User login
Developing guidance for patient movement requests
Clear guidelines in policy needed
In hospital medicine, inpatients often request more freedom to move within the hospital complex for a wide range of both benign and potentially concerning reasons, says Sara Stream, MD.

“Hospitalists are often confronted with a dilemma when considering these patient requests: how to promote patient-centered care and autonomy while balancing patient safety, concerns for hospital liability, and the delivery of timely, efficient medical care,” said Dr. Stream, a hospitalist at the VA New York Harbor Healthcare System. Guidance from medical literature and institutional policies on inpatient movement are lacking, so Dr. Stream coauthored an article seeking to develop a framework with which hospitalists can approach patient requests for liberalized movement.
The authors concluded that for a small subset of patients, liberalized movement within the hospital may be clinically feasible: those who are medically, physically, and psychiatrically stable enough to move off their assigned floors without inordinate risk. “For the rest of inpatients, movement outside their monitored inpatient settings may interfere with appropriate medical care and undermine the indications for acute hospitalization,” Dr. Stream said.
Creating institutional policy that identifies relevant clinical, legal and ethical considerations, while incorporating the varied perspectives of physicians, patients, nurses, and hospital administration/risk management will allow requests for increased movement to be evaluated systematically and transparently.
“When patients request liberalized movement, hospitalists should consider the requests systematically: first to identify the intent behind requests, and then to follow a framework to determine whether increased movement would be safe and allow appropriate medical care without creating additional risks,” Dr. Stream said.
Hospitalists should assess and compile individual patient requests for liberalized movement and work with other physicians, nurses, hospital administration, and risk management to devise pertinent policy on this issue that is specific to their institutions. “By eventually creating clear guidelines in policy, health care providers will spend less time managing each individual request to leave the floor because they have a systematic strategy for making consistent decisions about patient movement,” the authors concluded.
Reference
1. Stream S, Alfandre D. “Just Getting a Cup of Coffee” – Considering Best Practices for Patients’ Movement off the Hospital Floor. J Hosp Med. 2019 Nov. doi: 10.12788/jhm.3227.
Clear guidelines in policy needed
Clear guidelines in policy needed
In hospital medicine, inpatients often request more freedom to move within the hospital complex for a wide range of both benign and potentially concerning reasons, says Sara Stream, MD.
“Hospitalists are often confronted with a dilemma when considering these patient requests: how to promote patient-centered care and autonomy while balancing patient safety, concerns for hospital liability, and the delivery of timely, efficient medical care,” said Dr. Stream, a hospitalist at the VA New York Harbor Healthcare System. Guidance from medical literature and institutional policies on inpatient movement are lacking, so Dr. Stream coauthored an article seeking to develop a framework with which hospitalists can approach patient requests for liberalized movement.
The authors concluded that for a small subset of patients, liberalized movement within the hospital may be clinically feasible: those who are medically, physically, and psychiatrically stable enough to move off their assigned floors without inordinate risk. “For the rest of inpatients, movement outside their monitored inpatient settings may interfere with appropriate medical care and undermine the indications for acute hospitalization,” Dr. Stream said.
Creating institutional policy that identifies relevant clinical, legal and ethical considerations, while incorporating the varied perspectives of physicians, patients, nurses, and hospital administration/risk management will allow requests for increased movement to be evaluated systematically and transparently.
“When patients request liberalized movement, hospitalists should consider the requests systematically: first to identify the intent behind requests, and then to follow a framework to determine whether increased movement would be safe and allow appropriate medical care without creating additional risks,” Dr. Stream said.
Hospitalists should assess and compile individual patient requests for liberalized movement and work with other physicians, nurses, hospital administration, and risk management to devise pertinent policy on this issue that is specific to their institutions. “By eventually creating clear guidelines in policy, health care providers will spend less time managing each individual request to leave the floor because they have a systematic strategy for making consistent decisions about patient movement,” the authors concluded.
Reference
1. Stream S, Alfandre D. “Just Getting a Cup of Coffee” – Considering Best Practices for Patients’ Movement off the Hospital Floor. J Hosp Med. 2019 Nov. doi: 10.12788/jhm.3227.
In hospital medicine, inpatients often request more freedom to move within the hospital complex for a wide range of both benign and potentially concerning reasons, says Sara Stream, MD.
“Hospitalists are often confronted with a dilemma when considering these patient requests: how to promote patient-centered care and autonomy while balancing patient safety, concerns for hospital liability, and the delivery of timely, efficient medical care,” said Dr. Stream, a hospitalist at the VA New York Harbor Healthcare System. Guidance from medical literature and institutional policies on inpatient movement are lacking, so Dr. Stream coauthored an article seeking to develop a framework with which hospitalists can approach patient requests for liberalized movement.
The authors concluded that for a small subset of patients, liberalized movement within the hospital may be clinically feasible: those who are medically, physically, and psychiatrically stable enough to move off their assigned floors without inordinate risk. “For the rest of inpatients, movement outside their monitored inpatient settings may interfere with appropriate medical care and undermine the indications for acute hospitalization,” Dr. Stream said.
Creating institutional policy that identifies relevant clinical, legal and ethical considerations, while incorporating the varied perspectives of physicians, patients, nurses, and hospital administration/risk management will allow requests for increased movement to be evaluated systematically and transparently.
“When patients request liberalized movement, hospitalists should consider the requests systematically: first to identify the intent behind requests, and then to follow a framework to determine whether increased movement would be safe and allow appropriate medical care without creating additional risks,” Dr. Stream said.
Hospitalists should assess and compile individual patient requests for liberalized movement and work with other physicians, nurses, hospital administration, and risk management to devise pertinent policy on this issue that is specific to their institutions. “By eventually creating clear guidelines in policy, health care providers will spend less time managing each individual request to leave the floor because they have a systematic strategy for making consistent decisions about patient movement,” the authors concluded.
Reference
1. Stream S, Alfandre D. “Just Getting a Cup of Coffee” – Considering Best Practices for Patients’ Movement off the Hospital Floor. J Hosp Med. 2019 Nov. doi: 10.12788/jhm.3227.
CDC: Opioid prescribing and use rates down since 2010
Trends in opioid prescribing and use from 2010 to 2016 offer some encouragement, but opioid-attributable deaths continued to increase over that period, according to the Centers for Disease Control and Prevention.
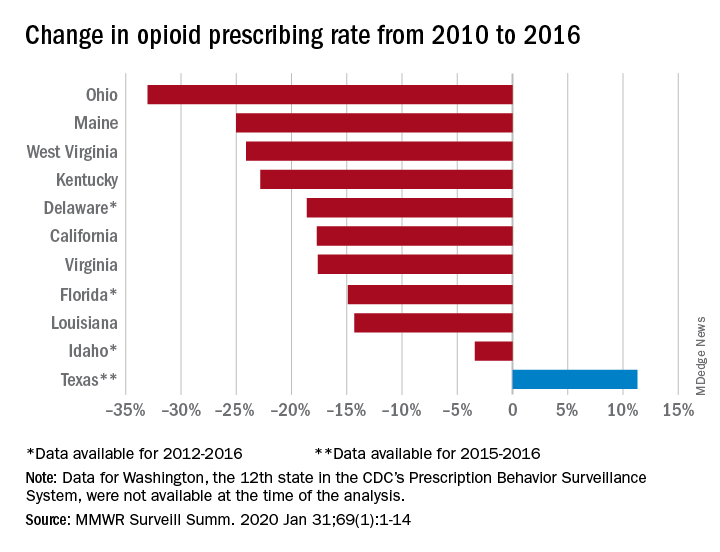
Prescribing rates dropped during that period, as did daily opioid dosage rates and the percentage of patients with high daily opioid dosages, Gail K. Strickler, PhD, of the Institute for Behavioral Health at Brandeis University in Waltham, Mass., and associates wrote in MMWR Surveillance Summaries.
