated and monitored by the health physician program (now HPSP) for several years. Physicians who agree to be monitored by these health programs have to pay their fees. The board finally agreed last July to end her HPSP participation.
Dr. Haney also filed a complaint in 2007 with the federal Department of Health & Human Services Office for Civil Rights, alleging that the board violated her civil rights under the Americans with Disabilities Act. She says that her lawsuit and the OCR investigation of the board enabled her to withdraw from the HPP in good standing in 2008..
What would she have done differently?
She regrets not hiring an attorney earlier because “most likely the board action would not have been made public. It snowballed after that -- any mistake I made in my career was viewed in the lens of potential impairment.”
She also regrets telling her employer about the nature of her illness and reporting it to the board. A psychiatrist she saw later shared advice he gives to other patients who want to remain anonymous: get help but go out of town, use a false name, and pay cash.
“I wish I had that advice when all this started. That was the best way to protect my career,” says Dr. Haney.
Protecting the public?
The Oregon Medical Board declined to comment on Dr. Haney’s experience because investigations are confidential, but the executive director, Nicole Krishnaswami, JD, answered questions in an email about how the current board operates.
She says the board has 11 medical professionals and employs a medical director and expert consultants in specialty-specific fields. MDs with mental health training are involved in investigating/reviewing cases involving doctors with mental illnesses.
“State medical boards have a responsibility to protect and inform the public. State laws further require state agencies to provide access and transparency regarding the board’s official actions. If the board receives a complaint that a licensee is impaired and thus unable to safely practice, the board has a responsibility to investigate and ensure the licensee is practicing medicine safely,” Ms. Krishnaswami said.
The HPSP is the monitoring program established by state law to provide oversight in order to ensure that licensees are not practicing while impaired. HPSP is separate from the board and the board adopted a statement outlining its perspective on the program in support of doctors with substance abuse and mental health disorder.
The board also founded the Oregon Wellness Program, which provides free, confidential counseling to all Oregon-licensed physicians and physician assistants.
Stigma continues
Dr. Haney feels there is huge stigma associated with mental illness in the medical profession. “If I had cancer twice, I wouldn’t have been put in this position and would be at the peak of my career,” she says.
Nearly half of the 862 emergency medicine physicians surveyed last October said they were reluctant to seek mental health treatment. The reasons included fear of professional repercussions and stigma in the workplace. Several physicians said they were concerned about potentially having to report the treatment on medical license applications in the future, according to a survey by the American College of Emergency Physicians.
In addition, 26% of the more than 12,000 physicians who responded to a Medscape survey last year said they didn’t want to risk disclosure (20%) or that they distrusted mental health professionals (6%).
Another physician fights back
Steven Miles, MD, an award-winning professor emeritus of medicine and bioethics at the Center for Bioethics at the University of Minnesota, in Minneapolis, understands their reluctance. In 1996, he disclosed on his license renewal application that he had recently been diagnosed with a mainly depressive type of bipolar disorder and was in treatment. He had already told his employer, who was supportive.
That set off a 14-month investigation of him by the Minnesota Board of Medical Practice. Dr. Miles and his psychiatrist refused to release his confidential records to a panel of physicians, most of whom had no expertise in mental health care. He also filed a federal claim that the board’s requests violated the ADA, and he won the case.
“Had the board given me evidence of impaired ability to practice with ordinary skill and safety, I would have cooperated. Instead, they proposed a course of action, which would have degraded the privacy of my relationship with my psychiatrist and arguably increased the barrier to getting proper care and the risk of impairment,” he said.
The board kept renewing his license, and Dr. Miles continued to work full time. “I was empowered and protected by my stature in the field at the time my mental illness was diagnosed. Early-career physicians do not yet have that protection and should be very careful of disclosing, given the still widespread stigma of mental illnesses,” he said.
His advocacy led to changes
Dr. Miles went public to mobilize support for his ADA claim. He wrote editorials that were published in JAMA and Minnesota Medicine that refer to the American Psychiatric Association’s 1984 position paper, which says that the mandatory disclosure of the physician’s confidential medical record is without merit. Dr. Miles adds that major newspapers ran stories based on his editorials.
The board backed down after Dr. Miles won his ADA case, and it met with him. “I said this is not good stewardship of the medical profession; you are injuring doctors by keeping them from psychiatric care, which is out of line with the medical view of the treatability of depression and that needs to change,” he says.
Dr. Miles says he won a victory because his practice continued. “I also won a victory in the way the board was handling these questions, which was an opening salvo in a process that continues to this day.”
The original form asked whether he had ever been diagnosed with or treated for manic depression, schizophrenia, compulsive gambling, or other psychiatric conditions.
The revised form asks, “Do you have a physical or mental condition that would affect your ability, with or without reasonable accommodation, to provide appropriate care to patients and otherwise perform the essential functions of a practitioner in your area of practice without posing a health or safety risk to your patients? If yes, what accommodations would help you provide appropriate care to patients and perform other essential functions?”
Dr. Miles says that the final wording wasn’t ideal and that it was confusing to physicians. He says this prompted additional changes in wording by the board. Starting in January, applicants will be asked, “Do you currently have any condition that is not being appropriately treated that is likely to impair or adversely affect your ability to practice medicine with reasonable skill and safety in a competent, ethical, and professional manner?” the medical board’s executive director, Ruth M. Martinez, said in an email.
When asked whether the board still investigates physicians who reveal mental illnesses on licensing applications, Ms. Martinez responded, “All disclosures are evaluated to assure that the practitioner is qualified and safe to practice.”
This article was updated 11/4/21.
A version of this article first appeared on Medscape.com.
3D vs 2D mammography for detecting cancer in dense breasts
Text copyright DenseBreast-info.org.
Answer
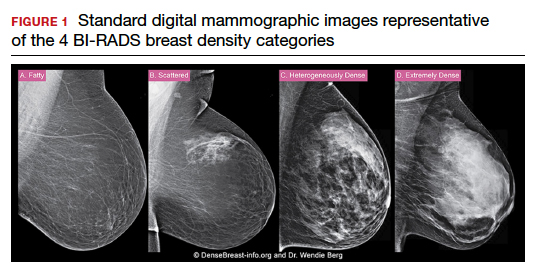
C. Overall, tomosynthesis depicts an additional 1 to 2 cancers per thousand women screened in the first round of screening when added to standard digital mammography;1-3 however, this improvement in cancer detection is only observed in women with fatty breasts (category A), scattered fibroglandular tissue (category B), and heterogeneously dense breasts (category C). Importantly, tomosynthesis does not significantly improve breast cancer detection in women with extremely dense breasts (category D).2,4
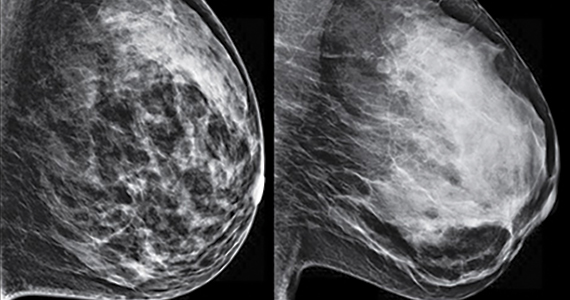
Digital breast tomosynthesis, also referred to as “3-dimensional mammography” (3D mammography) or tomosynthesis, uses a dedicated electronic detector system to obtain multiple projection images that are reconstructed by the computer to create thin slices or slabs of multiple slices of the breast. These slices can be individually “scrolled through” by the radiologist to reduce tissue overlap that may obscure breast cancers on a standard mammogram. While tomosynthesis improves breast cancer detection in women with fatty, scattered fibroglandular density, and heterogeneously dense breasts, there is very little soft tissue contrast in extremely dense breasts due to insufficient fat, and some cancers will remain hidden by dense tissue even on sliced images through the breast.
FIGURE 2 shows an example of cancer that was missed on tomosynthesis in a 51-year-old woman with extremely dense breasts and right breast pain. The cancer was masked by extremely dense tissue on standard digital mammography and tomosynthesis; no abnormalities were detected. Ultrasonography showed a 1.6-cm, irregular, hypoechoic mass at the site of pain, and biopsy revealed a grade 3 triple-receptor negative invasive ductal carcinoma.
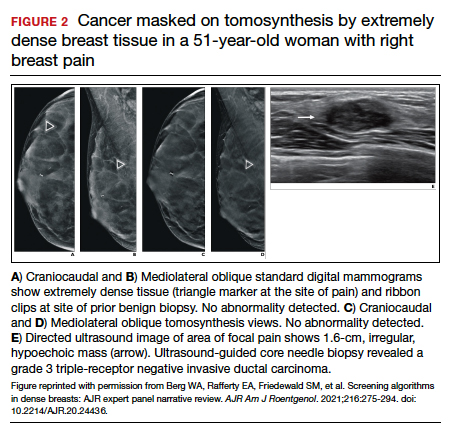
In women with dense breasts, especially extremely dense breasts, supplemental screening beyond tomosynthesis should be considered. Although tomosynthesis doesn’t improve cancer detection in extremely dense breasts, it does reduce callbacks for additional testing in all breast densities compared with standard digital mammography. Callbacks are reduced from approximately 100‒120 per 1,000 women screened with standard digital mammography alone1,5 to an average of 80 per 1,000 women when tomosynthesis and standard mammography are interpreted together.1-3 ●
For more information, visit medically sourced DenseBreast-info.org. Comprehensive resources include a free CME opportunity, Dense Breasts and Supplemental Screening.
- Conant EF, Zuckerman SP, McDonald ES, et al. Five consecutive years of screening with digital breast tomosynthesis: outcomes by screening year and round. Radiology. 2020;295:285-293.
- Rafferty EA, Durand MA, Conant EF, et al. Breast cancer screening using tomosynthesis and digital mammography in dense and nondense breasts. JAMA. 2016;315:1784-1786.
- Skaane P, Bandos AI, Niklason LT, et al. Digital mammography versus digital mammography plus tomosynthesis in breast cancer screening: the Oslo Tomosynthesis Screening Trial. Radiology. 2019;291:23-30.
- Lowry KP, Coley RY, Miglioretti DL, et al. Screening performance of digital breast tomosynthesis vs digital mammography in community practice by patient age, screening round, and breast density. JAMA Netw Open. 2020;3:e2011792.
- Lee CS, Sengupta D, Bhargavan-Chatfield M, et al. Association of patient age with outcomes of current-era, large-scale screening mammography: analysis of data from the National Mammography Database. JAMA Oncol. 2017;3:1134-1136.
Text copyright DenseBreast-info.org.
Answer
C. Overall, tomosynthesis depicts an additional 1 to 2 cancers per thousand women screened in the first round of screening when added to standard digital mammography;1-3 however, this improvement in cancer detection is only observed in women with fatty breasts (category A), scattered fibroglandular tissue (category B), and heterogeneously dense breasts (category C). Importantly, tomosynthesis does not significantly improve breast cancer detection in women with extremely dense breasts (category D).2,4
Digital breast tomosynthesis, also referred to as “3-dimensional mammography” (3D mammography) or tomosynthesis, uses a dedicated electronic detector system to obtain multiple projection images that are reconstructed by the computer to create thin slices or slabs of multiple slices of the breast. These slices can be individually “scrolled through” by the radiologist to reduce tissue overlap that may obscure breast cancers on a standard mammogram. While tomosynthesis improves breast cancer detection in women with fatty, scattered fibroglandular density, and heterogeneously dense breasts, there is very little soft tissue contrast in extremely dense breasts due to insufficient fat, and some cancers will remain hidden by dense tissue even on sliced images through the breast.
FIGURE 2 shows an example of cancer that was missed on tomosynthesis in a 51-year-old woman with extremely dense breasts and right breast pain. The cancer was masked by extremely dense tissue on standard digital mammography and tomosynthesis; no abnormalities were detected. Ultrasonography showed a 1.6-cm, irregular, hypoechoic mass at the site of pain, and biopsy revealed a grade 3 triple-receptor negative invasive ductal carcinoma.
In women with dense breasts, especially extremely dense breasts, supplemental screening beyond tomosynthesis should be considered. Although tomosynthesis doesn’t improve cancer detection in extremely dense breasts, it does reduce callbacks for additional testing in all breast densities compared with standard digital mammography. Callbacks are reduced from approximately 100‒120 per 1,000 women screened with standard digital mammography alone1,5 to an average of 80 per 1,000 women when tomosynthesis and standard mammography are interpreted together.1-3 ●
For more information, visit medically sourced DenseBreast-info.org. Comprehensive resources include a free CME opportunity, Dense Breasts and Supplemental Screening.
Text copyright DenseBreast-info.org.
Answer
C. Overall, tomosynthesis depicts an additional 1 to 2 cancers per thousand women screened in the first round of screening when added to standard digital mammography;1-3 however, this improvement in cancer detection is only observed in women with fatty breasts (category A), scattered fibroglandular tissue (category B), and heterogeneously dense breasts (category C). Importantly, tomosynthesis does not significantly improve breast cancer detection in women with extremely dense breasts (category D).2,4
Digital breast tomosynthesis, also referred to as “3-dimensional mammography” (3D mammography) or tomosynthesis, uses a dedicated electronic detector system to obtain multiple projection images that are reconstructed by the computer to create thin slices or slabs of multiple slices of the breast. These slices can be individually “scrolled through” by the radiologist to reduce tissue overlap that may obscure breast cancers on a standard mammogram. While tomosynthesis improves breast cancer detection in women with fatty, scattered fibroglandular density, and heterogeneously dense breasts, there is very little soft tissue contrast in extremely dense breasts due to insufficient fat, and some cancers will remain hidden by dense tissue even on sliced images through the breast.
FIGURE 2 shows an example of cancer that was missed on tomosynthesis in a 51-year-old woman with extremely dense breasts and right breast pain. The cancer was masked by extremely dense tissue on standard digital mammography and tomosynthesis; no abnormalities were detected. Ultrasonography showed a 1.6-cm, irregular, hypoechoic mass at the site of pain, and biopsy revealed a grade 3 triple-receptor negative invasive ductal carcinoma.
In women with dense breasts, especially extremely dense breasts, supplemental screening beyond tomosynthesis should be considered. Although tomosynthesis doesn’t improve cancer detection in extremely dense breasts, it does reduce callbacks for additional testing in all breast densities compared with standard digital mammography. Callbacks are reduced from approximately 100‒120 per 1,000 women screened with standard digital mammography alone1,5 to an average of 80 per 1,000 women when tomosynthesis and standard mammography are interpreted together.1-3 ●
For more information, visit medically sourced DenseBreast-info.org. Comprehensive resources include a free CME opportunity, Dense Breasts and Supplemental Screening.
- Conant EF, Zuckerman SP, McDonald ES, et al. Five consecutive years of screening with digital breast tomosynthesis: outcomes by screening year and round. Radiology. 2020;295:285-293.
- Rafferty EA, Durand MA, Conant EF, et al. Breast cancer screening using tomosynthesis and digital mammography in dense and nondense breasts. JAMA. 2016;315:1784-1786.
- Skaane P, Bandos AI, Niklason LT, et al. Digital mammography versus digital mammography plus tomosynthesis in breast cancer screening: the Oslo Tomosynthesis Screening Trial. Radiology. 2019;291:23-30.
- Lowry KP, Coley RY, Miglioretti DL, et al. Screening performance of digital breast tomosynthesis vs digital mammography in community practice by patient age, screening round, and breast density. JAMA Netw Open. 2020;3:e2011792.
- Lee CS, Sengupta D, Bhargavan-Chatfield M, et al. Association of patient age with outcomes of current-era, large-scale screening mammography: analysis of data from the National Mammography Database. JAMA Oncol. 2017;3:1134-1136.
- Conant EF, Zuckerman SP, McDonald ES, et al. Five consecutive years of screening with digital breast tomosynthesis: outcomes by screening year and round. Radiology. 2020;295:285-293.
- Rafferty EA, Durand MA, Conant EF, et al. Breast cancer screening using tomosynthesis and digital mammography in dense and nondense breasts. JAMA. 2016;315:1784-1786.
- Skaane P, Bandos AI, Niklason LT, et al. Digital mammography versus digital mammography plus tomosynthesis in breast cancer screening: the Oslo Tomosynthesis Screening Trial. Radiology. 2019;291:23-30.
- Lowry KP, Coley RY, Miglioretti DL, et al. Screening performance of digital breast tomosynthesis vs digital mammography in community practice by patient age, screening round, and breast density. JAMA Netw Open. 2020;3:e2011792.
- Lee CS, Sengupta D, Bhargavan-Chatfield M, et al. Association of patient age with outcomes of current-era, large-scale screening mammography: analysis of data from the National Mammography Database. JAMA Oncol. 2017;3:1134-1136.
Quiz developed in collaboration with ![]()
AGA Clinical Practice Guideline: Coagulation in cirrhosis
A clinical update from the American Gastroenterological Association focuses on bleeding and thrombosis-related questions in patients with cirrhosis. It provides guidance on test strategies for bleeding risk, preprocedure management of bleeding risk, venous thromboembolism (VTE) prophylaxis, screening for portal vein thrombosis (PVT), and anticoagulation therapies. It is aimed at primary care providers, gastroenterologists, and hepatologists, among other health care providers.
In cirrhosis, there are often changes to platelet (PLT) counts and prothrombin time/international normalized ratio (PT/INR), among other parameters, and historically these changes led to concerns that patients were at greater risk of bleeding or thrombosis. More recent evidence has led to a nuanced view. Neither factor necessarily suggests increased bleeding risk, and the severity of coagulopathy predicted by them does not predict the risk of bleeding complications.
Patients with cirrhosis are at greater risk of thrombosis, but clinicians may be hesitant to prescribe anticoagulants because of uncertain risk profiles, and test strategies employing PT/INR to estimate bleeding risk and track treatment endpoints in patients receiving vitamin K antagonists may not work in cirrhosis patients with alterations in procoagulant and anticoagulant measures. Recent efforts to address this led to testing of fibrin clot formation and lysis to better gauge the variety of abnormalities in cirrhosis patients.
The guideline, published in Gastroenterology, was informed by a technical review that focused on both bleeding-related and thrombosis-related questions. Bleeding-related questions included testing strategies and preprocedure prophylaxis to reduce bleeding risk. Thrombosis-related questions included whether VTE prophylaxis may be useful in hospitalized patients with cirrhosis, whether patients should be screened for PVT, potential therapies for nontumoral PVT, and whether or not anticoagulation is safe and effective when atrial fibrillation is present alongside cirrhosis.
Because of a lack of evidence, the guideline provides no recommendations on visco-elastic testing for bleeding risk in advance of common gastrointestinal procedures for patients with stable cirrhosis. It recommends against use of extensive preprocedural testing, such as repeated PT/INR or PLT count testing.
The guideline also looked at whether preprocedural efforts to correct coagulation parameters could reduce bleeding risk in patients with cirrhosis. It recommends against giving blood products ahead of the procedure for patients with stable cirrhosis without severe thrombocytopenia or severe coagulopathy. Such interventions can be considered for patients in the latter categories who are undergoing procedures with high bleeding risk after consideration of risks and benefits, and consultation with a hematologist.
Thrombopoietin receptor agonists (TPO-RAs) are also not recommended in patients with thrombocytopenia and stable cirrhosis undergoing common procedures, but they can be considered for patients who are more concerned about reduction of bleeding events and less concerned about the risk of PVT.
Patients who are hospitalized and meet the requirements should receive VTE prophylaxis. Although there is little available evidence about the effects of thromboprophylaxis in patients with cirrhosis, there is strong evidence of benefit in acutely ill hospitalized patients, and patients with cirrhosis are believed to be at a similar risk of VTE. There is evidence of increased bleed risk, but this is of very low certainty.
PVT should not be routinely tested for, but such testing can be offered to patients with a high level of concern over PVT and are not as worried about potential harms of treatment. This recommendation does not apply to patients waiting for a liver transplant.
Patients with non-umoral PVT should receive anticoagulation therapy, but patients who have high levels of concern about bleeding risk from anticoagulation and put a lower value on possible benefits of anticoagulation may choose not to receive it.
The guideline recommends anticoagulation for patients with atrial fibrillation and cirrhosis who are indicated for it. Patients with more concern about the bleeding risk of anticoagulation and place lower value on the reduction in stroke risk may choose to not receive anticoagulation. This is particularly true for those with more advanced cirrhosis (Child-Turcotte-Pugh Class C) and/or low CHA2DS2-VASC scores.
Nearly all of the recommendations in the guideline are conditional, reflecting a lack of data and a range of knowledge gaps that need filling. The authors call for additional research to identify specific patients who are at high risk for bleeding or thrombosis “to appropriately provide prophylaxis using blood product transfusion or TPO-RAs in patients at risk for clinically significant bleeding, to screen for and treat PVT, and to prevent clinically significant thromboembolic events.”
The development of the guideline was funded fully by the AGA. Members of the panel submitted conflict of interest information, and these statements are maintained at AGA headquarters.
A clinical update from the American Gastroenterological Association focuses on bleeding and thrombosis-related questions in patients with cirrhosis. It provides guidance on test strategies for bleeding risk, preprocedure management of bleeding risk, venous thromboembolism (VTE) prophylaxis, screening for portal vein thrombosis (PVT), and anticoagulation therapies. It is aimed at primary care providers, gastroenterologists, and hepatologists, among other health care providers.
In cirrhosis, there are often changes to platelet (PLT) counts and prothrombin time/international normalized ratio (PT/INR), among other parameters, and historically these changes led to concerns that patients were at greater risk of bleeding or thrombosis. More recent evidence has led to a nuanced view. Neither factor necessarily suggests increased bleeding risk, and the severity of coagulopathy predicted by them does not predict the risk of bleeding complications.
Patients with cirrhosis are at greater risk of thrombosis, but clinicians may be hesitant to prescribe anticoagulants because of uncertain risk profiles, and test strategies employing PT/INR to estimate bleeding risk and track treatment endpoints in patients receiving vitamin K antagonists may not work in cirrhosis patients with alterations in procoagulant and anticoagulant measures. Recent efforts to address this led to testing of fibrin clot formation and lysis to better gauge the variety of abnormalities in cirrhosis patients.
The guideline, published in Gastroenterology, was informed by a technical review that focused on both bleeding-related and thrombosis-related questions. Bleeding-related questions included testing strategies and preprocedure prophylaxis to reduce bleeding risk. Thrombosis-related questions included whether VTE prophylaxis may be useful in hospitalized patients with cirrhosis, whether patients should be screened for PVT, potential therapies for nontumoral PVT, and whether or not anticoagulation is safe and effective when atrial fibrillation is present alongside cirrhosis.
Because of a lack of evidence, the guideline provides no recommendations on visco-elastic testing for bleeding risk in advance of common gastrointestinal procedures for patients with stable cirrhosis. It recommends against use of extensive preprocedural testing, such as repeated PT/INR or PLT count testing.
The guideline also looked at whether preprocedural efforts to correct coagulation parameters could reduce bleeding risk in patients with cirrhosis. It recommends against giving blood products ahead of the procedure for patients with stable cirrhosis without severe thrombocytopenia or severe coagulopathy. Such interventions can be considered for patients in the latter categories who are undergoing procedures with high bleeding risk after consideration of risks and benefits, and consultation with a hematologist.
Thrombopoietin receptor agonists (TPO-RAs) are also not recommended in patients with thrombocytopenia and stable cirrhosis undergoing common procedures, but they can be considered for patients who are more concerned about reduction of bleeding events and less concerned about the risk of PVT.
Patients who are hospitalized and meet the requirements should receive VTE prophylaxis. Although there is little available evidence about the effects of thromboprophylaxis in patients with cirrhosis, there is strong evidence of benefit in acutely ill hospitalized patients, and patients with cirrhosis are believed to be at a similar risk of VTE. There is evidence of increased bleed risk, but this is of very low certainty.
PVT should not be routinely tested for, but such testing can be offered to patients with a high level of concern over PVT and are not as worried about potential harms of treatment. This recommendation does not apply to patients waiting for a liver transplant.
Patients with non-umoral PVT should receive anticoagulation therapy, but patients who have high levels of concern about bleeding risk from anticoagulation and put a lower value on possible benefits of anticoagulation may choose not to receive it.
The guideline recommends anticoagulation for patients with atrial fibrillation and cirrhosis who are indicated for it. Patients with more concern about the bleeding risk of anticoagulation and place lower value on the reduction in stroke risk may choose to not receive anticoagulation. This is particularly true for those with more advanced cirrhosis (Child-Turcotte-Pugh Class C) and/or low CHA2DS2-VASC scores.
Nearly all of the recommendations in the guideline are conditional, reflecting a lack of data and a range of knowledge gaps that need filling. The authors call for additional research to identify specific patients who are at high risk for bleeding or thrombosis “to appropriately provide prophylaxis using blood product transfusion or TPO-RAs in patients at risk for clinically significant bleeding, to screen for and treat PVT, and to prevent clinically significant thromboembolic events.”
The development of the guideline was funded fully by the AGA. Members of the panel submitted conflict of interest information, and these statements are maintained at AGA headquarters.
A clinical update from the American Gastroenterological Association focuses on bleeding and thrombosis-related questions in patients with cirrhosis. It provides guidance on test strategies for bleeding risk, preprocedure management of bleeding risk, venous thromboembolism (VTE) prophylaxis, screening for portal vein thrombosis (PVT), and anticoagulation therapies. It is aimed at primary care providers, gastroenterologists, and hepatologists, among other health care providers.
In cirrhosis, there are often changes to platelet (PLT) counts and prothrombin time/international normalized ratio (PT/INR), among other parameters, and historically these changes led to concerns that patients were at greater risk of bleeding or thrombosis. More recent evidence has led to a nuanced view. Neither factor necessarily suggests increased bleeding risk, and the severity of coagulopathy predicted by them does not predict the risk of bleeding complications.
Patients with cirrhosis are at greater risk of thrombosis, but clinicians may be hesitant to prescribe anticoagulants because of uncertain risk profiles, and test strategies employing PT/INR to estimate bleeding risk and track treatment endpoints in patients receiving vitamin K antagonists may not work in cirrhosis patients with alterations in procoagulant and anticoagulant measures. Recent efforts to address this led to testing of fibrin clot formation and lysis to better gauge the variety of abnormalities in cirrhosis patients.
The guideline, published in Gastroenterology, was informed by a technical review that focused on both bleeding-related and thrombosis-related questions. Bleeding-related questions included testing strategies and preprocedure prophylaxis to reduce bleeding risk. Thrombosis-related questions included whether VTE prophylaxis may be useful in hospitalized patients with cirrhosis, whether patients should be screened for PVT, potential therapies for nontumoral PVT, and whether or not anticoagulation is safe and effective when atrial fibrillation is present alongside cirrhosis.
Because of a lack of evidence, the guideline provides no recommendations on visco-elastic testing for bleeding risk in advance of common gastrointestinal procedures for patients with stable cirrhosis. It recommends against use of extensive preprocedural testing, such as repeated PT/INR or PLT count testing.
The guideline also looked at whether preprocedural efforts to correct coagulation parameters could reduce bleeding risk in patients with cirrhosis. It recommends against giving blood products ahead of the procedure for patients with stable cirrhosis without severe thrombocytopenia or severe coagulopathy. Such interventions can be considered for patients in the latter categories who are undergoing procedures with high bleeding risk after consideration of risks and benefits, and consultation with a hematologist.
Thrombopoietin receptor agonists (TPO-RAs) are also not recommended in patients with thrombocytopenia and stable cirrhosis undergoing common procedures, but they can be considered for patients who are more concerned about reduction of bleeding events and less concerned about the risk of PVT.
Patients who are hospitalized and meet the requirements should receive VTE prophylaxis. Although there is little available evidence about the effects of thromboprophylaxis in patients with cirrhosis, there is strong evidence of benefit in acutely ill hospitalized patients, and patients with cirrhosis are believed to be at a similar risk of VTE. There is evidence of increased bleed risk, but this is of very low certainty.
PVT should not be routinely tested for, but such testing can be offered to patients with a high level of concern over PVT and are not as worried about potential harms of treatment. This recommendation does not apply to patients waiting for a liver transplant.
Patients with non-umoral PVT should receive anticoagulation therapy, but patients who have high levels of concern about bleeding risk from anticoagulation and put a lower value on possible benefits of anticoagulation may choose not to receive it.
The guideline recommends anticoagulation for patients with atrial fibrillation and cirrhosis who are indicated for it. Patients with more concern about the bleeding risk of anticoagulation and place lower value on the reduction in stroke risk may choose to not receive anticoagulation. This is particularly true for those with more advanced cirrhosis (Child-Turcotte-Pugh Class C) and/or low CHA2DS2-VASC scores.
Nearly all of the recommendations in the guideline are conditional, reflecting a lack of data and a range of knowledge gaps that need filling. The authors call for additional research to identify specific patients who are at high risk for bleeding or thrombosis “to appropriately provide prophylaxis using blood product transfusion or TPO-RAs in patients at risk for clinically significant bleeding, to screen for and treat PVT, and to prevent clinically significant thromboembolic events.”
The development of the guideline was funded fully by the AGA. Members of the panel submitted conflict of interest information, and these statements are maintained at AGA headquarters.
FROM GASTROENTEROLOGY
Renal denervation remains only promising, per latest meta-analysis
Questions remain despite efficacy
According to the latest meta-analysis of sham-controlled randomized trials, catheter-based renal sympathetic denervation produces clinically meaningful reductions in blood pressure with acceptable safety, but the strategy is not yet regarded as ready for prime time, according to a summary of the results to be presented at the Transcatheter Cardiovascular Therapeutics annual meeting.
This meta-analysis was based on seven blinded trials, all of which associated denervation with a reduction in systolic ambulatory BP, according to Yousif Ahmad, BMBS, PhD, an interventional cardiologist at Yale University, New Haven, Conn.
Although the BP-lowering advantage in two of these studies did not reach statistical significance, the other five did, and all the data moved in the same direction.
For ambulatory diastolic pressure, the effect was more modest. One of the studies showed essentially a neutral effect. The reductions were statistically significant in only two, but, again, the data moved in the same direction in six of the studies, and a random-effects analysis suggested that the reductions, although modest, were potentially meaningful, according to Dr. Ahmad.
Overall, at a mean follow-up of 4.5 months, the reductions in ambulatory systolic and diastolic BPs were 3.61 and 1.85 mm Hg, respectively. The benefit was about the same whether renal denervation was or was not performed on the background of antihypertensive drugs, which was permitted in five of the seven trials. In the other two, all patients were off hypertensive medication.
Office-based systolic reduction: 6 mm Hg
When the same analysis was performed for office-based BP reductions, which were available for five of the seven trials, the overall reductions based on the meta-analysis were 5.86 and 3.63 mm Hg for the systolic and diastolic pressures, respectively. Again, background antihypertensive therapy was not a factor.
Of the seven trials, three randomized fewer than 100 patients. The largest, SYMPLICITY HTN-3, randomized 491 patients in 2:1 ratio to denervation or sham.
Three of the studies in the meta-analysis were trials of the Symplicity flex device. Another two evaluated the Symplicity Spyral catheter. Both deliver radiofrequency energy to for denervation. The Paradise device, the focus of the remaining two trials, employs energy in the form of ultrasound.
According to Dr. Ahmad, adverse events regardless of device were rare and not more common among those in the active treatment arm than in those treated with a sham procedure. Although one of these trials, RADIANCE-HTN SOLO associated denervation with efficacy and safety out to 12 months , Dr. Ahmad concluded that the mean follow-up of 4.5 months is not sufficient to consider long-term effects.
More than 20 meta-analyses published so far
By one count, there have been more than 20 meta-analyses of renal denervation published previously yet this intervention is still considered “controversial,” according to Dr. Ahmad. Relative to the previous meta-analyses, this included the RADIANCE-HTN TRIO trial, which is the latest such sham-controlled study and added 136 patients to the dataset of high-quality trials.
Basically, the results led Dr. Ahmad to conclude that, although the treatment effect is modest, it could be valuable in specific groups of patients, such as those reluctant or unable to take multiple medications or any medications at all. In addition to generating more data on efficacy and safety, he said longer follow-up is also needed for calculations of cost-effectiveness. Larger-scale observational studies might be one way of collecting these data, he reported.
The results of this study were published online in JACC Cardiovascular Interventions with an accompanying editorial by David E. Kandzari, MD, director of interventional cardiology, Piedmont Hart Institute, Atlanta.