Their analysis involved 11 of the 12 states (Washington was unable to provide data for the analysis) participating in the CDC’s Prescription Behavior Surveillance System, which uses data from the states’ prescription drug monitoring programs. The 11 states represented about 38% of the U.S. population in 2016.
The opioid prescribing rate fell in 10 of those 11 states, with declines varying from 3.4% in Idaho to 33.0% in Ohio. Prescribing went up in Texas by 11.3%, but the state only had data available for 2015 and 2016. Three other states – Delaware, Florida, and Idaho – were limited to data from 2012 to 2016, the investigators noted.
As for the other measures, all states showed declines for the mean daily opioid dosage. Texas had the smallest drop at 2.9% and Florida saw the largest, at 27.4%. All states also had reductions in the percentage of patients with high daily opioid dosage, with decreases varying from 5.7% in Idaho to 43.9% in Louisiana, Dr. Strickler and associates reported. A high daily dosage was defined as at least 90 morphine milligram equivalents for all class II-V opioid drugs.
“Despite these favorable trends ... opioid overdose deaths attributable to the most commonly prescribed opioids, the natural and semisynthetics (e.g., morphine and oxycodone), increased during 2010-2016,” they said.
It is possible that a change in mortality is lagging “behind changes in prescribing behaviors” or that “the trend in deaths related to these types of opioids has been driven by factors other than prescription opioid misuse rates, such as increasing mortality from heroin, which is frequently classified as morphine or found concomitantly with morphine postmortem, and a spike in deaths involving illicitly manufactured fentanyl combined with heroin and prescribed opioids since 2013,” the investigators suggested.
SOURCE: Strickler GK et al. MMWR Surveill Summ. 2020 Jan 31;69(1):1-14.
Trends in opioid prescribing and use from 2010 to 2016 offer some encouragement, but opioid-attributable deaths continued to increase over that period, according to the Centers for Disease Control and Prevention.
Prescribing rates dropped during that period, as did daily opioid dosage rates and the percentage of patients with high daily opioid dosages, Gail K. Strickler, PhD, of the Institute for Behavioral Health at Brandeis University in Waltham, Mass., and associates wrote in MMWR Surveillance Summaries.
Their analysis involved 11 of the 12 states (Washington was unable to provide data for the analysis) participating in the CDC’s Prescription Behavior Surveillance System, which uses data from the states’ prescription drug monitoring programs. The 11 states represented about 38% of the U.S. population in 2016.
The opioid prescribing rate fell in 10 of those 11 states, with declines varying from 3.4% in Idaho to 33.0% in Ohio. Prescribing went up in Texas by 11.3%, but the state only had data available for 2015 and 2016. Three other states – Delaware, Florida, and Idaho – were limited to data from 2012 to 2016, the investigators noted.
As for the other measures, all states showed declines for the mean daily opioid dosage. Texas had the smallest drop at 2.9% and Florida saw the largest, at 27.4%. All states also had reductions in the percentage of patients with high daily opioid dosage, with decreases varying from 5.7% in Idaho to 43.9% in Louisiana, Dr. Strickler and associates reported. A high daily dosage was defined as at least 90 morphine milligram equivalents for all class II-V opioid drugs.
“Despite these favorable trends ... opioid overdose deaths attributable to the most commonly prescribed opioids, the natural and semisynthetics (e.g., morphine and oxycodone), increased during 2010-2016,” they said.
It is possible that a change in mortality is lagging “behind changes in prescribing behaviors” or that “the trend in deaths related to these types of opioids has been driven by factors other than prescription opioid misuse rates, such as increasing mortality from heroin, which is frequently classified as morphine or found concomitantly with morphine postmortem, and a spike in deaths involving illicitly manufactured fentanyl combined with heroin and prescribed opioids since 2013,” the investigators suggested.
SOURCE: Strickler GK et al. MMWR Surveill Summ. 2020 Jan 31;69(1):1-14.
Trends in opioid prescribing and use from 2010 to 2016 offer some encouragement, but opioid-attributable deaths continued to increase over that period, according to the Centers for Disease Control and Prevention.
Prescribing rates dropped during that period, as did daily opioid dosage rates and the percentage of patients with high daily opioid dosages, Gail K. Strickler, PhD, of the Institute for Behavioral Health at Brandeis University in Waltham, Mass., and associates wrote in MMWR Surveillance Summaries.
Their analysis involved 11 of the 12 states (Washington was unable to provide data for the analysis) participating in the CDC’s Prescription Behavior Surveillance System, which uses data from the states’ prescription drug monitoring programs. The 11 states represented about 38% of the U.S. population in 2016.
The opioid prescribing rate fell in 10 of those 11 states, with declines varying from 3.4% in Idaho to 33.0% in Ohio. Prescribing went up in Texas by 11.3%, but the state only had data available for 2015 and 2016. Three other states – Delaware, Florida, and Idaho – were limited to data from 2012 to 2016, the investigators noted.
As for the other measures, all states showed declines for the mean daily opioid dosage. Texas had the smallest drop at 2.9% and Florida saw the largest, at 27.4%. All states also had reductions in the percentage of patients with high daily opioid dosage, with decreases varying from 5.7% in Idaho to 43.9% in Louisiana, Dr. Strickler and associates reported. A high daily dosage was defined as at least 90 morphine milligram equivalents for all class II-V opioid drugs.
“Despite these favorable trends ... opioid overdose deaths attributable to the most commonly prescribed opioids, the natural and semisynthetics (e.g., morphine and oxycodone), increased during 2010-2016,” they said.
It is possible that a change in mortality is lagging “behind changes in prescribing behaviors” or that “the trend in deaths related to these types of opioids has been driven by factors other than prescription opioid misuse rates, such as increasing mortality from heroin, which is frequently classified as morphine or found concomitantly with morphine postmortem, and a spike in deaths involving illicitly manufactured fentanyl combined with heroin and prescribed opioids since 2013,” the investigators suggested.
SOURCE: Strickler GK et al. MMWR Surveill Summ. 2020 Jan 31;69(1):1-14.
FROM MMWR SURVEILLANCE SUMMARIES
Systemic therapy options for pediatric skin diseases are improving
ORLANDO – Because Food and Drug Administration–approved treatment options for medications. However, this scenario is changing, A. Yasmine Kirkorian, MD, said at the ODAC Dermatology, Aesthetic & Surgical Conference.

“I really would like to emphasize that children with severe disease need to be treated,” added Dr. Kirkorian, a pediatric dermatologist at George Washington University, Washington, and Children’s National Health System, where she is interim chief of the division of dermatology.