Commenting on the large pile of meta-analyses, sometimes published months apart, Dr. Kandzari explained that their “short half-life” is a product of the continuous updating of data with new trials. For a procedure that remains controversial, he said these constant relooks are inevitable.
“My point is that, with more studies, we can expect to see more meta-analyses. It is just the way this is going to work,” Dr. Kandzari said in an interview.
Individual study data also relevant
Even as the authors of these analyses attempt to cull the best data from the most rigorously performed trials, “we are also going to have to look at the individual studies, because of the differences in the trial designs, particularly the devices used,” according to Dr. Kandzari, who was the principle investigator of the sham-controlled SPYRAL HTN-ON MED trial.
So far, the data, despite some inconsistencies, have supported “clinically meaningful” BP reductions and acceptable safety regardless of the device used, according to Dr. Kandzari. Although he also agrees with the basic premise that more long-term data are needed to better determine how renal denervation should be applied in management of hypertension, he does think it will eventually find a role that is “complimentary to, rather than a replacement for, drugs.”
“The effect is modest, but keep in mind that the effect size is similar to that of a single oral medication, and there are some features, such as an always-on 24-hour effect that could be useful,” he said.
“We have enough of a signal to start thinking of how this will be enveloped into routine care,” he said.

But it is not ready yet. This was the point made by Dr. Ahmad, and it was seconded by Dr. Kandzari. One of the senior authors of the meta-analysis, Deepak Bhatt, MD, executive director of interventional cardiovascular programs, Brigham and Women’s Health, Boston, was also asked to weigh on when it will be ready for prime time.
“At a minimum, I would recommend completion of ongoing sham-controlled randomized trials before considering clinical use of renal denervation. Longer term safety and durability data, as well as data on cost-effectiveness, are all still needed – preferably from randomized trials as opposed to registries,” he said.
“Ideally, larger sham-controlled trials with longer follow-up and clinical endpoints, as opposed to only blood pressure measurements, would be performed, although I am not aware of any plans at present,” he added.
Dr. Ahmad reported no financial relationships relevant to this research. Dr. Bhatt has financial relationships with more than 30 pharmaceutical companies, including those developing products relevant to hypertension and renal denervation. Dr. Kandzari reported financial relationships with Ablative Solutions and Medtronic.
Questions remain despite efficacy
Questions remain despite efficacy
According to the latest meta-analysis of sham-controlled randomized trials, catheter-based renal sympathetic denervation produces clinically meaningful reductions in blood pressure with acceptable safety, but the strategy is not yet regarded as ready for prime time, according to a summary of the results to be presented at the Transcatheter Cardiovascular Therapeutics annual meeting.
This meta-analysis was based on seven blinded trials, all of which associated denervation with a reduction in systolic ambulatory BP, according to Yousif Ahmad, BMBS, PhD, an interventional cardiologist at Yale University, New Haven, Conn.
Although the BP-lowering advantage in two of these studies did not reach statistical significance, the other five did, and all the data moved in the same direction.
For ambulatory diastolic pressure, the effect was more modest. One of the studies showed essentially a neutral effect. The reductions were statistically significant in only two, but, again, the data moved in the same direction in six of the studies, and a random-effects analysis suggested that the reductions, although modest, were potentially meaningful, according to Dr. Ahmad.
Overall, at a mean follow-up of 4.5 months, the reductions in ambulatory systolic and diastolic BPs were 3.61 and 1.85 mm Hg, respectively. The benefit was about the same whether renal denervation was or was not performed on the background of antihypertensive drugs, which was permitted in five of the seven trials. In the other two, all patients were off hypertensive medication.
Office-based systolic reduction: 6 mm Hg
When the same analysis was performed for office-based BP reductions, which were available for five of the seven trials, the overall reductions based on the meta-analysis were 5.86 and 3.63 mm Hg for the systolic and diastolic pressures, respectively. Again, background antihypertensive therapy was not a factor.
Of the seven trials, three randomized fewer than 100 patients. The largest, SYMPLICITY HTN-3, randomized 491 patients in 2:1 ratio to denervation or sham.
Three of the studies in the meta-analysis were trials of the Symplicity flex device. Another two evaluated the Symplicity Spyral catheter. Both deliver radiofrequency energy to for denervation. The Paradise device, the focus of the remaining two trials, employs energy in the form of ultrasound.
According to Dr. Ahmad, adverse events regardless of device were rare and not more common among those in the active treatment arm than in those treated with a sham procedure. Although one of these trials, RADIANCE-HTN SOLO associated denervation with efficacy and safety out to 12 months , Dr. Ahmad concluded that the mean follow-up of 4.5 months is not sufficient to consider long-term effects.
More than 20 meta-analyses published so far
By one count, there have been more than 20 meta-analyses of renal denervation published previously yet this intervention is still considered “controversial,” according to Dr. Ahmad. Relative to the previous meta-analyses, this included the RADIANCE-HTN TRIO trial, which is the latest such sham-controlled study and added 136 patients to the dataset of high-quality trials.
Basically, the results led Dr. Ahmad to conclude that, although the treatment effect is modest, it could be valuable in specific groups of patients, such as those reluctant or unable to take multiple medications or any medications at all. In addition to generating more data on efficacy and safety, he said longer follow-up is also needed for calculations of cost-effectiveness. Larger-scale observational studies might be one way of collecting these data, he reported.
The results of this study were published online in JACC Cardiovascular Interventions with an accompanying editorial by David E. Kandzari, MD, director of interventional cardiology, Piedmont Hart Institute, Atlanta.
Commenting on the large pile of meta-analyses, sometimes published months apart, Dr. Kandzari explained that their “short half-life” is a product of the continuous updating of data with new trials. For a procedure that remains controversial, he said these constant relooks are inevitable.
“My point is that, with more studies, we can expect to see more meta-analyses. It is just the way this is going to work,” Dr. Kandzari said in an interview.
Individual study data also relevant
Even as the authors of these analyses attempt to cull the best data from the most rigorously performed trials, “we are also going to have to look at the individual studies, because of the differences in the trial designs, particularly the devices used,” according to Dr. Kandzari, who was the principle investigator of the sham-controlled SPYRAL HTN-ON MED trial.
So far, the data, despite some inconsistencies, have supported “clinically meaningful” BP reductions and acceptable safety regardless of the device used, according to Dr. Kandzari. Although he also agrees with the basic premise that more long-term data are needed to better determine how renal denervation should be applied in management of hypertension, he does think it will eventually find a role that is “complimentary to, rather than a replacement for, drugs.”
“The effect is modest, but keep in mind that the effect size is similar to that of a single oral medication, and there are some features, such as an always-on 24-hour effect that could be useful,” he said.
“We have enough of a signal to start thinking of how this will be enveloped into routine care,” he said.
But it is not ready yet. This was the point made by Dr. Ahmad, and it was seconded by Dr. Kandzari. One of the senior authors of the meta-analysis, Deepak Bhatt, MD, executive director of interventional cardiovascular programs, Brigham and Women’s Health, Boston, was also asked to weigh on when it will be ready for prime time.
“At a minimum, I would recommend completion of ongoing sham-controlled randomized trials before considering clinical use of renal denervation. Longer term safety and durability data, as well as data on cost-effectiveness, are all still needed – preferably from randomized trials as opposed to registries,” he said.
“Ideally, larger sham-controlled trials with longer follow-up and clinical endpoints, as opposed to only blood pressure measurements, would be performed, although I am not aware of any plans at present,” he added.
Dr. Ahmad reported no financial relationships relevant to this research. Dr. Bhatt has financial relationships with more than 30 pharmaceutical companies, including those developing products relevant to hypertension and renal denervation. Dr. Kandzari reported financial relationships with Ablative Solutions and Medtronic.
According to the latest meta-analysis of sham-controlled randomized trials, catheter-based renal sympathetic denervation produces clinically meaningful reductions in blood pressure with acceptable safety, but the strategy is not yet regarded as ready for prime time, according to a summary of the results to be presented at the Transcatheter Cardiovascular Therapeutics annual meeting.
This meta-analysis was based on seven blinded trials, all of which associated denervation with a reduction in systolic ambulatory BP, according to Yousif Ahmad, BMBS, PhD, an interventional cardiologist at Yale University, New Haven, Conn.
Although the BP-lowering advantage in two of these studies did not reach statistical significance, the other five did, and all the data moved in the same direction.
For ambulatory diastolic pressure, the effect was more modest. One of the studies showed essentially a neutral effect. The reductions were statistically significant in only two, but, again, the data moved in the same direction in six of the studies, and a random-effects analysis suggested that the reductions, although modest, were potentially meaningful, according to Dr. Ahmad.
Overall, at a mean follow-up of 4.5 months, the reductions in ambulatory systolic and diastolic BPs were 3.61 and 1.85 mm Hg, respectively. The benefit was about the same whether renal denervation was or was not performed on the background of antihypertensive drugs, which was permitted in five of the seven trials. In the other two, all patients were off hypertensive medication.
Office-based systolic reduction: 6 mm Hg
When the same analysis was performed for office-based BP reductions, which were available for five of the seven trials, the overall reductions based on the meta-analysis were 5.86 and 3.63 mm Hg for the systolic and diastolic pressures, respectively. Again, background antihypertensive therapy was not a factor.
Of the seven trials, three randomized fewer than 100 patients. The largest, SYMPLICITY HTN-3, randomized 491 patients in 2:1 ratio to denervation or sham.
Three of the studies in the meta-analysis were trials of the Symplicity flex device. Another two evaluated the Symplicity Spyral catheter. Both deliver radiofrequency energy to for denervation. The Paradise device, the focus of the remaining two trials, employs energy in the form of ultrasound.
According to Dr. Ahmad, adverse events regardless of device were rare and not more common among those in the active treatment arm than in those treated with a sham procedure. Although one of these trials, RADIANCE-HTN SOLO associated denervation with efficacy and safety out to 12 months , Dr. Ahmad concluded that the mean follow-up of 4.5 months is not sufficient to consider long-term effects.
More than 20 meta-analyses published so far
By one count, there have been more than 20 meta-analyses of renal denervation published previously yet this intervention is still considered “controversial,” according to Dr. Ahmad. Relative to the previous meta-analyses, this included the RADIANCE-HTN TRIO trial, which is the latest such sham-controlled study and added 136 patients to the dataset of high-quality trials.
Basically, the results led Dr. Ahmad to conclude that, although the treatment effect is modest, it could be valuable in specific groups of patients, such as those reluctant or unable to take multiple medications or any medications at all. In addition to generating more data on efficacy and safety, he said longer follow-up is also needed for calculations of cost-effectiveness. Larger-scale observational studies might be one way of collecting these data, he reported.
The results of this study were published online in JACC Cardiovascular Interventions with an accompanying editorial by David E. Kandzari, MD, director of interventional cardiology, Piedmont Hart Institute, Atlanta.
Commenting on the large pile of meta-analyses, sometimes published months apart, Dr. Kandzari explained that their “short half-life” is a product of the continuous updating of data with new trials. For a procedure that remains controversial, he said these constant relooks are inevitable.
“My point is that, with more studies, we can expect to see more meta-analyses. It is just the way this is going to work,” Dr. Kandzari said in an interview.
Individual study data also relevant
Even as the authors of these analyses attempt to cull the best data from the most rigorously performed trials, “we are also going to have to look at the individual studies, because of the differences in the trial designs, particularly the devices used,” according to Dr. Kandzari, who was the principle investigator of the sham-controlled SPYRAL HTN-ON MED trial.
So far, the data, despite some inconsistencies, have supported “clinically meaningful” BP reductions and acceptable safety regardless of the device used, according to Dr. Kandzari. Although he also agrees with the basic premise that more long-term data are needed to better determine how renal denervation should be applied in management of hypertension, he does think it will eventually find a role that is “complimentary to, rather than a replacement for, drugs.”
“The effect is modest, but keep in mind that the effect size is similar to that of a single oral medication, and there are some features, such as an always-on 24-hour effect that could be useful,” he said.
“We have enough of a signal to start thinking of how this will be enveloped into routine care,” he said.
But it is not ready yet. This was the point made by Dr. Ahmad, and it was seconded by Dr. Kandzari. One of the senior authors of the meta-analysis, Deepak Bhatt, MD, executive director of interventional cardiovascular programs, Brigham and Women’s Health, Boston, was also asked to weigh on when it will be ready for prime time.
“At a minimum, I would recommend completion of ongoing sham-controlled randomized trials before considering clinical use of renal denervation. Longer term safety and durability data, as well as data on cost-effectiveness, are all still needed – preferably from randomized trials as opposed to registries,” he said.
“Ideally, larger sham-controlled trials with longer follow-up and clinical endpoints, as opposed to only blood pressure measurements, would be performed, although I am not aware of any plans at present,” he added.
Dr. Ahmad reported no financial relationships relevant to this research. Dr. Bhatt has financial relationships with more than 30 pharmaceutical companies, including those developing products relevant to hypertension and renal denervation. Dr. Kandzari reported financial relationships with Ablative Solutions and Medtronic.
FROM TCT 2021
James Bond taken down by an epidemiologist
No, Mr. Bond, I expect you to die
Movie watching usually requires a certain suspension of disbelief, and it’s safe to say James Bond movies require this more than most. Between the impossible gadgets and ludicrous doomsday plans, very few have ever stopped to consider the health risks of the James Bond universe.

Now, however, Bond, James Bond, has met his most formidable opponent: Wouter Graumans, a graduate student in epidemiology from the Netherlands. During a foray to Burkina Faso to study infectious diseases, Mr. Graumans came down with a case of food poisoning, which led him to wonder how 007 is able to trot across this big world of ours without contracting so much as a sinus infection.
Because Mr. Graumans is a man of science and conviction, mere speculation wasn’t enough. He and a group of coauthors wrote an entire paper on the health risks of the James Bond universe.
Doing so required watching over 3,000 minutes of numerous movies and analyzing Bond’s 86 total trips to 46 different countries based on current Centers for Disease Control and Prevention advice for travel to those countries. Time which, the authors state in the abstract, “could easily have been spent on more pressing societal issues or forms of relaxation that are more acceptable in academic circles.”
Naturally, Mr. Bond’s line of work entails exposure to unpleasant things, such as poison, dehydration, heatstroke, and dangerous wildlife (everything from ticks to crocodiles), though oddly enough he never succumbs to any of it. He’s also curiously immune to hangovers, despite rarely drinking anything nonalcoholic. There are also less obvious risks: For one, 007 rarely washes his hands. During one movie, he handles raw chicken to lure away a pack of crocodiles but fails to wash his hands afterward, leaving him at risk for multiple food-borne illnesses.
Of course, we must address the elephant in the bedroom: Mr. Bond’s numerous, er, encounters with women. One would imagine the biggest risk to those women would be from the various STDs that likely course through Bond’s body, but of the 27% who died shortly after … encountering … him, all involved violence, with disease playing no obvious role. Who knows, maybe he’s clean? Stranger things have happened.
The timing of this article may seem a bit suspicious. Was it a PR stunt by the studio? Rest assured, the authors addressed this, noting that they received no funding for the study, and that, “given the futility of its academic value, this is deemed entirely appropriate by all authors.” We love when a punchline writes itself.
How to see Atlanta on $688.35 a day
The world is always changing, so we have to change with it. This week, LOTME becomes a travel guide, and our first stop is the Big A, the Big Peach, Dogwood City, Empire City of the South, Wakanda.
There’s lots to do in Atlanta: Celebrate a World Series win, visit the College Football Hall of Fame or the World of Coca Cola, or take the Stranger Things/Upside Down film locations tour. Serious adventurers, however, get out of the city and go to Emory Decatur Hospital in – you guessed it – Decatur (unofficial motto: “Everything is Greater in Decatur”).
Find the emergency room and ask for Taylor Davis, who will be your personal guide. She’ll show you how to check in at the desk, sit in the waiting room for 7 hours, and then leave without seeing any medical personnel or receiving any sort of attention whatsoever. All the things she did when she went there in July for a head injury.
Ms. Davis told Fox5 Atlanta: “I didn’t get my vitals taken, nobody called my name. I wasn’t seen at all.”
But wait! There’s more! By booking your trip through LOTMEgo* and using the code “Decatur,” you’ll get the Taylor Davis special, which includes a bill/cover charge for $688.35 from the hospital. An Emory Healthcare patient financial services employee told Ms. Davis that “you get charged before you are seen. Not for being seen.”
If all this has you ready to hop in your car (really?), then check out LOTMEgo* on Twittbook and InstaTok. You’ll also find trick-or-treating tips and discounts on haunted hospital tours.
*Does not actually exist
Breaking down the hot flash
Do you ever wonder why we scramble for cold things when we’re feeling nauseous? Whether it’s the cool air that needs to hit your face in the car or a cold, damp towel on the back of your neck, scientists think it could possibly be an evolutionary mechanism at the cellular level.

Motion sickness it’s actually a battle of body temperature, according to an article from LiveScience. Capillaries in the skin dilate, allowing for more blood flow near the skin’s surface and causing core temperature to fall. Once body temperature drops, the hypothalamus, which regulates temperature, tries to do its job by raising body temperature. Thus the hot flash!
The cold compress and cool air help fight the battle by counteracting the hypothalamus, but why the drop in body temperature to begin with?
There are a few theories. Dr. Robert Glatter, an emergency physician at Lenox Hill Hospital in New York, told LiveScience that the lack of oxygen needed in body tissue to survive at lower temperatures could be making it difficult to get oxygen to the body when a person is ill, and is “more likely an adaptive response influenced by poorly understood mechanisms at the cellular level.”
Another theory is that the nausea and body temperature shift is the body’s natural response to help people vomit.
Then there’s the theory of “defensive hypothermia,” which suggests that cold sweats are a possible mechanism to conserve energy so the body can fight off an intruder, which was supported by a 2014 study and a 2016 review.
It’s another one of the body’s many survival tricks.
Teachers were right: Pupils can do the math
Teachers liked to preach that we wouldn’t have calculators with us all the time, but that wound up not being true. Our phones have calculators at the press of a button. But maybe even calculators aren’t always needed because our pupils do more math than you think.

The pupil light reflex – constrict in light and dilate in darkness – is well known, but recent work shows that pupil size is also regulated by cognitive and perceptual factors. By presenting subjects with images of various numbers of dots and measuring pupil size, the investigators were able to show “that numerical information is intrinsically related to perception,” lead author Dr. Elisa Castaldi of Florence University noted in a written statement.
The researchers found that pupils are responsible for important survival techniques. Coauthor David Burr of the University of Sydney and the University of Florence gave an evolutionary perspective: “When we look around, we spontaneously perceive the form, size, movement and colour of a scene. Equally spontaneously, we perceive the number of items before us. This ability, shared with most other animals, is an evolutionary fundamental: It reveals immediately important quantities, such as how many apples there are on the tree, or how many enemies are attacking.”
Useful information, indeed, but our pupils seem to be more interested in the quantity of beers in the refrigerator.
No, Mr. Bond, I expect you to die
Movie watching usually requires a certain suspension of disbelief, and it’s safe to say James Bond movies require this more than most. Between the impossible gadgets and ludicrous doomsday plans, very few have ever stopped to consider the health risks of the James Bond universe.
Now, however, Bond, James Bond, has met his most formidable opponent: Wouter Graumans, a graduate student in epidemiology from the Netherlands. During a foray to Burkina Faso to study infectious diseases, Mr. Graumans came down with a case of food poisoning, which led him to wonder how 007 is able to trot across this big world of ours without contracting so much as a sinus infection.
Because Mr. Graumans is a man of science and conviction, mere speculation wasn’t enough. He and a group of coauthors wrote an entire paper on the health risks of the James Bond universe.
Doing so required watching over 3,000 minutes of numerous movies and analyzing Bond’s 86 total trips to 46 different countries based on current Centers for Disease Control and Prevention advice for travel to those countries. Time which, the authors state in the abstract, “could easily have been spent on more pressing societal issues or forms of relaxation that are more acceptable in academic circles.”
Naturally, Mr. Bond’s line of work entails exposure to unpleasant things, such as poison, dehydration, heatstroke, and dangerous wildlife (everything from ticks to crocodiles), though oddly enough he never succumbs to any of it. He’s also curiously immune to hangovers, despite rarely drinking anything nonalcoholic. There are also less obvious risks: For one, 007 rarely washes his hands. During one movie, he handles raw chicken to lure away a pack of crocodiles but fails to wash his hands afterward, leaving him at risk for multiple food-borne illnesses.
Of course, we must address the elephant in the bedroom: Mr. Bond’s numerous, er, encounters with women. One would imagine the biggest risk to those women would be from the various STDs that likely course through Bond’s body, but of the 27% who died shortly after … encountering … him, all involved violence, with disease playing no obvious role. Who knows, maybe he’s clean? Stranger things have happened.
The timing of this article may seem a bit suspicious. Was it a PR stunt by the studio? Rest assured, the authors addressed this, noting that they received no funding for the study, and that, “given the futility of its academic value, this is deemed entirely appropriate by all authors.” We love when a punchline writes itself.
How to see Atlanta on $688.35 a day
The world is always changing, so we have to change with it. This week, LOTME becomes a travel guide, and our first stop is the Big A, the Big Peach, Dogwood City, Empire City of the South, Wakanda.
There’s lots to do in Atlanta: Celebrate a World Series win, visit the College Football Hall of Fame or the World of Coca Cola, or take the Stranger Things/Upside Down film locations tour. Serious adventurers, however, get out of the city and go to Emory Decatur Hospital in – you guessed it – Decatur (unofficial motto: “Everything is Greater in Decatur”).
Find the emergency room and ask for Taylor Davis, who will be your personal guide. She’ll show you how to check in at the desk, sit in the waiting room for 7 hours, and then leave without seeing any medical personnel or receiving any sort of attention whatsoever. All the things she did when she went there in July for a head injury.
Ms. Davis told Fox5 Atlanta: “I didn’t get my vitals taken, nobody called my name. I wasn’t seen at all.”
But wait! There’s more! By booking your trip through LOTMEgo* and using the code “Decatur,” you’ll get the Taylor Davis special, which includes a bill/cover charge for $688.35 from the hospital. An Emory Healthcare patient financial services employee told Ms. Davis that “you get charged before you are seen. Not for being seen.”
If all this has you ready to hop in your car (really?), then check out LOTMEgo* on Twittbook and InstaTok. You’ll also find trick-or-treating tips and discounts on haunted hospital tours.
*Does not actually exist
Breaking down the hot flash
Do you ever wonder why we scramble for cold things when we’re feeling nauseous? Whether it’s the cool air that needs to hit your face in the car or a cold, damp towel on the back of your neck, scientists think it could possibly be an evolutionary mechanism at the cellular level.
Motion sickness it’s actually a battle of body temperature, according to an article from LiveScience. Capillaries in the skin dilate, allowing for more blood flow near the skin’s surface and causing core temperature to fall. Once body temperature drops, the hypothalamus, which regulates temperature, tries to do its job by raising body temperature. Thus the hot flash!
The cold compress and cool air help fight the battle by counteracting the hypothalamus, but why the drop in body temperature to begin with?
There are a few theories. Dr. Robert Glatter, an emergency physician at Lenox Hill Hospital in New York, told LiveScience that the lack of oxygen needed in body tissue to survive at lower temperatures could be making it difficult to get oxygen to the body when a person is ill, and is “more likely an adaptive response influenced by poorly understood mechanisms at the cellular level.”
Another theory is that the nausea and body temperature shift is the body’s natural response to help people vomit.
Then there’s the theory of “defensive hypothermia,” which suggests that cold sweats are a possible mechanism to conserve energy so the body can fight off an intruder, which was supported by a 2014 study and a 2016 review.
It’s another one of the body’s many survival tricks.
Teachers were right: Pupils can do the math
Teachers liked to preach that we wouldn’t have calculators with us all the time, but that wound up not being true. Our phones have calculators at the press of a button. But maybe even calculators aren’t always needed because our pupils do more math than you think.
The pupil light reflex – constrict in light and dilate in darkness – is well known, but recent work shows that pupil size is also regulated by cognitive and perceptual factors. By presenting subjects with images of various numbers of dots and measuring pupil size, the investigators were able to show “that numerical information is intrinsically related to perception,” lead author Dr. Elisa Castaldi of Florence University noted in a written statement.
The researchers found that pupils are responsible for important survival techniques. Coauthor David Burr of the University of Sydney and the University of Florence gave an evolutionary perspective: “When we look around, we spontaneously perceive the form, size, movement and colour of a scene. Equally spontaneously, we perceive the number of items before us. This ability, shared with most other animals, is an evolutionary fundamental: It reveals immediately important quantities, such as how many apples there are on the tree, or how many enemies are attacking.”
Useful information, indeed, but our pupils seem to be more interested in the quantity of beers in the refrigerator.
No, Mr. Bond, I expect you to die
Movie watching usually requires a certain suspension of disbelief, and it’s safe to say James Bond movies require this more than most. Between the impossible gadgets and ludicrous doomsday plans, very few have ever stopped to consider the health risks of the James Bond universe.
Now, however, Bond, James Bond, has met his most formidable opponent: Wouter Graumans, a graduate student in epidemiology from the Netherlands. During a foray to Burkina Faso to study infectious diseases, Mr. Graumans came down with a case of food poisoning, which led him to wonder how 007 is able to trot across this big world of ours without contracting so much as a sinus infection.
Because Mr. Graumans is a man of science and conviction, mere speculation wasn’t enough. He and a group of coauthors wrote an entire paper on the health risks of the James Bond universe.
Doing so required watching over 3,000 minutes of numerous movies and analyzing Bond’s 86 total trips to 46 different countries based on current Centers for Disease Control and Prevention advice for travel to those countries. Time which, the authors state in the abstract, “could easily have been spent on more pressing societal issues or forms of relaxation that are more acceptable in academic circles.”
Naturally, Mr. Bond’s line of work entails exposure to unpleasant things, such as poison, dehydration, heatstroke, and dangerous wildlife (everything from ticks to crocodiles), though oddly enough he never succumbs to any of it. He’s also curiously immune to hangovers, despite rarely drinking anything nonalcoholic. There are also less obvious risks: For one, 007 rarely washes his hands. During one movie, he handles raw chicken to lure away a pack of crocodiles but fails to wash his hands afterward, leaving him at risk for multiple food-borne illnesses.
Of course, we must address the elephant in the bedroom: Mr. Bond’s numerous, er, encounters with women. One would imagine the biggest risk to those women would be from the various STDs that likely course through Bond’s body, but of the 27% who died shortly after … encountering … him, all involved violence, with disease playing no obvious role. Who knows, maybe he’s clean? Stranger things have happened.
The timing of this article may seem a bit suspicious. Was it a PR stunt by the studio? Rest assured, the authors addressed this, noting that they received no funding for the study, and that, “given the futility of its academic value, this is deemed entirely appropriate by all authors.” We love when a punchline writes itself.
How to see Atlanta on $688.35 a day
The world is always changing, so we have to change with it. This week, LOTME becomes a travel guide, and our first stop is the Big A, the Big Peach, Dogwood City, Empire City of the South, Wakanda.
There’s lots to do in Atlanta: Celebrate a World Series win, visit the College Football Hall of Fame or the World of Coca Cola, or take the Stranger Things/Upside Down film locations tour. Serious adventurers, however, get out of the city and go to Emory Decatur Hospital in – you guessed it – Decatur (unofficial motto: “Everything is Greater in Decatur”).
Find the emergency room and ask for Taylor Davis, who will be your personal guide. She’ll show you how to check in at the desk, sit in the waiting room for 7 hours, and then leave without seeing any medical personnel or receiving any sort of attention whatsoever. All the things she did when she went there in July for a head injury.
Ms. Davis told Fox5 Atlanta: “I didn’t get my vitals taken, nobody called my name. I wasn’t seen at all.”
But wait! There’s more! By booking your trip through LOTMEgo* and using the code “Decatur,” you’ll get the Taylor Davis special, which includes a bill/cover charge for $688.35 from the hospital. An Emory Healthcare patient financial services employee told Ms. Davis that “you get charged before you are seen. Not for being seen.”
If all this has you ready to hop in your car (really?), then check out LOTMEgo* on Twittbook and InstaTok. You’ll also find trick-or-treating tips and discounts on haunted hospital tours.
*Does not actually exist
Breaking down the hot flash
Do you ever wonder why we scramble for cold things when we’re feeling nauseous? Whether it’s the cool air that needs to hit your face in the car or a cold, damp towel on the back of your neck, scientists think it could possibly be an evolutionary mechanism at the cellular level.
Motion sickness it’s actually a battle of body temperature, according to an article from LiveScience. Capillaries in the skin dilate, allowing for more blood flow near the skin’s surface and causing core temperature to fall. Once body temperature drops, the hypothalamus, which regulates temperature, tries to do its job by raising body temperature. Thus the hot flash!
The cold compress and cool air help fight the battle by counteracting the hypothalamus, but why the drop in body temperature to begin with?
There are a few theories. Dr. Robert Glatter, an emergency physician at Lenox Hill Hospital in New York, told LiveScience that the lack of oxygen needed in body tissue to survive at lower temperatures could be making it difficult to get oxygen to the body when a person is ill, and is “more likely an adaptive response influenced by poorly understood mechanisms at the cellular level.”
Another theory is that the nausea and body temperature shift is the body’s natural response to help people vomit.
Then there’s the theory of “defensive hypothermia,” which suggests that cold sweats are a possible mechanism to conserve energy so the body can fight off an intruder, which was supported by a 2014 study and a 2016 review.
It’s another one of the body’s many survival tricks.
Teachers were right: Pupils can do the math
Teachers liked to preach that we wouldn’t have calculators with us all the time, but that wound up not being true. Our phones have calculators at the press of a button. But maybe even calculators aren’t always needed because our pupils do more math than you think.
The pupil light reflex – constrict in light and dilate in darkness – is well known, but recent work shows that pupil size is also regulated by cognitive and perceptual factors. By presenting subjects with images of various numbers of dots and measuring pupil size, the investigators were able to show “that numerical information is intrinsically related to perception,” lead author Dr. Elisa Castaldi of Florence University noted in a written statement.
The researchers found that pupils are responsible for important survival techniques. Coauthor David Burr of the University of Sydney and the University of Florence gave an evolutionary perspective: “When we look around, we spontaneously perceive the form, size, movement and colour of a scene. Equally spontaneously, we perceive the number of items before us. This ability, shared with most other animals, is an evolutionary fundamental: It reveals immediately important quantities, such as how many apples there are on the tree, or how many enemies are attacking.”
Useful information, indeed, but our pupils seem to be more interested in the quantity of beers in the refrigerator.
New study ‘changes understanding of bone loss after menopause’
In the longest study of bone loss in postmenopausal women to date, on average, bone mineral density (BMD) at the femoral neck (the most common location for a hip fracture) had dropped by 10% in 25 years – less than expected based on shorter studies.
Specifically, average BMD loss at the femoral neck was 0.4% per year during 25 years in this new study from Finland, compared with a drop of 1.6% per year over 15 years reported in other cohorts.
Five-year BMD change appeared to predict long-term bone loss. However, certain women had faster bone loss, indicating that they should be followed more closely.
“Although the average bone loss was 10.1% ... there is a significant variation in the bone loss rate” among women in the study, senior author Joonas Sirola, MD, PhD, associate professor, University of Eastern Finland, and coauthor Heikki Kröger, MD, PhD, a professor at the same university, explained to this news organization in an email, so “women with fast bone loss should receive special attention.
The findings from the Kuopio Osteoporosis Risk Factor and Prevention study by Anna Moilanen and colleagues were published online October 19 in the Journal of Bone and Mineral Research.
Several factors might explain the lower than expected drop in femoral neck BMD (the site that is used to diagnose osteoporosis), Dr. Sirola and Dr. Kröger said. BMD depends on a person’s age, race, sex, and genes. And compared with other countries, people in Finland consume more dairy products, and more postmenopausal women there take hormone replacement therapy (HRT).
“If otherwise indicated, HRT seemed to effectively protect from bone loss,” the researchers noted.
Also, the number of women who smoked or used corticosteroids was low, so bone loss in other populations may be higher. Moreover, the women who completed the study may have been healthier to start with, so the results should be interpreted with caution, they urge.
Nevertheless, the study sheds light on long-term changes in BMD in postmenopausal women and “stresses the importance of high peak bone mass before menopause and keeping a healthy weight” during aging to protect bone health, they say.