Current on-label systemic therapies for pediatric skin disease include etanercept for psoriasis (4 years and older), ustekinumab for psoriasis (12 years and older), adalimumab for hidradenitis suppurativa (12 years and older), and omalizumab for chronic idiopathic urticaria (12 years and older). A new addition to the list is dupilumab, which was approved for children and adolescents with atopic dermatitis (AD) aged 12 years and older in 2019, she noted.
Dupilumab is currently being studied in children aged 6 months to 12 years, and other clinical trials are evaluating more options for pediatric patients with AD, alopecia areata, and psoriasis. They include a clinical trial of the oral Janus kinase 3 (JAK3) inhibitor PF-06651600 in patients aged 12 years and older with alopecia areata. Six biologic therapies are being evaluated for psoriasis in patients beginning at 6 years: ixekizumab, secukinumab, ustekinumab, guselkumab, brodalumab, and apremilast.
Some systemic therapies are off-label “but used all the time” for dermatologic diseases in pediatrics, Dr. Kirkorian noted. One example is methotrexate, which is approved by the FDA for acute lymphoblastic leukemia, meningeal leukemia, and juvenile idiopathic arthritis down to infancy. Having existing efficacy and safety data for a medication in a pediatric population, even for a different disease, can be helpful when counseling parents of children with severe dermatologic disease. “If you have something, even in an older population of children, it can be reassuring, or you can use evidence from other diseases,” she said.
While methotrexate is a cheap option and approved by the FDA for other pediatric indications down to infancy, the cons of using it to treat AD in pediatric patients are numerous. Treatment requires a number of blood draws for lab testing, which can be discouraging for younger patients, and the reported adverse effect profile may be concerning to some parents, while “in practice doesn’t really occur,” she said. Methotrexate is a teratogen so is not appropriate for teenagers who are sexually active and not using contraception.
The “biggest problem,” though, is the issue of whether methotrexate is effective, since it doesn’t always work for AD, Dr. Kirkorian said. “Even at the highest doses, I often feel that we fail the atopic children,” as opposed to using it to treat psoriasis, “where you know I’m going to get you on something that works.”
In contrast, cyclosporine is FDA approved down to infancy, and works quickly as a bridge to other therapy, and is not expensive, Dr. Kirkorian said. Cons include the need for blood draws, blood pressure checks, drug interactions, and adverse effects, she noted, adding that she tries to use cyclosporine as a bridge to on-label and off-label dupilumab.
Even with FDA approval for dupilumab down to age 12 years, she said it can be difficult to get insurance approval for the on-label treatment for patients in this age group with AD, before they first fail other therapies (even with off-label systemic drugs). For patients under age 12 years, getting approval is even more challenging and requires rigorous documentation of what therapies the child has failed, and how it has affected their quality of life, she said.
“If you send in a letter to the insurance company without an IGA [Investigator Global Assessment] or SCORAD, you’re going to get rejected,” Dr. Kirkorian said. In addition to those two measures, she provides “everything else,” including the impact of the disease on quality of life of patients, and school, she said, adding, “Did they miss school, did they get hospitalized for infections? And do they have comorbid diseases that might help you get approval?”
In pediatric patients with psoriasis, common issues are more likely to be about how insurance dictates step therapy. She has often found that young children may stop responding to etanercept after a few years, which can justify a switch to ustekinumab or a new treatment in a clinical trial, she said. Adolescents with psoriasis can receive ustekinumab, which is approved for psoriasis in patients aged 12-17 years, she said, noting that the infrequent ustekinumab dosing schedule is often beneficial in this population.
When all other approved options fail for young patients with psoriasis, justifying off-label use isn’t always easy. “You just have to make a justification based on the literature, even though it’s off label,” citing available safety information for other diseases, and “demonstrate over and over the impact on quality of life,” which works “most of the time,” Dr. Kirkorian said.
She reported having no conflicts of interest.
ORLANDO – Because Food and Drug Administration–approved treatment options for medications. However, this scenario is changing, A. Yasmine Kirkorian, MD, said at the ODAC Dermatology, Aesthetic & Surgical Conference.
“I really would like to emphasize that children with severe disease need to be treated,” added Dr. Kirkorian, a pediatric dermatologist at George Washington University, Washington, and Children’s National Health System, where she is interim chief of the division of dermatology.
Current on-label systemic therapies for pediatric skin disease include etanercept for psoriasis (4 years and older), ustekinumab for psoriasis (12 years and older), adalimumab for hidradenitis suppurativa (12 years and older), and omalizumab for chronic idiopathic urticaria (12 years and older). A new addition to the list is dupilumab, which was approved for children and adolescents with atopic dermatitis (AD) aged 12 years and older in 2019, she noted.
Dupilumab is currently being studied in children aged 6 months to 12 years, and other clinical trials are evaluating more options for pediatric patients with AD, alopecia areata, and psoriasis. They include a clinical trial of the oral Janus kinase 3 (JAK3) inhibitor PF-06651600 in patients aged 12 years and older with alopecia areata. Six biologic therapies are being evaluated for psoriasis in patients beginning at 6 years: ixekizumab, secukinumab, ustekinumab, guselkumab, brodalumab, and apremilast.
Some systemic therapies are off-label “but used all the time” for dermatologic diseases in pediatrics, Dr. Kirkorian noted. One example is methotrexate, which is approved by the FDA for acute lymphoblastic leukemia, meningeal leukemia, and juvenile idiopathic arthritis down to infancy. Having existing efficacy and safety data for a medication in a pediatric population, even for a different disease, can be helpful when counseling parents of children with severe dermatologic disease. “If you have something, even in an older population of children, it can be reassuring, or you can use evidence from other diseases,” she said.
While methotrexate is a cheap option and approved by the FDA for other pediatric indications down to infancy, the cons of using it to treat AD in pediatric patients are numerous. Treatment requires a number of blood draws for lab testing, which can be discouraging for younger patients, and the reported adverse effect profile may be concerning to some parents, while “in practice doesn’t really occur,” she said. Methotrexate is a teratogen so is not appropriate for teenagers who are sexually active and not using contraception.
The “biggest problem,” though, is the issue of whether methotrexate is effective, since it doesn’t always work for AD, Dr. Kirkorian said. “Even at the highest doses, I often feel that we fail the atopic children,” as opposed to using it to treat psoriasis, “where you know I’m going to get you on something that works.”
In contrast, cyclosporine is FDA approved down to infancy, and works quickly as a bridge to other therapy, and is not expensive, Dr. Kirkorian said. Cons include the need for blood draws, blood pressure checks, drug interactions, and adverse effects, she noted, adding that she tries to use cyclosporine as a bridge to on-label and off-label dupilumab.
Even with FDA approval for dupilumab down to age 12 years, she said it can be difficult to get insurance approval for the on-label treatment for patients in this age group with AD, before they first fail other therapies (even with off-label systemic drugs). For patients under age 12 years, getting approval is even more challenging and requires rigorous documentation of what therapies the child has failed, and how it has affected their quality of life, she said.