Indeed the work “changes our understanding of bone loss in older women,” said Dr. Kröger in a press release from the university.
Check BMD every 5 years after menopause
Invited to comment, American Society of Bone and Mineral Research President Peter R. Ebeling, MD, who was not involved with the research, noted key findings are that the rate of femoral neck bone loss after perimenopause was far less than previously expected, and 5-year BMD change appeared to predict long-term bone loss in postmenopausal women.
“We know bone loss begins 1 year before menopause and accelerates over the next 5 years,” Dr. Ebeling, from Monash University, Melbourne, added in an email. “This study indicates some stabilization of bone loss thereafter with lesser effects of low estrogen levels on bone.”
“It probably means bone density does not need to be measured as frequently following the menopause transition and could be every 5 years, rather than every 2 years, if there was concern about continuing bone loss.”
Baseline risk factors and long-term changes in BMD
For the study, researchers examined the association between risk factors for bone loss and long-term changes in femoral neck BMD in 2,695 women living in Kuopio who were 47 to 56 years old in 1989. The women were a mean age of 53 years, and 62% were postmenopausal.
They answered questionnaires and had femoral neck BMD measured by DEXA every 5 years.
A total of 2,695, 2,583, 2,482, 2,135, 1,305, and 686 women were assessed at baseline and 5-, 10-, 15-, 20- and 25-year follow-ups, respectively, indicating significant study drop-out by 25 years.
By then, 17% of patients had died, 9% needed long-term care, some were unwilling to continue in the study, and others had factors that would have resulted in DEXA measurement errors (for example, hip implants, spine degeneration).
Researchers divided participants into quartiles of mean initial femoral neck BMD: 1.09 g/cm2, 0.97 g/cm2, 0.89 g/cm2, and 0.79 g/cm2, corresponding with quartiles 1 to 4 respectively (where quartile 1 had the highest initial femoral BMD and quartile 4 the lowest).
At 25 years, the mean femoral BMD had dropped to 0.97 g/cm2, 0.87 g/cm2, 0.80 g/cm2, and 0.73 g/cm2 in these respective quartiles.
Women lost 0.9%, 0.5%, 3.0%, and 1.0% of their initial BMD each year in quartiles 1 to 4, respectively.
And at 25 years, the women had lost 22.5%, 12.5%, 7.5%, and 2.5% of their initial BMD in the four quartiles, respectively.
Women in quartile 1 had the greatest drop in femoral BMD at 25 years, although their mean BMD at 25 years was higher than the mean initial BMD of the other women.
The prevalence of bone-affecting diseases, smoking, and use of vitamin D/calcium supplementation, corticosteroids, or alcohol was similar in the four quartiles and was not associated with significant differences in annual bone loss.
The most important protective factor was HRT
However, body mass index (BMI) and HRT were significantly different in the four quartiles.
On average, women in quartile 1 had a mean BMI of 26.7 kg/m2 at baseline and 27.8 kg/m2 at 25 years. Women in quartile 4 (lowest initial BMD and lowest drop in BMD) had a mean BMI of 24.9 kg/m2 at baseline and 28.4 kg/m2 at 25 years.
Women in quartile 4 (lowest initial BMD and lowest drop in BMD) were more likely to take HRT than women in quartile 1 (highest initial BMD and highest drop in BMD), at 41% versus 26%, respectively.
“The average decrease in bone mineral density was lower than has been assumed on the basis of earlier, shorter follow-ups where the bone loss rate at the femoral neck has been estimated to be even more than 20%,” Dr. Sirola commented in the press release.
“There were also surprisingly few risk factors affecting bone mineral density. The most significant factor protecting against bone loss was hormone replacement therapy. Weight gain during the follow-up also protected against bone loss,” Dr. Sirola added.
The study was funded by the Academy of Finland, Finnish Ministry of Education and Culture, and the Päivikki and Sakari Sohlberg Foundation. The authors and Dr. Ebeling have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
In the longest study of bone loss in postmenopausal women to date, on average, bone mineral density (BMD) at the femoral neck (the most common location for a hip fracture) had dropped by 10% in 25 years – less than expected based on shorter studies.
Specifically, average BMD loss at the femoral neck was 0.4% per year during 25 years in this new study from Finland, compared with a drop of 1.6% per year over 15 years reported in other cohorts.
Five-year BMD change appeared to predict long-term bone loss. However, certain women had faster bone loss, indicating that they should be followed more closely.
“Although the average bone loss was 10.1% ... there is a significant variation in the bone loss rate” among women in the study, senior author Joonas Sirola, MD, PhD, associate professor, University of Eastern Finland, and coauthor Heikki Kröger, MD, PhD, a professor at the same university, explained to this news organization in an email, so “women with fast bone loss should receive special attention.
The findings from the Kuopio Osteoporosis Risk Factor and Prevention study by Anna Moilanen and colleagues were published online October 19 in the Journal of Bone and Mineral Research.
Several factors might explain the lower than expected drop in femoral neck BMD (the site that is used to diagnose osteoporosis), Dr. Sirola and Dr. Kröger said. BMD depends on a person’s age, race, sex, and genes. And compared with other countries, people in Finland consume more dairy products, and more postmenopausal women there take hormone replacement therapy (HRT).
“If otherwise indicated, HRT seemed to effectively protect from bone loss,” the researchers noted.
Also, the number of women who smoked or used corticosteroids was low, so bone loss in other populations may be higher. Moreover, the women who completed the study may have been healthier to start with, so the results should be interpreted with caution, they urge.
Nevertheless, the study sheds light on long-term changes in BMD in postmenopausal women and “stresses the importance of high peak bone mass before menopause and keeping a healthy weight” during aging to protect bone health, they say.
Indeed the work “changes our understanding of bone loss in older women,” said Dr. Kröger in a press release from the university.
Check BMD every 5 years after menopause
Invited to comment, American Society of Bone and Mineral Research President Peter R. Ebeling, MD, who was not involved with the research, noted key findings are that the rate of femoral neck bone loss after perimenopause was far less than previously expected, and 5-year BMD change appeared to predict long-term bone loss in postmenopausal women.
“We know bone loss begins 1 year before menopause and accelerates over the next 5 years,” Dr. Ebeling, from Monash University, Melbourne, added in an email. “This study indicates some stabilization of bone loss thereafter with lesser effects of low estrogen levels on bone.”
“It probably means bone density does not need to be measured as frequently following the menopause transition and could be every 5 years, rather than every 2 years, if there was concern about continuing bone loss.”
Baseline risk factors and long-term changes in BMD
For the study, researchers examined the association between risk factors for bone loss and long-term changes in femoral neck BMD in 2,695 women living in Kuopio who were 47 to 56 years old in 1989. The women were a mean age of 53 years, and 62% were postmenopausal.
They answered questionnaires and had femoral neck BMD measured by DEXA every 5 years.
A total of 2,695, 2,583, 2,482, 2,135, 1,305, and 686 women were assessed at baseline and 5-, 10-, 15-, 20- and 25-year follow-ups, respectively, indicating significant study drop-out by 25 years.
By then, 17% of patients had died, 9% needed long-term care, some were unwilling to continue in the study, and others had factors that would have resulted in DEXA measurement errors (for example, hip implants, spine degeneration).
Researchers divided participants into quartiles of mean initial femoral neck BMD: 1.09 g/cm2, 0.97 g/cm2, 0.89 g/cm2, and 0.79 g/cm2, corresponding with quartiles 1 to 4 respectively (where quartile 1 had the highest initial femoral BMD and quartile 4 the lowest).
At 25 years, the mean femoral BMD had dropped to 0.97 g/cm2, 0.87 g/cm2, 0.80 g/cm2, and 0.73 g/cm2 in these respective quartiles.
Women lost 0.9%, 0.5%, 3.0%, and 1.0% of their initial BMD each year in quartiles 1 to 4, respectively.
And at 25 years, the women had lost 22.5%, 12.5%, 7.5%, and 2.5% of their initial BMD in the four quartiles, respectively.
Women in quartile 1 had the greatest drop in femoral BMD at 25 years, although their mean BMD at 25 years was higher than the mean initial BMD of the other women.
The prevalence of bone-affecting diseases, smoking, and use of vitamin D/calcium supplementation, corticosteroids, or alcohol was similar in the four quartiles and was not associated with significant differences in annual bone loss.
The most important protective factor was HRT
However, body mass index (BMI) and HRT were significantly different in the four quartiles.
On average, women in quartile 1 had a mean BMI of 26.7 kg/m2 at baseline and 27.8 kg/m2 at 25 years. Women in quartile 4 (lowest initial BMD and lowest drop in BMD) had a mean BMI of 24.9 kg/m2 at baseline and 28.4 kg/m2 at 25 years.
Women in quartile 4 (lowest initial BMD and lowest drop in BMD) were more likely to take HRT than women in quartile 1 (highest initial BMD and highest drop in BMD), at 41% versus 26%, respectively.
“The average decrease in bone mineral density was lower than has been assumed on the basis of earlier, shorter follow-ups where the bone loss rate at the femoral neck has been estimated to be even more than 20%,” Dr. Sirola commented in the press release.
“There were also surprisingly few risk factors affecting bone mineral density. The most significant factor protecting against bone loss was hormone replacement therapy. Weight gain during the follow-up also protected against bone loss,” Dr. Sirola added.
The study was funded by the Academy of Finland, Finnish Ministry of Education and Culture, and the Päivikki and Sakari Sohlberg Foundation. The authors and Dr. Ebeling have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
In the longest study of bone loss in postmenopausal women to date, on average, bone mineral density (BMD) at the femoral neck (the most common location for a hip fracture) had dropped by 10% in 25 years – less than expected based on shorter studies.
Specifically, average BMD loss at the femoral neck was 0.4% per year during 25 years in this new study from Finland, compared with a drop of 1.6% per year over 15 years reported in other cohorts.
Five-year BMD change appeared to predict long-term bone loss. However, certain women had faster bone loss, indicating that they should be followed more closely.
“Although the average bone loss was 10.1% ... there is a significant variation in the bone loss rate” among women in the study, senior author Joonas Sirola, MD, PhD, associate professor, University of Eastern Finland, and coauthor Heikki Kröger, MD, PhD, a professor at the same university, explained to this news organization in an email, so “women with fast bone loss should receive special attention.
The findings from the Kuopio Osteoporosis Risk Factor and Prevention study by Anna Moilanen and colleagues were published online October 19 in the Journal of Bone and Mineral Research.
Several factors might explain the lower than expected drop in femoral neck BMD (the site that is used to diagnose osteoporosis), Dr. Sirola and Dr. Kröger said. BMD depends on a person’s age, race, sex, and genes. And compared with other countries, people in Finland consume more dairy products, and more postmenopausal women there take hormone replacement therapy (HRT).
“If otherwise indicated, HRT seemed to effectively protect from bone loss,” the researchers noted.
Also, the number of women who smoked or used corticosteroids was low, so bone loss in other populations may be higher. Moreover, the women who completed the study may have been healthier to start with, so the results should be interpreted with caution, they urge.
Nevertheless, the study sheds light on long-term changes in BMD in postmenopausal women and “stresses the importance of high peak bone mass before menopause and keeping a healthy weight” during aging to protect bone health, they say.
Indeed the work “changes our understanding of bone loss in older women,” said Dr. Kröger in a press release from the university.
Check BMD every 5 years after menopause
Invited to comment, American Society of Bone and Mineral Research President Peter R. Ebeling, MD, who was not involved with the research, noted key findings are that the rate of femoral neck bone loss after perimenopause was far less than previously expected, and 5-year BMD change appeared to predict long-term bone loss in postmenopausal women.
“We know bone loss begins 1 year before menopause and accelerates over the next 5 years,” Dr. Ebeling, from Monash University, Melbourne, added in an email. “This study indicates some stabilization of bone loss thereafter with lesser effects of low estrogen levels on bone.”
“It probably means bone density does not need to be measured as frequently following the menopause transition and could be every 5 years, rather than every 2 years, if there was concern about continuing bone loss.”
Baseline risk factors and long-term changes in BMD
For the study, researchers examined the association between risk factors for bone loss and long-term changes in femoral neck BMD in 2,695 women living in Kuopio who were 47 to 56 years old in 1989. The women were a mean age of 53 years, and 62% were postmenopausal.
They answered questionnaires and had femoral neck BMD measured by DEXA every 5 years.
A total of 2,695, 2,583, 2,482, 2,135, 1,305, and 686 women were assessed at baseline and 5-, 10-, 15-, 20- and 25-year follow-ups, respectively, indicating significant study drop-out by 25 years.
By then, 17% of patients had died, 9% needed long-term care, some were unwilling to continue in the study, and others had factors that would have resulted in DEXA measurement errors (for example, hip implants, spine degeneration).
Researchers divided participants into quartiles of mean initial femoral neck BMD: 1.09 g/cm2, 0.97 g/cm2, 0.89 g/cm2, and 0.79 g/cm2, corresponding with quartiles 1 to 4 respectively (where quartile 1 had the highest initial femoral BMD and quartile 4 the lowest).
At 25 years, the mean femoral BMD had dropped to 0.97 g/cm2, 0.87 g/cm2, 0.80 g/cm2, and 0.73 g/cm2 in these respective quartiles.
Women lost 0.9%, 0.5%, 3.0%, and 1.0% of their initial BMD each year in quartiles 1 to 4, respectively.
And at 25 years, the women had lost 22.5%, 12.5%, 7.5%, and 2.5% of their initial BMD in the four quartiles, respectively.
Women in quartile 1 had the greatest drop in femoral BMD at 25 years, although their mean BMD at 25 years was higher than the mean initial BMD of the other women.
The prevalence of bone-affecting diseases, smoking, and use of vitamin D/calcium supplementation, corticosteroids, or alcohol was similar in the four quartiles and was not associated with significant differences in annual bone loss.
The most important protective factor was HRT
However, body mass index (BMI) and HRT were significantly different in the four quartiles.
On average, women in quartile 1 had a mean BMI of 26.7 kg/m2 at baseline and 27.8 kg/m2 at 25 years. Women in quartile 4 (lowest initial BMD and lowest drop in BMD) had a mean BMI of 24.9 kg/m2 at baseline and 28.4 kg/m2 at 25 years.
Women in quartile 4 (lowest initial BMD and lowest drop in BMD) were more likely to take HRT than women in quartile 1 (highest initial BMD and highest drop in BMD), at 41% versus 26%, respectively.
“The average decrease in bone mineral density was lower than has been assumed on the basis of earlier, shorter follow-ups where the bone loss rate at the femoral neck has been estimated to be even more than 20%,” Dr. Sirola commented in the press release.
“There were also surprisingly few risk factors affecting bone mineral density. The most significant factor protecting against bone loss was hormone replacement therapy. Weight gain during the follow-up also protected against bone loss,” Dr. Sirola added.
The study was funded by the Academy of Finland, Finnish Ministry of Education and Culture, and the Päivikki and Sakari Sohlberg Foundation. The authors and Dr. Ebeling have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Enlarging purple plaque on leg
Clinical and dermoscopic features were consistent with a sporadic angiokeratoma, a benign ectasia of vessels associated with keratinization. Diagnosis was confirmed with shave biopsy.
Sporadic angiokeratomas are common and increase with age. They may occur anywhere on the skin, including mucous membranes. Dermoscopy of an angiokeratoma will reveal vascular lacunae—small pools of red or near black blood—as well as keratin scale, which appears as a white veil or shiny white lines.1
Other subtypes of angiokeratomas may be more widespread, including angiokeratoma of Fordyce, which manifests on the vulva and scrotum (usually in adulthood), or angiokeratoma circumscriptum, which occurs congenitally. There are some rare syndromic conditions associated with widespread angiokeratomas, such as Fabry disease.
The differential diagnosis for this solitary, vascular-appearing papule or plaque includes cherry angioma, pyogenic granuloma, and melanoma. Over time, the keratinization may increase, and lesions may look like a wart. A punch or shave biopsy will distinguish an angiokeratoma from other types of lesions. When a lesion is small enough, it may also be curative.
Angiokeratomas do not require treatment. However, because they may occur in cosmetically sensitive areas or bleed when traumatized, remedies are often sought. Excision, cryotherapy, electrocautery, and vascular laser are all possible treatments.
In this case, the patient was treated with curettage and light electrocautery, which left a small scar.
Text courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. Photos courtesy of Jonathan Karnes, MD (copyright retained).
1. Zaballos P, Daufí C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325. doi: 10.1001/archderm.143.3.318
Clinical and dermoscopic features were consistent with a sporadic angiokeratoma, a benign ectasia of vessels associated with keratinization. Diagnosis was confirmed with shave biopsy.
Sporadic angiokeratomas are common and increase with age. They may occur anywhere on the skin, including mucous membranes. Dermoscopy of an angiokeratoma will reveal vascular lacunae—small pools of red or near black blood—as well as keratin scale, which appears as a white veil or shiny white lines.1
Other subtypes of angiokeratomas may be more widespread, including angiokeratoma of Fordyce, which manifests on the vulva and scrotum (usually in adulthood), or angiokeratoma circumscriptum, which occurs congenitally. There are some rare syndromic conditions associated with widespread angiokeratomas, such as Fabry disease.
The differential diagnosis for this solitary, vascular-appearing papule or plaque includes cherry angioma, pyogenic granuloma, and melanoma. Over time, the keratinization may increase, and lesions may look like a wart. A punch or shave biopsy will distinguish an angiokeratoma from other types of lesions. When a lesion is small enough, it may also be curative.
Angiokeratomas do not require treatment. However, because they may occur in cosmetically sensitive areas or bleed when traumatized, remedies are often sought. Excision, cryotherapy, electrocautery, and vascular laser are all possible treatments.
In this case, the patient was treated with curettage and light electrocautery, which left a small scar.
Text courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. Photos courtesy of Jonathan Karnes, MD (copyright retained).
Clinical and dermoscopic features were consistent with a sporadic angiokeratoma, a benign ectasia of vessels associated with keratinization. Diagnosis was confirmed with shave biopsy.
Sporadic angiokeratomas are common and increase with age. They may occur anywhere on the skin, including mucous membranes. Dermoscopy of an angiokeratoma will reveal vascular lacunae—small pools of red or near black blood—as well as keratin scale, which appears as a white veil or shiny white lines.1
Other subtypes of angiokeratomas may be more widespread, including angiokeratoma of Fordyce, which manifests on the vulva and scrotum (usually in adulthood), or angiokeratoma circumscriptum, which occurs congenitally. There are some rare syndromic conditions associated with widespread angiokeratomas, such as Fabry disease.
The differential diagnosis for this solitary, vascular-appearing papule or plaque includes cherry angioma, pyogenic granuloma, and melanoma. Over time, the keratinization may increase, and lesions may look like a wart. A punch or shave biopsy will distinguish an angiokeratoma from other types of lesions. When a lesion is small enough, it may also be curative.
Angiokeratomas do not require treatment. However, because they may occur in cosmetically sensitive areas or bleed when traumatized, remedies are often sought. Excision, cryotherapy, electrocautery, and vascular laser are all possible treatments.
In this case, the patient was treated with curettage and light electrocautery, which left a small scar.
Text courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. Photos courtesy of Jonathan Karnes, MD (copyright retained).
1. Zaballos P, Daufí C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325. doi: 10.1001/archderm.143.3.318
1. Zaballos P, Daufí C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325. doi: 10.1001/archderm.143.3.318
Phototoxicity Secondary to Home Fireplace Exposure After Photodynamic Therapy for Actinic Keratosis
To the Editor:
Photodynamic therapy (PDT) is a US Food and Drug Administration–approved treatment for actinic keratosis (AK). It also commonly is administered off label for basal cell carcinoma, Bowen disease, photoaging, and acne vulgaris and is being investigated for other applications.1,2 In the context of treating AK, the mechanism employed in PDT most commonly involves the application of exogenous aminolevulinic acid (ALA), which is metabolized to the endogenous photosensitizer protoporphyrin IX (PpIX) in skin cells by enzymes in the heme biosynthetic pathway.3 The preferential uptake of ALA and conversion to PpIX is due to the altered and increased permeability of abnormal keratin layers of aging, sun-damaged cells, and skin tumors. Selectivity of ALA also occurs due to the preferential intracellular accumulation of PpIX in proliferating, relatively iron–deficient, precancerous and cancerous cells. The therapeutic effect is achieved with light exposure to blue light wavelength at 417 nm and corresponds to the excitation peak of PpIX,4 which activates PpIX and forms reactive oxygen species in the presence of oxygen that ultimately cause cell necrosis and apoptosis.5 Because it takes approximately 24 hours for PpIX to be completely metabolized from the skin, patients are counseled to avoid sun or artificial light exposure in the first 24 hours post-PDT, regardless of the indication, to avoid a severe phototoxic reaction.3,6,7 Although it is normal and desirable for patients to experience some form of a phototoxic reaction, which may include erythema, edema, crusting, vesiculation, or erosion in most patients, these types of reactions most often are secondary to the intended exposure and incidental natural or artificial light exposures.6 We report a case of a severe phototoxic reaction in which a patient experienced painful erythema and purulence on the left side of the chin after being within an arm’s length of a flame in a fireplace following PDT treatment.
A 59-year-old man presented to our dermatology clinic for his second of 3 PDT sessions to treat AKs on the face. He had a history of a basal cell carcinoma on the left nasolabial fold that previously was treated with Mohs micrographic surgery and melanoma on the left ear that was previously treated with excision. The patient received the initial PDT session 1 month prior and experienced a mild reaction with minimal redness and peeling that resolved in 4 to 5 days. For the second treatment, per standard protocol at our clinic, ALA was applied to the face, after which the patient incubated for 1 hour prior to blue light exposure (mean [SD] peak output of 417 [5] nm for 1000 seconds and 10 J/cm2).
After blue light exposure, broad-spectrum sunscreen (sun protection factor 47) was applied to our patient’s face, and he wore a wide-brimmed hat upon leaving the clinic and walking to his car. Similar to the first PDT session 1 month prior, he experienced minimal pain immediately after treatment. Once home and approximately 4 to 5 hours after PDT, he tended to a fire using his left hand and leaned into the fireplace with the left side of his face, which was within an arm’s length of the flames. Although his skin did not come in direct contact with the flames, the brief 2- to 3-minute exposure to the flame’s light and heat produced an immediate intense burning pain that the patient likened to the pain of blue light exposure. Within 24 hours, he developed a severe inflammatory reaction that included erythema, edema, desquamation, and pustules on the left side of the chin and cheek that produced a purulent discharge (Figure). The purulence resolved the next day; however, the other clinical manifestations persisted for 1 week. Despite the discomfort and symptoms, our patient did not seek medical attention and instead managed his symptoms conservatively with cold compresses. Although he noticed an overall subjective improvement in the appearance of his face after this second treatment, he received a third treatment with PDT approximately 1 month later, which resulted in a response that was similar to his first visit.
Photodynamic therapy is an increasingly accepted treatment modality for a plethora of benign and malignant dermatologic conditions. Although blue and red light are the most common light sources utilized with PDT because their wavelengths (404–420 nm and 635 nm, respectively) correspond to the excitation peaks of photosensitizers, alternative light sources increasingly are being explored. There is increasing interest in utilizing infrared (IR) light sources (700–1,000,000 nm) to penetrate deeper into the skin in the treatment of precancerous and cancerous lesions. Exposure to IR radiation is known to raise skin temperature via inside-out dermal water absorption and is thought to be useful in PDT-ALA by promoting ALA penetration and its conversion to PpIX.8 In a randomized controlled trial by Giehl et al,9 visible light plus water-filtered IR-A light was shown to produce considerably less pain in ALA-PDT compared to placebo, though efficacy was not statistically affected. There are burgeoning trials examining the role of IR in treating dermatologic conditions such as acne, but research is still needed on ALA-PDT activated by IR radiation to target AKs.
Although the PDT side-effect profile of phototoxicity, dyspigmentation, and hypersensitivity is well documented, phototoxicity secondary to flame exposure is rare. In our patient, the synergistic effect of light and heat produced an exuberant phototoxic reaction. As the applications for PDT continue to broaden, this case may represent the importance of addressing additional precautions, such as avoiding open flames in the house or while camping, in the PDT aftercare instructions to maximize patient safety.
- Fritsch C, Ruzicka T. Fluorescence diagnosis and photodynamic therapy in dermatology from experimental state to clinic standard methods. J Environ Pathol Toxicol Oncol. 2006;25:425-439.
- Lang K, Schulte KW, Ruzicka T, et al. Aminolevulinic acid (Levulan)in photodynamic therapy of actinic keratoses. Skin Therapy Lett. 2001;6:1-2, 5.
- Kennedy JC, Pottier RH. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J Photochem Photobiol B. 1992;14:275-292.
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163.
- Gad F, Viau G, Boushira M, et al. Photodynamic therapy with 5-aminolevulinic acid induces apoptosis and caspase activation in malignant T cells. J Cutan Med Surg. 2001;5:8-13.
- Piacquadio DJ, Chen DM, Farber HF, et al. Photodynamic therapy with aminolevulinic acid topical solution and visible blue light in the treatment of multiple actinic keratoses of the face and scalp: investigator-blinded, phase 3, multicenter trials. Arch Dermatol. 2004;140:41-46.
- Rhodes LE, Tsoukas MM, Anderson RR, et al. Iontophoretic delivery of ALA provides a quantitative model for ALA pharmacokinetics and PpIX phototoxicity in human skin. J Invest Dermatol. 1997;108:87-91.
- Dover JS, Phillips TJ, Arndt KA. Cutaneous effects and therapeutic uses of heat with emphasis on infrared radiation. J Am Acad Dermatol. 1989;20(2, pt 1):278-286.
- Giehl KA, Kriz M, Grahovac M, et al. A controlled trial of photodynamic therapy of actinic keratosis comparing different red light sources. Eur J Dermatol. 2014;24:335-341.
To the Editor:
Photodynamic therapy (PDT) is a US Food and Drug Administration–approved treatment for actinic keratosis (AK). It also commonly is administered off label for basal cell carcinoma, Bowen disease, photoaging, and acne vulgaris and is being investigated for other applications.1,2 In the context of treating AK, the mechanism employed in PDT most commonly involves the application of exogenous aminolevulinic acid (ALA), which is metabolized to the endogenous photosensitizer protoporphyrin IX (PpIX) in skin cells by enzymes in the heme biosynthetic pathway.3 The preferential uptake of ALA and conversion to PpIX is due to the altered and increased permeability of abnormal keratin layers of aging, sun-damaged cells, and skin tumors. Selectivity of ALA also occurs due to the preferential intracellular accumulation of PpIX in proliferating, relatively iron–deficient, precancerous and cancerous cells. The therapeutic effect is achieved with light exposure to blue light wavelength at 417 nm and corresponds to the excitation peak of PpIX,4 which activates PpIX and forms reactive oxygen species in the presence of oxygen that ultimately cause cell necrosis and apoptosis.5 Because it takes approximately 24 hours for PpIX to be completely metabolized from the skin, patients are counseled to avoid sun or artificial light exposure in the first 24 hours post-PDT, regardless of the indication, to avoid a severe phototoxic reaction.3,6,7 Although it is normal and desirable for patients to experience some form of a phototoxic reaction, which may include erythema, edema, crusting, vesiculation, or erosion in most patients, these types of reactions most often are secondary to the intended exposure and incidental natural or artificial light exposures.6 We report a case of a severe phototoxic reaction in which a patient experienced painful erythema and purulence on the left side of the chin after being within an arm’s length of a flame in a fireplace following PDT treatment.
A 59-year-old man presented to our dermatology clinic for his second of 3 PDT sessions to treat AKs on the face. He had a history of a basal cell carcinoma on the left nasolabial fold that previously was treated with Mohs micrographic surgery and melanoma on the left ear that was previously treated with excision. The patient received the initial PDT session 1 month prior and experienced a mild reaction with minimal redness and peeling that resolved in 4 to 5 days. For the second treatment, per standard protocol at our clinic, ALA was applied to the face, after which the patient incubated for 1 hour prior to blue light exposure (mean [SD] peak output of 417 [5] nm for 1000 seconds and 10 J/cm2).
After blue light exposure, broad-spectrum sunscreen (sun protection factor 47) was applied to our patient’s face, and he wore a wide-brimmed hat upon leaving the clinic and walking to his car. Similar to the first PDT session 1 month prior, he experienced minimal pain immediately after treatment. Once home and approximately 4 to 5 hours after PDT, he tended to a fire using his left hand and leaned into the fireplace with the left side of his face, which was within an arm’s length of the flames. Although his skin did not come in direct contact with the flames, the brief 2- to 3-minute exposure to the flame’s light and heat produced an immediate intense burning pain that the patient likened to the pain of blue light exposure. Within 24 hours, he developed a severe inflammatory reaction that included erythema, edema, desquamation, and pustules on the left side of the chin and cheek that produced a purulent discharge (Figure). The purulence resolved the next day; however, the other clinical manifestations persisted for 1 week. Despite the discomfort and symptoms, our patient did not seek medical attention and instead managed his symptoms conservatively with cold compresses. Although he noticed an overall subjective improvement in the appearance of his face after this second treatment, he received a third treatment with PDT approximately 1 month later, which resulted in a response that was similar to his first visit.
Photodynamic therapy is an increasingly accepted treatment modality for a plethora of benign and malignant dermatologic conditions. Although blue and red light are the most common light sources utilized with PDT because their wavelengths (404–420 nm and 635 nm, respectively) correspond to the excitation peaks of photosensitizers, alternative light sources increasingly are being explored. There is increasing interest in utilizing infrared (IR) light sources (700–1,000,000 nm) to penetrate deeper into the skin in the treatment of precancerous and cancerous lesions. Exposure to IR radiation is known to raise skin temperature via inside-out dermal water absorption and is thought to be useful in PDT-ALA by promoting ALA penetration and its conversion to PpIX.8 In a randomized controlled trial by Giehl et al,9 visible light plus water-filtered IR-A light was shown to produce considerably less pain in ALA-PDT compared to placebo, though efficacy was not statistically affected. There are burgeoning trials examining the role of IR in treating dermatologic conditions such as acne, but research is still needed on ALA-PDT activated by IR radiation to target AKs.
Although the PDT side-effect profile of phototoxicity, dyspigmentation, and hypersensitivity is well documented, phototoxicity secondary to flame exposure is rare. In our patient, the synergistic effect of light and heat produced an exuberant phototoxic reaction. As the applications for PDT continue to broaden, this case may represent the importance of addressing additional precautions, such as avoiding open flames in the house or while camping, in the PDT aftercare instructions to maximize patient safety.
To the Editor:
Photodynamic therapy (PDT) is a US Food and Drug Administration–approved treatment for actinic keratosis (AK). It also commonly is administered off label for basal cell carcinoma, Bowen disease, photoaging, and acne vulgaris and is being investigated for other applications.1,2 In the context of treating AK, the mechanism employed in PDT most commonly involves the application of exogenous aminolevulinic acid (ALA), which is metabolized to the endogenous photosensitizer protoporphyrin IX (PpIX) in skin cells by enzymes in the heme biosynthetic pathway.3 The preferential uptake of ALA and conversion to PpIX is due to the altered and increased permeability of abnormal keratin layers of aging, sun-damaged cells, and skin tumors. Selectivity of ALA also occurs due to the preferential intracellular accumulation of PpIX in proliferating, relatively iron–deficient, precancerous and cancerous cells. The therapeutic effect is achieved with light exposure to blue light wavelength at 417 nm and corresponds to the excitation peak of PpIX,4 which activates PpIX and forms reactive oxygen species in the presence of oxygen that ultimately cause cell necrosis and apoptosis.5 Because it takes approximately 24 hours for PpIX to be completely metabolized from the skin, patients are counseled to avoid sun or artificial light exposure in the first 24 hours post-PDT, regardless of the indication, to avoid a severe phototoxic reaction.3,6,7 Although it is normal and desirable for patients to experience some form of a phototoxic reaction, which may include erythema, edema, crusting, vesiculation, or erosion in most patients, these types of reactions most often are secondary to the intended exposure and incidental natural or artificial light exposures.6 We report a case of a severe phototoxic reaction in which a patient experienced painful erythema and purulence on the left side of the chin after being within an arm’s length of a flame in a fireplace following PDT treatment.