“If you send in a letter to the insurance company without an IGA [Investigator Global Assessment] or SCORAD, you’re going to get rejected,” Dr. Kirkorian said. In addition to those two measures, she provides “everything else,” including the impact of the disease on quality of life of patients, and school, she said, adding, “Did they miss school, did they get hospitalized for infections? And do they have comorbid diseases that might help you get approval?”
In pediatric patients with psoriasis, common issues are more likely to be about how insurance dictates step therapy. She has often found that young children may stop responding to etanercept after a few years, which can justify a switch to ustekinumab or a new treatment in a clinical trial, she said. Adolescents with psoriasis can receive ustekinumab, which is approved for psoriasis in patients aged 12-17 years, she said, noting that the infrequent ustekinumab dosing schedule is often beneficial in this population.
When all other approved options fail for young patients with psoriasis, justifying off-label use isn’t always easy. “You just have to make a justification based on the literature, even though it’s off label,” citing available safety information for other diseases, and “demonstrate over and over the impact on quality of life,” which works “most of the time,” Dr. Kirkorian said.
She reported having no conflicts of interest.
ORLANDO – Because Food and Drug Administration–approved treatment options for medications. However, this scenario is changing, A. Yasmine Kirkorian, MD, said at the ODAC Dermatology, Aesthetic & Surgical Conference.
“I really would like to emphasize that children with severe disease need to be treated,” added Dr. Kirkorian, a pediatric dermatologist at George Washington University, Washington, and Children’s National Health System, where she is interim chief of the division of dermatology.
Current on-label systemic therapies for pediatric skin disease include etanercept for psoriasis (4 years and older), ustekinumab for psoriasis (12 years and older), adalimumab for hidradenitis suppurativa (12 years and older), and omalizumab for chronic idiopathic urticaria (12 years and older). A new addition to the list is dupilumab, which was approved for children and adolescents with atopic dermatitis (AD) aged 12 years and older in 2019, she noted.
Dupilumab is currently being studied in children aged 6 months to 12 years, and other clinical trials are evaluating more options for pediatric patients with AD, alopecia areata, and psoriasis. They include a clinical trial of the oral Janus kinase 3 (JAK3) inhibitor PF-06651600 in patients aged 12 years and older with alopecia areata. Six biologic therapies are being evaluated for psoriasis in patients beginning at 6 years: ixekizumab, secukinumab, ustekinumab, guselkumab, brodalumab, and apremilast.
Some systemic therapies are off-label “but used all the time” for dermatologic diseases in pediatrics, Dr. Kirkorian noted. One example is methotrexate, which is approved by the FDA for acute lymphoblastic leukemia, meningeal leukemia, and juvenile idiopathic arthritis down to infancy. Having existing efficacy and safety data for a medication in a pediatric population, even for a different disease, can be helpful when counseling parents of children with severe dermatologic disease. “If you have something, even in an older population of children, it can be reassuring, or you can use evidence from other diseases,” she said.
While methotrexate is a cheap option and approved by the FDA for other pediatric indications down to infancy, the cons of using it to treat AD in pediatric patients are numerous. Treatment requires a number of blood draws for lab testing, which can be discouraging for younger patients, and the reported adverse effect profile may be concerning to some parents, while “in practice doesn’t really occur,” she said. Methotrexate is a teratogen so is not appropriate for teenagers who are sexually active and not using contraception.
The “biggest problem,” though, is the issue of whether methotrexate is effective, since it doesn’t always work for AD, Dr. Kirkorian said. “Even at the highest doses, I often feel that we fail the atopic children,” as opposed to using it to treat psoriasis, “where you know I’m going to get you on something that works.”
In contrast, cyclosporine is FDA approved down to infancy, and works quickly as a bridge to other therapy, and is not expensive, Dr. Kirkorian said. Cons include the need for blood draws, blood pressure checks, drug interactions, and adverse effects, she noted, adding that she tries to use cyclosporine as a bridge to on-label and off-label dupilumab.
Even with FDA approval for dupilumab down to age 12 years, she said it can be difficult to get insurance approval for the on-label treatment for patients in this age group with AD, before they first fail other therapies (even with off-label systemic drugs). For patients under age 12 years, getting approval is even more challenging and requires rigorous documentation of what therapies the child has failed, and how it has affected their quality of life, she said.
“If you send in a letter to the insurance company without an IGA [Investigator Global Assessment] or SCORAD, you’re going to get rejected,” Dr. Kirkorian said. In addition to those two measures, she provides “everything else,” including the impact of the disease on quality of life of patients, and school, she said, adding, “Did they miss school, did they get hospitalized for infections? And do they have comorbid diseases that might help you get approval?”
In pediatric patients with psoriasis, common issues are more likely to be about how insurance dictates step therapy. She has often found that young children may stop responding to etanercept after a few years, which can justify a switch to ustekinumab or a new treatment in a clinical trial, she said. Adolescents with psoriasis can receive ustekinumab, which is approved for psoriasis in patients aged 12-17 years, she said, noting that the infrequent ustekinumab dosing schedule is often beneficial in this population.
When all other approved options fail for young patients with psoriasis, justifying off-label use isn’t always easy. “You just have to make a justification based on the literature, even though it’s off label,” citing available safety information for other diseases, and “demonstrate over and over the impact on quality of life,” which works “most of the time,” Dr. Kirkorian said.
She reported having no conflicts of interest.
EXPERT ANALYSIS FROM ODAC 2020
Streamlining the transition from pediatric to adult care
Diabetes is a complex disease with a range of nuanced therapy options and a plethora of risk factors that could significantly affect patient quality of life and long-term outcomes. From the outset, after diagnosis, a selected regimen has to be meticulously tailored to a patient’s clinical needs and monitored over time, and many other nonclinical variables, such as patient preference, social history, access to care, and support systems, as well as the cost of the drugs and its impact on the patient, must also be considered.
The increase in the incidence of youth-onset diabetes means that more young adults are making the transition from pediatric to adult care, and careful care coordination is paramount at the handover point to ensure that a full and complete account of the history gets transferred to the adult-care provider.