A 59-year-old man presented to our dermatology clinic for his second of 3 PDT sessions to treat AKs on the face. He had a history of a basal cell carcinoma on the left nasolabial fold that previously was treated with Mohs micrographic surgery and melanoma on the left ear that was previously treated with excision. The patient received the initial PDT session 1 month prior and experienced a mild reaction with minimal redness and peeling that resolved in 4 to 5 days. For the second treatment, per standard protocol at our clinic, ALA was applied to the face, after which the patient incubated for 1 hour prior to blue light exposure (mean [SD] peak output of 417 [5] nm for 1000 seconds and 10 J/cm2).
After blue light exposure, broad-spectrum sunscreen (sun protection factor 47) was applied to our patient’s face, and he wore a wide-brimmed hat upon leaving the clinic and walking to his car. Similar to the first PDT session 1 month prior, he experienced minimal pain immediately after treatment. Once home and approximately 4 to 5 hours after PDT, he tended to a fire using his left hand and leaned into the fireplace with the left side of his face, which was within an arm’s length of the flames. Although his skin did not come in direct contact with the flames, the brief 2- to 3-minute exposure to the flame’s light and heat produced an immediate intense burning pain that the patient likened to the pain of blue light exposure. Within 24 hours, he developed a severe inflammatory reaction that included erythema, edema, desquamation, and pustules on the left side of the chin and cheek that produced a purulent discharge (Figure). The purulence resolved the next day; however, the other clinical manifestations persisted for 1 week. Despite the discomfort and symptoms, our patient did not seek medical attention and instead managed his symptoms conservatively with cold compresses. Although he noticed an overall subjective improvement in the appearance of his face after this second treatment, he received a third treatment with PDT approximately 1 month later, which resulted in a response that was similar to his first visit.
Photodynamic therapy is an increasingly accepted treatment modality for a plethora of benign and malignant dermatologic conditions. Although blue and red light are the most common light sources utilized with PDT because their wavelengths (404–420 nm and 635 nm, respectively) correspond to the excitation peaks of photosensitizers, alternative light sources increasingly are being explored. There is increasing interest in utilizing infrared (IR) light sources (700–1,000,000 nm) to penetrate deeper into the skin in the treatment of precancerous and cancerous lesions. Exposure to IR radiation is known to raise skin temperature via inside-out dermal water absorption and is thought to be useful in PDT-ALA by promoting ALA penetration and its conversion to PpIX.8 In a randomized controlled trial by Giehl et al,9 visible light plus water-filtered IR-A light was shown to produce considerably less pain in ALA-PDT compared to placebo, though efficacy was not statistically affected. There are burgeoning trials examining the role of IR in treating dermatologic conditions such as acne, but research is still needed on ALA-PDT activated by IR radiation to target AKs.
Although the PDT side-effect profile of phototoxicity, dyspigmentation, and hypersensitivity is well documented, phototoxicity secondary to flame exposure is rare. In our patient, the synergistic effect of light and heat produced an exuberant phototoxic reaction. As the applications for PDT continue to broaden, this case may represent the importance of addressing additional precautions, such as avoiding open flames in the house or while camping, in the PDT aftercare instructions to maximize patient safety.
- Fritsch C, Ruzicka T. Fluorescence diagnosis and photodynamic therapy in dermatology from experimental state to clinic standard methods. J Environ Pathol Toxicol Oncol. 2006;25:425-439.
- Lang K, Schulte KW, Ruzicka T, et al. Aminolevulinic acid (Levulan)in photodynamic therapy of actinic keratoses. Skin Therapy Lett. 2001;6:1-2, 5.
- Kennedy JC, Pottier RH. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J Photochem Photobiol B. 1992;14:275-292.
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163.
- Gad F, Viau G, Boushira M, et al. Photodynamic therapy with 5-aminolevulinic acid induces apoptosis and caspase activation in malignant T cells. J Cutan Med Surg. 2001;5:8-13.
- Piacquadio DJ, Chen DM, Farber HF, et al. Photodynamic therapy with aminolevulinic acid topical solution and visible blue light in the treatment of multiple actinic keratoses of the face and scalp: investigator-blinded, phase 3, multicenter trials. Arch Dermatol. 2004;140:41-46.
- Rhodes LE, Tsoukas MM, Anderson RR, et al. Iontophoretic delivery of ALA provides a quantitative model for ALA pharmacokinetics and PpIX phototoxicity in human skin. J Invest Dermatol. 1997;108:87-91.
- Dover JS, Phillips TJ, Arndt KA. Cutaneous effects and therapeutic uses of heat with emphasis on infrared radiation. J Am Acad Dermatol. 1989;20(2, pt 1):278-286.
- Giehl KA, Kriz M, Grahovac M, et al. A controlled trial of photodynamic therapy of actinic keratosis comparing different red light sources. Eur J Dermatol. 2014;24:335-341.
- Fritsch C, Ruzicka T. Fluorescence diagnosis and photodynamic therapy in dermatology from experimental state to clinic standard methods. J Environ Pathol Toxicol Oncol. 2006;25:425-439.
- Lang K, Schulte KW, Ruzicka T, et al. Aminolevulinic acid (Levulan)in photodynamic therapy of actinic keratoses. Skin Therapy Lett. 2001;6:1-2, 5.
- Kennedy JC, Pottier RH. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J Photochem Photobiol B. 1992;14:275-292.
- Wan MT, Lin JY. Current evidence and applications of photodynamic therapy in dermatology. Clin Cosmet Investig Dermatol. 2014;7:145-163.
- Gad F, Viau G, Boushira M, et al. Photodynamic therapy with 5-aminolevulinic acid induces apoptosis and caspase activation in malignant T cells. J Cutan Med Surg. 2001;5:8-13.
- Piacquadio DJ, Chen DM, Farber HF, et al. Photodynamic therapy with aminolevulinic acid topical solution and visible blue light in the treatment of multiple actinic keratoses of the face and scalp: investigator-blinded, phase 3, multicenter trials. Arch Dermatol. 2004;140:41-46.
- Rhodes LE, Tsoukas MM, Anderson RR, et al. Iontophoretic delivery of ALA provides a quantitative model for ALA pharmacokinetics and PpIX phototoxicity in human skin. J Invest Dermatol. 1997;108:87-91.
- Dover JS, Phillips TJ, Arndt KA. Cutaneous effects and therapeutic uses of heat with emphasis on infrared radiation. J Am Acad Dermatol. 1989;20(2, pt 1):278-286.
- Giehl KA, Kriz M, Grahovac M, et al. A controlled trial of photodynamic therapy of actinic keratosis comparing different red light sources. Eur J Dermatol. 2014;24:335-341.
Practice Points
- As the applications of photodynamic therapy (PDT) in dermatology continue to expand, it is imperative for providers and patients alike to be knowledgeable with aftercare instructions and potential adverse effects.
- Avoid open flames in the house or while camping following PDT to maximize patient safety and prevent phototoxicity.
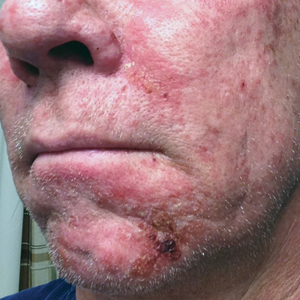
Early Pilomatrix Carcinoma: A Case Report With Emphasis on Molecular Pathology and Review of the Literature
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
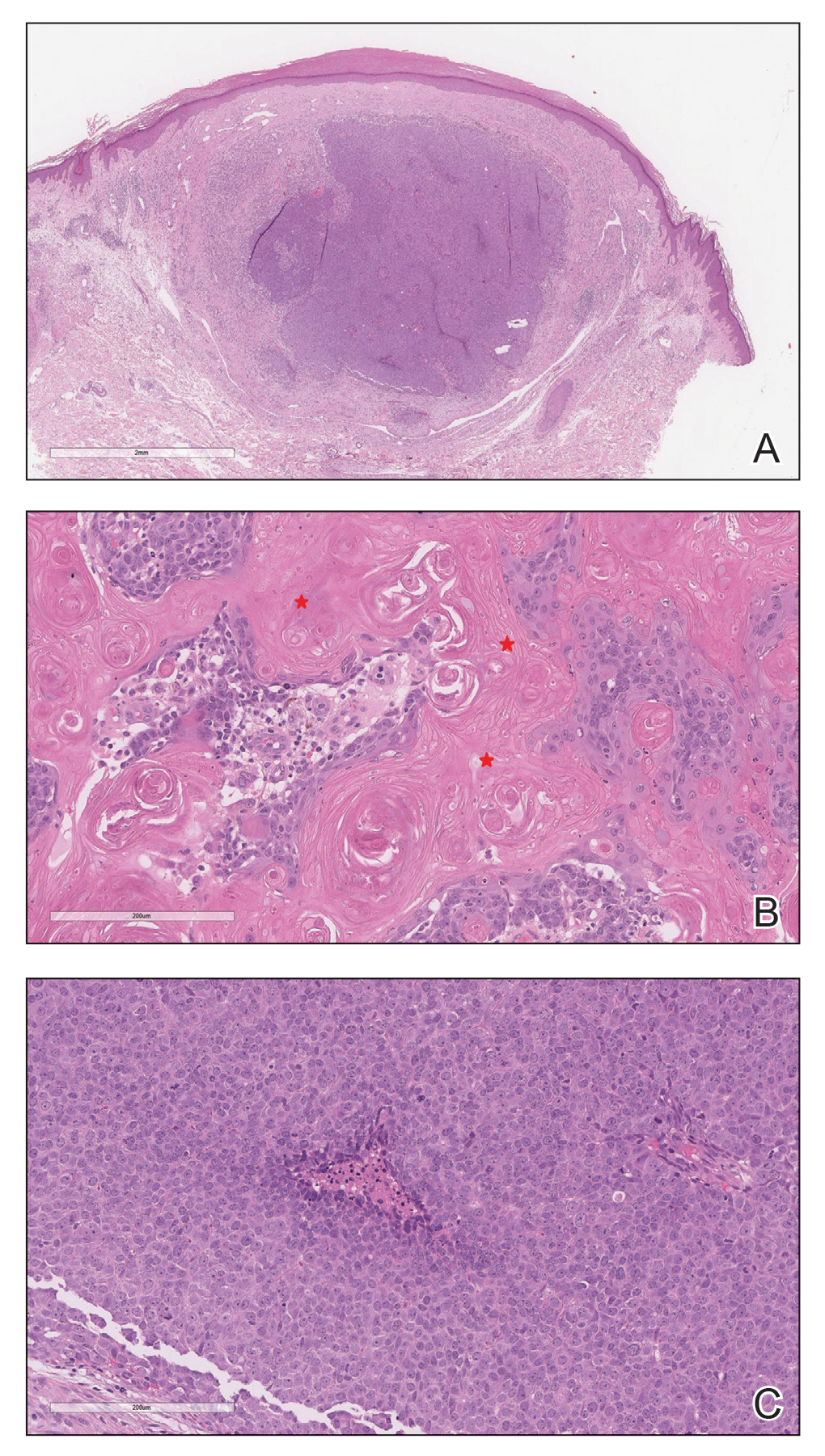
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.

Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.

Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).

The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
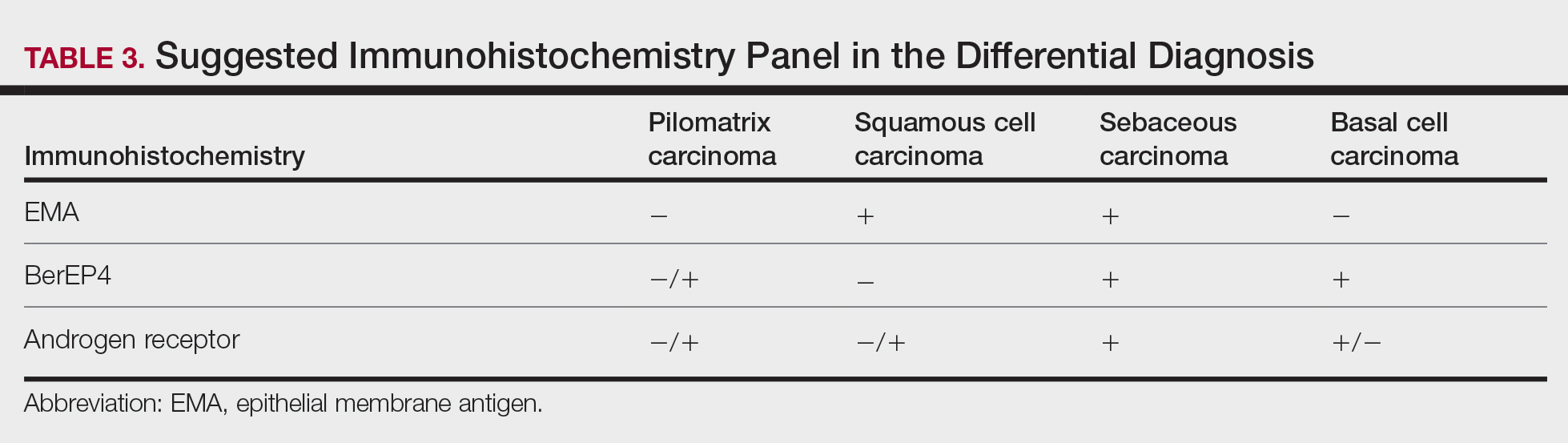
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.
Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.
Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).
The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.
Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.
Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).
The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
Practice Points
- Clinicians and pathologists should be aware of pilomatrix carcinoma to facilitate early detection.
- Early diagnosis and prompt treatment of pilomatrix carcinoma is crucial in lowering recurrence rate and avoiding a poor outcome.
- Caudal-related homeobox transcription factor 2 and β-catenin components of the Wnt signaling pathway play an important role in the pathogenesis of pilomatrix carcinoma.
- Although controversial, wide local excision is the treatment of choice for pilomatrix carcinoma.

TANS Syndrome: Tanorexia, Anorexia, and Nonmelanoma Skin Cancer
The term tanorexia describes compulsive use of a tanning bed, a disorder often identified in White patients. This compulsion is driven by underlying psychological distress that typically correlates with another psychiatric disorder, such as anxiety, body dysmorphic disorder, or an eating disorder. 1 Severe anorexia combined with excessive indoor tanning led to a notable burden of cutaneous squamous cell carcinomas (SCCs) and keratoacanthomas in one of our patients. We discuss the management and approach to patient care in this difficult situation, which we have coined TANS syndrome (for T anorexia, A norexia, and N onmelanoma s kin cancer).
A Patient With TANS Syndrome
A 35-year-old cachectic woman, who appeared much older than her chronologic age, presented for management of numerous painful bleeding skin lesions. Diffuse, erythematous, tender nodules with central keratotic cores, some several centimeters in diameter, were scattered on the abdomen, chest, and extremities (Figure 1); similar lesions were noted on the neck (Figure 2). Numerous erythematous scaly papules and plaques consistent with actinic keratoses were noted throughout the body.
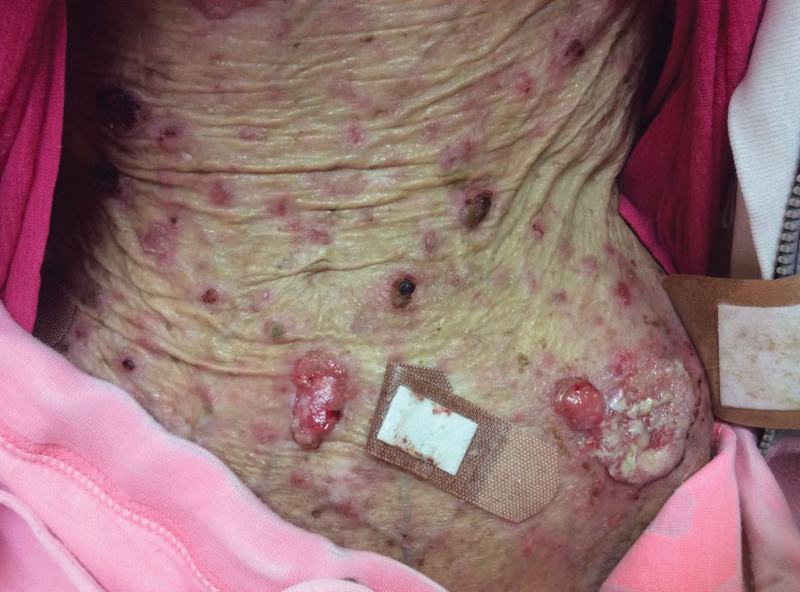
The patient reported that the cutaneous SCCs presented over the last few years, whereas her eating disorder began in adolescence and persisted despite multiple intensive outpatient and inpatient programs. The patient adamantly refused repeat hospitalization, against repeated suggestions by health care providers and her family. Comorbidities related to her anorexia included severe renal insufficiency, iron deficiency anemia, hypertriglyceridemia, kwashiorkor, and pellagra.
Within the last year, the patient had several biopsies showing SCC, keratoacanthoma type. The largest tumors had been treated by Mohs micrographic surgery, excision, and electrodesiccation or curettage. Adjuvant therapy over the last 2 years consisted of tazarotene cream 0.1%, imiquimod cream 5%, oral nicotinamide 500 mg twice daily, and acitretin 10 to 20 mg daily. Human papillomavirus 9-valent vaccine, recombinant, also had been tried as a chemopreventive and treatment, based on a published report of 2 patients in whom keratinocytic carcinomas decreased after such vaccination.2 The dose of acitretin was kept low because of the patient’s severe renal insufficiency and lack of supporting data for its use in this setting. Despite these modalities, our patient continued to develop new cutaneous SCCs.
We considered starting intralesional methotrexate but deferred this course of action, given the patient’s deteriorating renal function. Our plan was to initiate intralesional 5-fluorouracil; however, the patient was admitted to the hospital and subsequently died due to cardiovascular complications of anorexia.
UV Radiation in the Setting of Immune Compromise
Habitual tanning bed use has been recognized as a psychologic addiction.3,4 After exposure to UV radiation, damaged DNA upregulates pro-opiomelanocortin, which posttranslationally generates β-endorphins to elevate mood.3,5
Tanning beds deliver a higher dose of UVA radiation than UVB radiation and cause darkening of pigmentation by oxidation of preformed melanin and redistribution of melanosomes.3 UVA radiation (320–400 nm) emitted from a tanning bed is 10- to 15-times higher than the radiation emitted by the midday sun and causes DNA damage through generation of reactive oxygen species. UVA penetrates the dermis; its harmful effect on DNA contributes to the pathogenesis of melanoma.
UVB radiation (290–320 nm) is mainly restricted to the epidermis and is largely responsible for erythema of the skin. UVB specifically causes direct damage to DNA by forming pyrimidine dimers, superficially causing sunburn. Excessive exposure to UVB radiation increases the risk for nonmelanoma skin cancer.6
Severe starvation and chronic malnutrition, as seen in anorexia nervosa, also are known to lead to immunosuppression.7 Exposure to UV radiation has been shown to impair the function of antigen-presenting cells, cytokines, and suppressor T cells, and is classified as a Group 1 carcinogen by the World Health Organization.3,8 Combining a compromised immune system in anorexia with DNA damage from frequent indoor tanning provides a dangerous milieu for carcinogenesis.8 Without immune surveillance, as occurs with adequate nutrition, treatment of cutaneous SCC is, at best, challenging.
Primary care physicians, dermatologists, psychiatrists, nutritionists, and public health officials should educate high-risk patients to prevent TANS syndrome.
- Petit A, Karila L, Chalmin F, et al. Phenomenology and psychopathology of excessive indoor tanning. Int J Dermatol. 2014;53:664-672. doi:10.1111/ijd.12336
- Nichols AJ, Allen AH, Shareef S, et al. Association of human papillomavirus vaccine with the development of keratinocyte carcinomas. JAMA Dermatol. 2017;153:571-574. doi:10.1001/jamadermatol.2016.5703
- Madigan LM, Lim HW. Tanning beds: impact on health, and recent regulations. Clin Dermatol. 2016;34:640-648. doi:10.1016/j.clindermatol.2016.05.016
- Schwebel DC. Adolescent tanning, disordered eating, and risk taking. J Dev Behav Pediatr. 2014;35:225-227. doi:10.1097/DBP.0000000000000045
- Friedman B, English JC 3rd, Ferris LK. Indoor tanning, skin cancer and the young female patient: a review of the literature. J Pediatr Adolesc Gynecol. 2015;28:275-283. doi:10.1016/j.jpag.2014.07.015
- Armstrong BK, Kricker A. Epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18. doi:10.1016/s1011-1344(01)00198-1
- Hanachi M, Bohem V, Bemer P, et al. Negative role of malnutrition in cell-mediated immune response: Pneumocystis jirovecii pneumonia (PCP) in a severely malnourished, HIV-negative patient with anorexia nervosa. Clin Nutr ESPEN. 2018;25:163-165. doi:10.1016/j.clnesp.2018.03.121
- Schwarz T, Beissert S. Milestones in photoimmunology. J Invest Dermatol. 2013;133:E7-E10. doi:10.1038/skinbio.2013.177
The term tanorexia describes compulsive use of a tanning bed, a disorder often identified in White patients. This compulsion is driven by underlying psychological distress that typically correlates with another psychiatric disorder, such as anxiety, body dysmorphic disorder, or an eating disorder. 1 Severe anorexia combined with excessive indoor tanning led to a notable burden of cutaneous squamous cell carcinomas (SCCs) and keratoacanthomas in one of our patients. We discuss the management and approach to patient care in this difficult situation, which we have coined TANS syndrome (for T anorexia, A norexia, and N onmelanoma s kin cancer).
A Patient With TANS Syndrome
A 35-year-old cachectic woman, who appeared much older than her chronologic age, presented for management of numerous painful bleeding skin lesions. Diffuse, erythematous, tender nodules with central keratotic cores, some several centimeters in diameter, were scattered on the abdomen, chest, and extremities (Figure 1); similar lesions were noted on the neck (Figure 2). Numerous erythematous scaly papules and plaques consistent with actinic keratoses were noted throughout the body.
The patient reported that the cutaneous SCCs presented over the last few years, whereas her eating disorder began in adolescence and persisted despite multiple intensive outpatient and inpatient programs. The patient adamantly refused repeat hospitalization, against repeated suggestions by health care providers and her family. Comorbidities related to her anorexia included severe renal insufficiency, iron deficiency anemia, hypertriglyceridemia, kwashiorkor, and pellagra.
Within the last year, the patient had several biopsies showing SCC, keratoacanthoma type. The largest tumors had been treated by Mohs micrographic surgery, excision, and electrodesiccation or curettage. Adjuvant therapy over the last 2 years consisted of tazarotene cream 0.1%, imiquimod cream 5%, oral nicotinamide 500 mg twice daily, and acitretin 10 to 20 mg daily. Human papillomavirus 9-valent vaccine, recombinant, also had been tried as a chemopreventive and treatment, based on a published report of 2 patients in whom keratinocytic carcinomas decreased after such vaccination.2 The dose of acitretin was kept low because of the patient’s severe renal insufficiency and lack of supporting data for its use in this setting. Despite these modalities, our patient continued to develop new cutaneous SCCs.
We considered starting intralesional methotrexate but deferred this course of action, given the patient’s deteriorating renal function. Our plan was to initiate intralesional 5-fluorouracil; however, the patient was admitted to the hospital and subsequently died due to cardiovascular complications of anorexia.
UV Radiation in the Setting of Immune Compromise
Habitual tanning bed use has been recognized as a psychologic addiction.3,4 After exposure to UV radiation, damaged DNA upregulates pro-opiomelanocortin, which posttranslationally generates β-endorphins to elevate mood.3,5
Tanning beds deliver a higher dose of UVA radiation than UVB radiation and cause darkening of pigmentation by oxidation of preformed melanin and redistribution of melanosomes.3 UVA radiation (320–400 nm) emitted from a tanning bed is 10- to 15-times higher than the radiation emitted by the midday sun and causes DNA damage through generation of reactive oxygen species. UVA penetrates the dermis; its harmful effect on DNA contributes to the pathogenesis of melanoma.
UVB radiation (290–320 nm) is mainly restricted to the epidermis and is largely responsible for erythema of the skin. UVB specifically causes direct damage to DNA by forming pyrimidine dimers, superficially causing sunburn. Excessive exposure to UVB radiation increases the risk for nonmelanoma skin cancer.6
Severe starvation and chronic malnutrition, as seen in anorexia nervosa, also are known to lead to immunosuppression.7 Exposure to UV radiation has been shown to impair the function of antigen-presenting cells, cytokines, and suppressor T cells, and is classified as a Group 1 carcinogen by the World Health Organization.3,8 Combining a compromised immune system in anorexia with DNA damage from frequent indoor tanning provides a dangerous milieu for carcinogenesis.8 Without immune surveillance, as occurs with adequate nutrition, treatment of cutaneous SCC is, at best, challenging.
Primary care physicians, dermatologists, psychiatrists, nutritionists, and public health officials should educate high-risk patients to prevent TANS syndrome.
The term tanorexia describes compulsive use of a tanning bed, a disorder often identified in White patients. This compulsion is driven by underlying psychological distress that typically correlates with another psychiatric disorder, such as anxiety, body dysmorphic disorder, or an eating disorder. 1 Severe anorexia combined with excessive indoor tanning led to a notable burden of cutaneous squamous cell carcinomas (SCCs) and keratoacanthomas in one of our patients. We discuss the management and approach to patient care in this difficult situation, which we have coined TANS syndrome (for T anorexia, A norexia, and N onmelanoma s kin cancer).
A Patient With TANS Syndrome
A 35-year-old cachectic woman, who appeared much older than her chronologic age, presented for management of numerous painful bleeding skin lesions. Diffuse, erythematous, tender nodules with central keratotic cores, some several centimeters in diameter, were scattered on the abdomen, chest, and extremities (Figure 1); similar lesions were noted on the neck (Figure 2). Numerous erythematous scaly papules and plaques consistent with actinic keratoses were noted throughout the body.
The patient reported that the cutaneous SCCs presented over the last few years, whereas her eating disorder began in adolescence and persisted despite multiple intensive outpatient and inpatient programs. The patient adamantly refused repeat hospitalization, against repeated suggestions by health care providers and her family. Comorbidities related to her anorexia included severe renal insufficiency, iron deficiency anemia, hypertriglyceridemia, kwashiorkor, and pellagra.
Within the last year, the patient had several biopsies showing SCC, keratoacanthoma type. The largest tumors had been treated by Mohs micrographic surgery, excision, and electrodesiccation or curettage. Adjuvant therapy over the last 2 years consisted of tazarotene cream 0.1%, imiquimod cream 5%, oral nicotinamide 500 mg twice daily, and acitretin 10 to 20 mg daily. Human papillomavirus 9-valent vaccine, recombinant, also had been tried as a chemopreventive and treatment, based on a published report of 2 patients in whom keratinocytic carcinomas decreased after such vaccination.2 The dose of acitretin was kept low because of the patient’s severe renal insufficiency and lack of supporting data for its use in this setting. Despite these modalities, our patient continued to develop new cutaneous SCCs.
We considered starting intralesional methotrexate but deferred this course of action, given the patient’s deteriorating renal function. Our plan was to initiate intralesional 5-fluorouracil; however, the patient was admitted to the hospital and subsequently died due to cardiovascular complications of anorexia.
UV Radiation in the Setting of Immune Compromise
Habitual tanning bed use has been recognized as a psychologic addiction.3,4 After exposure to UV radiation, damaged DNA upregulates pro-opiomelanocortin, which posttranslationally generates β-endorphins to elevate mood.3,5
Tanning beds deliver a higher dose of UVA radiation than UVB radiation and cause darkening of pigmentation by oxidation of preformed melanin and redistribution of melanosomes.3 UVA radiation (320–400 nm) emitted from a tanning bed is 10- to 15-times higher than the radiation emitted by the midday sun and causes DNA damage through generation of reactive oxygen species. UVA penetrates the dermis; its harmful effect on DNA contributes to the pathogenesis of melanoma.
UVB radiation (290–320 nm) is mainly restricted to the epidermis and is largely responsible for erythema of the skin. UVB specifically causes direct damage to DNA by forming pyrimidine dimers, superficially causing sunburn. Excessive exposure to UVB radiation increases the risk for nonmelanoma skin cancer.6
Severe starvation and chronic malnutrition, as seen in anorexia nervosa, also are known to lead to immunosuppression.7 Exposure to UV radiation has been shown to impair the function of antigen-presenting cells, cytokines, and suppressor T cells, and is classified as a Group 1 carcinogen by the World Health Organization.3,8 Combining a compromised immune system in anorexia with DNA damage from frequent indoor tanning provides a dangerous milieu for carcinogenesis.8 Without immune surveillance, as occurs with adequate nutrition, treatment of cutaneous SCC is, at best, challenging.
Primary care physicians, dermatologists, psychiatrists, nutritionists, and public health officials should educate high-risk patients to prevent TANS syndrome.
- Petit A, Karila L, Chalmin F, et al. Phenomenology and psychopathology of excessive indoor tanning. Int J Dermatol. 2014;53:664-672. doi:10.1111/ijd.12336
- Nichols AJ, Allen AH, Shareef S, et al. Association of human papillomavirus vaccine with the development of keratinocyte carcinomas. JAMA Dermatol. 2017;153:571-574. doi:10.1001/jamadermatol.2016.5703
- Madigan LM, Lim HW. Tanning beds: impact on health, and recent regulations. Clin Dermatol. 2016;34:640-648. doi:10.1016/j.clindermatol.2016.05.016
- Schwebel DC. Adolescent tanning, disordered eating, and risk taking. J Dev Behav Pediatr. 2014;35:225-227. doi:10.1097/DBP.0000000000000045
- Friedman B, English JC 3rd, Ferris LK. Indoor tanning, skin cancer and the young female patient: a review of the literature. J Pediatr Adolesc Gynecol. 2015;28:275-283. doi:10.1016/j.jpag.2014.07.015
- Armstrong BK, Kricker A. Epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18. doi:10.1016/s1011-1344(01)00198-1
- Hanachi M, Bohem V, Bemer P, et al. Negative role of malnutrition in cell-mediated immune response: Pneumocystis jirovecii pneumonia (PCP) in a severely malnourished, HIV-negative patient with anorexia nervosa. Clin Nutr ESPEN. 2018;25:163-165. doi:10.1016/j.clnesp.2018.03.121
- Schwarz T, Beissert S. Milestones in photoimmunology. J Invest Dermatol. 2013;133:E7-E10. doi:10.1038/skinbio.2013.177
- Petit A, Karila L, Chalmin F, et al. Phenomenology and psychopathology of excessive indoor tanning. Int J Dermatol. 2014;53:664-672. doi:10.1111/ijd.12336
- Nichols AJ, Allen AH, Shareef S, et al. Association of human papillomavirus vaccine with the development of keratinocyte carcinomas. JAMA Dermatol. 2017;153:571-574. doi:10.1001/jamadermatol.2016.5703
- Madigan LM, Lim HW. Tanning beds: impact on health, and recent regulations. Clin Dermatol. 2016;34:640-648. doi:10.1016/j.clindermatol.2016.05.016
- Schwebel DC. Adolescent tanning, disordered eating, and risk taking. J Dev Behav Pediatr. 2014;35:225-227. doi:10.1097/DBP.0000000000000045
- Friedman B, English JC 3rd, Ferris LK. Indoor tanning, skin cancer and the young female patient: a review of the literature. J Pediatr Adolesc Gynecol. 2015;28:275-283. doi:10.1016/j.jpag.2014.07.015
- Armstrong BK, Kricker A. Epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18. doi:10.1016/s1011-1344(01)00198-1
- Hanachi M, Bohem V, Bemer P, et al. Negative role of malnutrition in cell-mediated immune response: Pneumocystis jirovecii pneumonia (PCP) in a severely malnourished, HIV-negative patient with anorexia nervosa. Clin Nutr ESPEN. 2018;25:163-165. doi:10.1016/j.clnesp.2018.03.121
- Schwarz T, Beissert S. Milestones in photoimmunology. J Invest Dermatol. 2013;133:E7-E10. doi:10.1038/skinbio.2013.177
Practice Points
- Primary care physicians, dermatologists, psychiatrists, nutritionists, and public health officials should educate high-risk patients to prevent TANS syndrome.
- Combining a compromised immune system in anorexia with DNA damage from frequent indoor tanning provides a dangerous milieu for carcinogenesis.
- Comorbidities related to TANS syndrome make it challenging to effectively treat cutaneous squamous cell carcinoma.