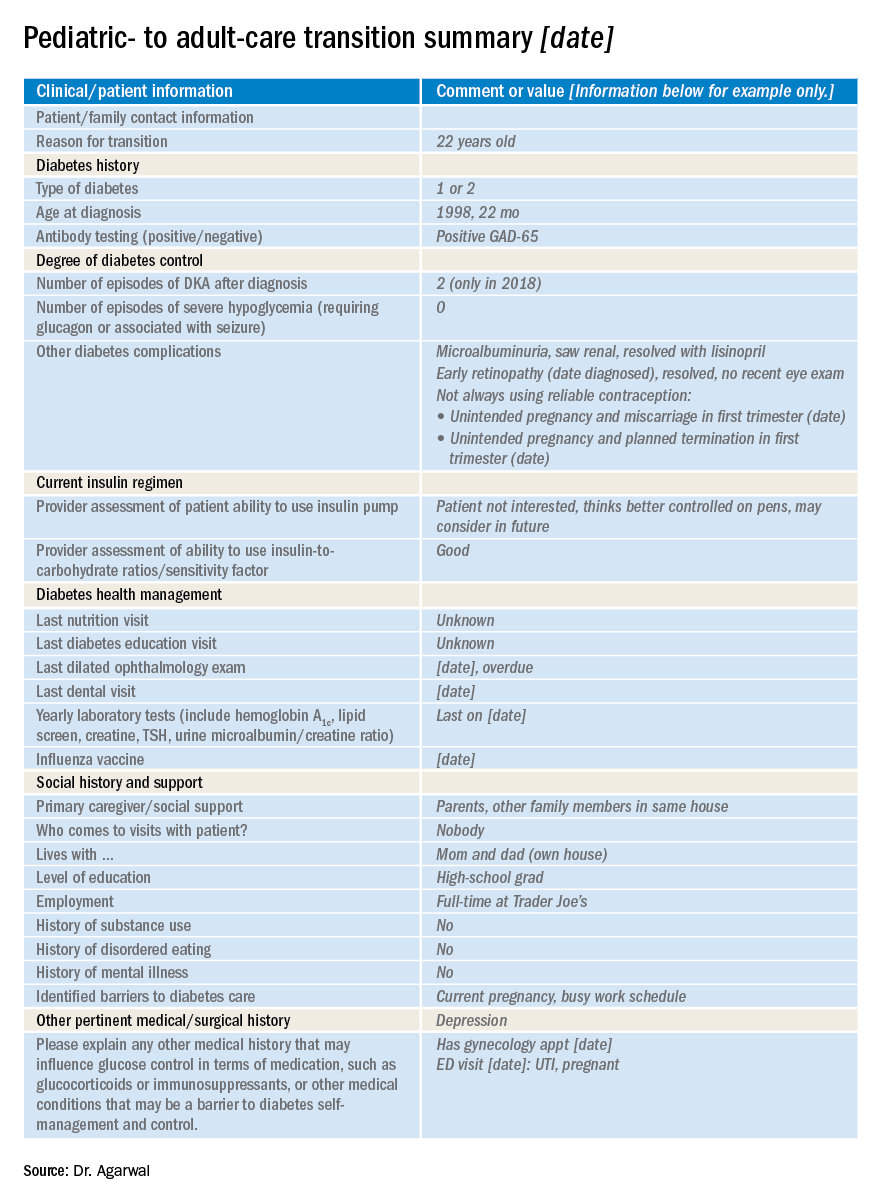
So how do you distill the information from all those records (on paper and online) that you’ve accumulated during the time you’ve been treating a young adult who is now transitioning to adult care?
Transition summary
One resource that can facilitate this handover is the transition summary. It effectively consolidates and packages the aforementioned aspects of care and patient history so that the adult-care provider does not have to collect the patient’s history from the start. The transition summary should not be confused with the discharge or medical summary, which focuses only on the preceding clinical care.
It is important to stress at this stage that collaboration between the pediatric- and adult-care providers is crucial to the success of such a summary, from its creation, to its implementation, and through the subsequent and inevitable revisions and updates.
Benefits all around
After we introduced the transition summary at my institution, we found that the average initial patient visit with the new adult-care provider decreased by 12 minutes (with a range of 6-19 min). The adult-care providers welcomed receiving such detailed, important patient information packaged in a concise and readily accessible format. It helped them identify the preceding care team members, which facilitated continuity of care, and it also helped them forge a better therapeutic relationship with the patient earlier on in their engagement.

We also learned that patients were more comfortable with the transition, and the referring providers were relieved and reassured that their patients would continue to receive personalized care with the new adult-care provider.
At a personal level, I found I was less stressed as I could spend better-quality clinical time with patients. And I got to eliminate those unwieldy stacks of medical records since getting buy-in from divisional and IT leadership enabled us to automate the entire process of information transfer.
It is important to note that the patient has to consent to release of medical records to other institutions.
Setting up the summary
At our clinic, I started out by adapting the transition summary from guidelines provided by the Endocrine Society to make a template. Then, in collaboration with my pediatric colleagues, I removed and added information so that the revised document would contain information that is vitally important and not readily available in the chart and would be feasible to fill out. For example, we included details such as the patient’s psychosocial history, an estimation of the patient barriers to diabetes management, family relationship issues, and the patient’s reasons for not adopting advanced diabetes technology (see accompanying example of a transition summary) .
I kept the summary brief, at two pages, and piloted it with referring providers who were interested in using the summary and with related supporting services. I also sought buy-in from my institution. This meant that I needed pediatric and adult divisional leadership support, which offered me information technology, resources, and expertise to automate the summary within the electronic health record. Once I had feedback from would-be users, we revised and updated the summary. We set up training for staff, including pediatric providers, nurse practitioners, social workers, and nurses who could fill out the summary, and ultimately succeeded in making it mandatory that the adult-care provider receive a summary before scheduling or seeing the transfer patient.
I started out with a paper version, and once we’d refined the questions, we incorporated it into the electronic medical record.
The information we use in our summary is grouped under the following headings:
- Reason for transition.
- Diabetes type.
- Degree of diabetes control.
- Type of insulin therapy and supplies.
- Current and former insulin regimen: reasons for discontinuation of any therapies or reluctance to start any therapies.
- Diabetes health maintenance.
- Social history and support, including living situation, main social support network, child protective services involvement.
- Other pertinent medical surgical history, including psychiatric disease.
Tips and takeaways
Top of the list of takeaways is that you should make the final document work for you, your colleagues, and ultimately, your patients – customize it as you see fit, but be sure to keep it short and easy to fill out. Make a note as you start using it in practice of what you think might be missing from the chart and whether updates are needed. If you can, it’s a great idea to fold the transfer summary into the electronic medical record, though it’s not imperative. Care coordination is key to successful transfer of patients, whether from pediatric to adult care or hospital to home. A small change to work flow can result in a huge change in patient and provider satisfaction, as well as a reduction in visit times.
Dr. Agarwal is director of the Supporting Emerging Adults With Diabetes (SEAD) program at Montefiore Medical Center and assistant professor of medicine at Albert Einstein College of Medicine, New York. She reports no disclosures or financial conflicts of interest. Write to her at cenews@mdedge.com.
Diabetes is a complex disease with a range of nuanced therapy options and a plethora of risk factors that could significantly affect patient quality of life and long-term outcomes. From the outset, after diagnosis, a selected regimen has to be meticulously tailored to a patient’s clinical needs and monitored over time, and many other nonclinical variables, such as patient preference, social history, access to care, and support systems, as well as the cost of the drugs and its impact on the patient, must also be considered.
The increase in the incidence of youth-onset diabetes means that more young adults are making the transition from pediatric to adult care, and careful care coordination is paramount at the handover point to ensure that a full and complete account of the history gets transferred to the adult-care provider.
So how do you distill the information from all those records (on paper and online) that you’ve accumulated during the time you’ve been treating a young adult who is now transitioning to adult care?
Transition summary
One resource that can facilitate this handover is the transition summary. It effectively consolidates and packages the aforementioned aspects of care and patient history so that the adult-care provider does not have to collect the patient’s history from the start. The transition summary should not be confused with the discharge or medical summary, which focuses only on the preceding clinical care.
It is important to stress at this stage that collaboration between the pediatric- and adult-care providers is crucial to the success of such a summary, from its creation, to its implementation, and through the subsequent and inevitable revisions and updates.
Benefits all around
After we introduced the transition summary at my institution, we found that the average initial patient visit with the new adult-care provider decreased by 12 minutes (with a range of 6-19 min). The adult-care providers welcomed receiving such detailed, important patient information packaged in a concise and readily accessible format. It helped them identify the preceding care team members, which facilitated continuity of care, and it also helped them forge a better therapeutic relationship with the patient earlier on in their engagement.
We also learned that patients were more comfortable with the transition, and the referring providers were relieved and reassured that their patients would continue to receive personalized care with the new adult-care provider.
At a personal level, I found I was less stressed as I could spend better-quality clinical time with patients. And I got to eliminate those unwieldy stacks of medical records since getting buy-in from divisional and IT leadership enabled us to automate the entire process of information transfer.
It is important to note that the patient has to consent to release of medical records to other institutions.
Setting up the summary
At our clinic, I started out by adapting the transition summary from guidelines provided by the Endocrine Society to make a template. Then, in collaboration with my pediatric colleagues, I removed and added information so that the revised document would contain information that is vitally important and not readily available in the chart and would be feasible to fill out. For example, we included details such as the patient’s psychosocial history, an estimation of the patient barriers to diabetes management, family relationship issues, and the patient’s reasons for not adopting advanced diabetes technology (see accompanying example of a transition summary) .
I kept the summary brief, at two pages, and piloted it with referring providers who were interested in using the summary and with related supporting services. I also sought buy-in from my institution. This meant that I needed pediatric and adult divisional leadership support, which offered me information technology, resources, and expertise to automate the summary within the electronic health record. Once I had feedback from would-be users, we revised and updated the summary. We set up training for staff, including pediatric providers, nurse practitioners, social workers, and nurses who could fill out the summary, and ultimately succeeded in making it mandatory that the adult-care provider receive a summary before scheduling or seeing the transfer patient.
I started out with a paper version, and once we’d refined the questions, we incorporated it into the electronic medical record.
The information we use in our summary is grouped under the following headings:
- Reason for transition.
- Diabetes type.
- Degree of diabetes control.
- Type of insulin therapy and supplies.
- Current and former insulin regimen: reasons for discontinuation of any therapies or reluctance to start any therapies.
- Diabetes health maintenance.
- Social history and support, including living situation, main social support network, child protective services involvement.
- Other pertinent medical surgical history, including psychiatric disease.
Tips and takeaways
Top of the list of takeaways is that you should make the final document work for you, your colleagues, and ultimately, your patients – customize it as you see fit, but be sure to keep it short and easy to fill out. Make a note as you start using it in practice of what you think might be missing from the chart and whether updates are needed. If you can, it’s a great idea to fold the transfer summary into the electronic medical record, though it’s not imperative. Care coordination is key to successful transfer of patients, whether from pediatric to adult care or hospital to home. A small change to work flow can result in a huge change in patient and provider satisfaction, as well as a reduction in visit times.
Dr. Agarwal is director of the Supporting Emerging Adults With Diabetes (SEAD) program at Montefiore Medical Center and assistant professor of medicine at Albert Einstein College of Medicine, New York. She reports no disclosures or financial conflicts of interest. Write to her at cenews@mdedge.com.
Diabetes is a complex disease with a range of nuanced therapy options and a plethora of risk factors that could significantly affect patient quality of life and long-term outcomes. From the outset, after diagnosis, a selected regimen has to be meticulously tailored to a patient’s clinical needs and monitored over time, and many other nonclinical variables, such as patient preference, social history, access to care, and support systems, as well as the cost of the drugs and its impact on the patient, must also be considered.
The increase in the incidence of youth-onset diabetes means that more young adults are making the transition from pediatric to adult care, and careful care coordination is paramount at the handover point to ensure that a full and complete account of the history gets transferred to the adult-care provider.
So how do you distill the information from all those records (on paper and online) that you’ve accumulated during the time you’ve been treating a young adult who is now transitioning to adult care?
Transition summary
One resource that can facilitate this handover is the transition summary. It effectively consolidates and packages the aforementioned aspects of care and patient history so that the adult-care provider does not have to collect the patient’s history from the start. The transition summary should not be confused with the discharge or medical summary, which focuses only on the preceding clinical care.
It is important to stress at this stage that collaboration between the pediatric- and adult-care providers is crucial to the success of such a summary, from its creation, to its implementation, and through the subsequent and inevitable revisions and updates.
Benefits all around
After we introduced the transition summary at my institution, we found that the average initial patient visit with the new adult-care provider decreased by 12 minutes (with a range of 6-19 min). The adult-care providers welcomed receiving such detailed, important patient information packaged in a concise and readily accessible format. It helped them identify the preceding care team members, which facilitated continuity of care, and it also helped them forge a better therapeutic relationship with the patient earlier on in their engagement.
We also learned that patients were more comfortable with the transition, and the referring providers were relieved and reassured that their patients would continue to receive personalized care with the new adult-care provider.
At a personal level, I found I was less stressed as I could spend better-quality clinical time with patients. And I got to eliminate those unwieldy stacks of medical records since getting buy-in from divisional and IT leadership enabled us to automate the entire process of information transfer.
It is important to note that the patient has to consent to release of medical records to other institutions.
Setting up the summary
At our clinic, I started out by adapting the transition summary from guidelines provided by the Endocrine Society to make a template. Then, in collaboration with my pediatric colleagues, I removed and added information so that the revised document would contain information that is vitally important and not readily available in the chart and would be feasible to fill out. For example, we included details such as the patient’s psychosocial history, an estimation of the patient barriers to diabetes management, family relationship issues, and the patient’s reasons for not adopting advanced diabetes technology (see accompanying example of a transition summary) .
I kept the summary brief, at two pages, and piloted it with referring providers who were interested in using the summary and with related supporting services. I also sought buy-in from my institution. This meant that I needed pediatric and adult divisional leadership support, which offered me information technology, resources, and expertise to automate the summary within the electronic health record. Once I had feedback from would-be users, we revised and updated the summary. We set up training for staff, including pediatric providers, nurse practitioners, social workers, and nurses who could fill out the summary, and ultimately succeeded in making it mandatory that the adult-care provider receive a summary before scheduling or seeing the transfer patient.
I started out with a paper version, and once we’d refined the questions, we incorporated it into the electronic medical record.
The information we use in our summary is grouped under the following headings:
- Reason for transition.
- Diabetes type.
- Degree of diabetes control.
- Type of insulin therapy and supplies.
- Current and former insulin regimen: reasons for discontinuation of any therapies or reluctance to start any therapies.
- Diabetes health maintenance.
- Social history and support, including living situation, main social support network, child protective services involvement.
- Other pertinent medical surgical history, including psychiatric disease.
Tips and takeaways
Top of the list of takeaways is that you should make the final document work for you, your colleagues, and ultimately, your patients – customize it as you see fit, but be sure to keep it short and easy to fill out. Make a note as you start using it in practice of what you think might be missing from the chart and whether updates are needed. If you can, it’s a great idea to fold the transfer summary into the electronic medical record, though it’s not imperative. Care coordination is key to successful transfer of patients, whether from pediatric to adult care or hospital to home. A small change to work flow can result in a huge change in patient and provider satisfaction, as well as a reduction in visit times.
Dr. Agarwal is director of the Supporting Emerging Adults With Diabetes (SEAD) program at Montefiore Medical Center and assistant professor of medicine at Albert Einstein College of Medicine, New York. She reports no disclosures or financial conflicts of interest. Write to her at cenews@mdedge.com.
Acknowledging Disparities in Dementia Care for Increasingly Diverse Ethnoracial Patient Populations
Alzheimer disease and related dementias are a global health concern, affecting nearly 47 million people worldwide. Alzheimer disease and related dementias were among the top 10 causes of death worldwide in 2015 and are expected to increase by 10 million cases annually.1 Despite the ethnic diversity of the US, there are considerable gaps in the literature regarding dementia and how it is diagnosed and treated among many ethnic and racial groups.
In 2012, President Barack Obama signed a declaration with the intention of decreasing ethnoracial disparities in Alzheimer disease research and treatment by increasing clinical care, research, and services targeted to racial and ethnic minorities.2 Despite that declaration, in the US there are gaps in access to care for the geriatric population in general. The American Geriatrics Society estimates that the US has fewer than half the needed number of practicing geriatricians. In 2016, there was 1 geriatrician for every 1,924 Americans aged ≥ 65 years.3 Furthermore, health care providers (HCPs) are often not of the same ethnicity or adequately trained to assess and build relationships with ethnically and racially diverse populations.2 Given the projected growth in the numbers of individuals worldwide with dementia, we have a responsibility to continue to develop strategies to provide more inclusive care.
By 2060, minority populations aged ≥ 65 years are expected to represent 45% of the US population, up from 22% in 2014.4 The growth of racial and ethnic minority groups are expected to exceed the growth of the non-Hispanic white population in the next few decades. By 2060, it is estimated that the US population will increase by 75% for non-Hispanic whites, 172% for African Americans, 270% for Asian and Pacific Islanders, 274% for American Indian and Alaska Natives, and 391% for Hispanics.4
A growing body of evidence suggests that Alzheimer disease and related dementias may disproportionately afflict minority groups in the US, which will become quite significant in the years ahead. The Alzheimer’s Association estimates that the prevalence of Alzheimer disease and other dementias among those aged > 65 years, is about twice the rate in African Americans and about 1.5 times the rate in Hispanics when compared with non-Hispanic whites.5 While increases in the incidence of Alzheimer disease and related dementias in non-Hispanic whites is expected to plateau around 2050, its incidence in ethnic and racial minority groups will continue to grow, especially among Hispanics.4 This stark realization provides additional compelling reasons for the US to develop preventative interventions or treatment options that may help delay the onset of the disease and to improve the quality of life of those with the disease or caregiving for those afflicted with it. Culturally competent care of these individuals is paramount.
Diagnosis
Early and accurate diagnosis of individuals with dementia confers many benefits, including early treatment; clinical trial participation; management of comorbid conditions; training, education, and support for patients and families; and legal, financial, and end of life care planning.3 Beyond the logistical concerns (such as HCP shortages), one of the challenges of assessing minority groups is finding staff who are culturally competent or speak the language necessary to accurately communicate and interact with these subgroups. Hispanics and African Americans often receive delayed or inadequate health care services or are diagnosed in an emergency department or other nontraditional setting.5
Even those individuals seeking or receiving care in primary care settings are not always forthcoming about their cognitive status. Only 56% of respondents in a recent survey of patients who had experienced subjective cognitive decline reported that they had discussed it with their HCP.4 This reticence is thought to be influenced by multiple factors, including distrust of the medical establishment, religious or spiritual beliefs, cultural or family beliefs and expectations about geriatric care, and lack of understanding about normal aging vs cognitive disorders. Furthermore, the sensitivity and specificity of current diagnostic tests for dementia have been questioned for nonwhite populations given the clinical presentation of dementia can vary across ethnoracial groups.5
As Luria noted, cognitive assessment tools developed and validated for use with one culture frequently results in experimental failures and are not valid for use with other cultural groups.1 Cognitive testing results are influenced by educational and cultural factors, and this is one of the challenges in correctly diagnosing those of differing ethnoracial backgrounds. Individuals in racial and ethnic minorities may have limited formal education and/or high illiteracy rates and/or cultural nuances to problem solving, thinking, and memory that may not be reflected in current assessment tools.1
There is hope that testing bias could be altered or eliminated using neuroimaging or biomarkers. However, the Alzheimer’s Disease Neuroimaging Initiative study of patients in the US and Canada included < 5% African American or Hispanic participants in its total sample. Few studies have systematically examined ethnoracial differences in amyloid positron emission tomography, and none have been published to date in ethnoracially diverse groups that assess the more recently developed tau imaging agents.1
Diversity Among Caregivers
The research community must make greater efforts to improve recruitment of more diverse populations into clinical trials. Recent efforts by the National Institute on Aging in conjunction with the Alzheimer’s Association include developing a national strategy for clinical research recruitment and retention with an emphasis on local and diverse populations. This strategy should include various training modules, webinars, and similar educational opportunities for researchers and clinical HCPs, including HCPs from diverse ethnoracial backgrounds, to implement culturally appropriate research methodologies across these diverse groups. It is important that these educational materials be disseminated to caregivers in a way they can comprehend, as the impact on caregivers of those with Alzheimer disease and related dementias is considerable.
The US currently has 7 unpaid caregivers for every adult in the high-risk group of patients aged ≥ 65 years, but this will decline to a ratio of 4:1 by 2030.4 More than two-thirds of caregivers are non-Hispanic white, while 10% are African American, 8% are Hispanic, and 5% are Asian.3 About 34% of caregivers are themselves aged ≥ 65 years and are at risk for declines in their own health given the time and financial requirements of caring for someone else.3 In 2017, the 16.1 million family and other unpaid caregivers of people with dementia provided an estimated 18.4 billion hours of unpaid care, often resulting in considerable financial strain for these individuals. More than half of the caregivers report providing ≥ 21 hours of care per week; and 42% reported providing an average of 9 hours of care per day for people with dementia.
Caregivers report increased stress, sleep deprivation, depression and anxiety, and uncertainty in their ability to provide quality care to the individual with Alzheimer or a related dementia.3 The disproportionate prevalence of Alzheimer disease and other dementias in racially and ethnically diverse populations could further magnify already existing socioeconomic and other disparities and potentially lead to worsening of health outcomes in these groups.4 Given that minority populations tend to cluster geographically, community partnerships with local churches, senior centers, community centers, and other nontraditional settings may offer better opportunities for connecting with caregivers.
Conclusions
The growth and increasing diversity of the US older adult population in the coming decades require us as HCPs, researchers, and educators to dedicate more resources to ethnoracially diverse populations. There are still a great many unknowns about Alzheimer disease and dementia, most especially among nonwhites. Research, clinical care, and education must focus on outreach to marginalized groups so we may better be able to diagnose and treat the fastest growing older adult populations in the US. A complex combination of educational, cultural, social, and environmental factors likely contribute to delayed diagnosis and care of these groups, as well as lack of access to medical care, research venues, and trust issues between minority groups and the medical establishment. We all have an obligation to acknowledge these disparities and elicit the support of our colleagues and workplaces to raise awareness and dedicate necessary resources to this growing concern.
1. Babulal GM, Quiroz YT, Albensi BC, et al; International Society to Advance Alzheimer’s Research and Treatment, Alzheimer’s Association. Perspectives on ethnic and racial disparities in Alzheimer’s disease and related dementias: update and areas of immediate need. Alzheimers Dement. 2019;15(2):292-312.
2. Brewster P, Barnes L, Haan M, et al. Progress and future challenges in aging and diversity research in the United States. Alzheimers Dement. 2019;15(7):995-1003.
3. Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019;15(3):321-387.
4. Matthews KA, Xu W, Gaglioti AH, et al. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015-2060) in adults aged ≥65 years. Alzheimers Dement. 2019;15(1):17-24.
5. Chin AL, Negash S, Hamilton R. Diversity and disparity in dementia: the impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25(3):187-195.
Alzheimer disease and related dementias are a global health concern, affecting nearly 47 million people worldwide. Alzheimer disease and related dementias were among the top 10 causes of death worldwide in 2015 and are expected to increase by 10 million cases annually.1 Despite the ethnic diversity of the US, there are considerable gaps in the literature regarding dementia and how it is diagnosed and treated among many ethnic and racial groups.
In 2012, President Barack Obama signed a declaration with the intention of decreasing ethnoracial disparities in Alzheimer disease research and treatment by increasing clinical care, research, and services targeted to racial and ethnic minorities.2 Despite that declaration, in the US there are gaps in access to care for the geriatric population in general. The American Geriatrics Society estimates that the US has fewer than half the needed number of practicing geriatricians. In 2016, there was 1 geriatrician for every 1,924 Americans aged ≥ 65 years.3 Furthermore, health care providers (HCPs) are often not of the same ethnicity or adequately trained to assess and build relationships with ethnically and racially diverse populations.2 Given the projected growth in the numbers of individuals worldwide with dementia, we have a responsibility to continue to develop strategies to provide more inclusive care.
By 2060, minority populations aged ≥ 65 years are expected to represent 45% of the US population, up from 22% in 2014.4 The growth of racial and ethnic minority groups are expected to exceed the growth of the non-Hispanic white population in the next few decades. By 2060, it is estimated that the US population will increase by 75% for non-Hispanic whites, 172% for African Americans, 270% for Asian and Pacific Islanders, 274% for American Indian and Alaska Natives, and 391% for Hispanics.4
A growing body of evidence suggests that Alzheimer disease and related dementias may disproportionately afflict minority groups in the US, which will become quite significant in the years ahead. The Alzheimer’s Association estimates that the prevalence of Alzheimer disease and other dementias among those aged > 65 years, is about twice the rate in African Americans and about 1.5 times the rate in Hispanics when compared with non-Hispanic whites.5 While increases in the incidence of Alzheimer disease and related dementias in non-Hispanic whites is expected to plateau around 2050, its incidence in ethnic and racial minority groups will continue to grow, especially among Hispanics.4 This stark realization provides additional compelling reasons for the US to develop preventative interventions or treatment options that may help delay the onset of the disease and to improve the quality of life of those with the disease or caregiving for those afflicted with it. Culturally competent care of these individuals is paramount.
Diagnosis
Early and accurate diagnosis of individuals with dementia confers many benefits, including early treatment; clinical trial participation; management of comorbid conditions; training, education, and support for patients and families; and legal, financial, and end of life care planning.3 Beyond the logistical concerns (such as HCP shortages), one of the challenges of assessing minority groups is finding staff who are culturally competent or speak the language necessary to accurately communicate and interact with these subgroups. Hispanics and African Americans often receive delayed or inadequate health care services or are diagnosed in an emergency department or other nontraditional setting.5
Even those individuals seeking or receiving care in primary care settings are not always forthcoming about their cognitive status. Only 56% of respondents in a recent survey of patients who had experienced subjective cognitive decline reported that they had discussed it with their HCP.4 This reticence is thought to be influenced by multiple factors, including distrust of the medical establishment, religious or spiritual beliefs, cultural or family beliefs and expectations about geriatric care, and lack of understanding about normal aging vs cognitive disorders. Furthermore, the sensitivity and specificity of current diagnostic tests for dementia have been questioned for nonwhite populations given the clinical presentation of dementia can vary across ethnoracial groups.5
As Luria noted, cognitive assessment tools developed and validated for use with one culture frequently results in experimental failures and are not valid for use with other cultural groups.1 Cognitive testing results are influenced by educational and cultural factors, and this is one of the challenges in correctly diagnosing those of differing ethnoracial backgrounds. Individuals in racial and ethnic minorities may have limited formal education and/or high illiteracy rates and/or cultural nuances to problem solving, thinking, and memory that may not be reflected in current assessment tools.1
There is hope that testing bias could be altered or eliminated using neuroimaging or biomarkers. However, the Alzheimer’s Disease Neuroimaging Initiative study of patients in the US and Canada included < 5% African American or Hispanic participants in its total sample. Few studies have systematically examined ethnoracial differences in amyloid positron emission tomography, and none have been published to date in ethnoracially diverse groups that assess the more recently developed tau imaging agents.1
Diversity Among Caregivers
The research community must make greater efforts to improve recruitment of more diverse populations into clinical trials. Recent efforts by the National Institute on Aging in conjunction with the Alzheimer’s Association include developing a national strategy for clinical research recruitment and retention with an emphasis on local and diverse populations. This strategy should include various training modules, webinars, and similar educational opportunities for researchers and clinical HCPs, including HCPs from diverse ethnoracial backgrounds, to implement culturally appropriate research methodologies across these diverse groups. It is important that these educational materials be disseminated to caregivers in a way they can comprehend, as the impact on caregivers of those with Alzheimer disease and related dementias is considerable.
The US currently has 7 unpaid caregivers for every adult in the high-risk group of patients aged ≥ 65 years, but this will decline to a ratio of 4:1 by 2030.4 More than two-thirds of caregivers are non-Hispanic white, while 10% are African American, 8% are Hispanic, and 5% are Asian.3 About 34% of caregivers are themselves aged ≥ 65 years and are at risk for declines in their own health given the time and financial requirements of caring for someone else.3 In 2017, the 16.1 million family and other unpaid caregivers of people with dementia provided an estimated 18.4 billion hours of unpaid care, often resulting in considerable financial strain for these individuals. More than half of the caregivers report providing ≥ 21 hours of care per week; and 42% reported providing an average of 9 hours of care per day for people with dementia.
Caregivers report increased stress, sleep deprivation, depression and anxiety, and uncertainty in their ability to provide quality care to the individual with Alzheimer or a related dementia.3 The disproportionate prevalence of Alzheimer disease and other dementias in racially and ethnically diverse populations could further magnify already existing socioeconomic and other disparities and potentially lead to worsening of health outcomes in these groups.4 Given that minority populations tend to cluster geographically, community partnerships with local churches, senior centers, community centers, and other nontraditional settings may offer better opportunities for connecting with caregivers.
Conclusions
The growth and increasing diversity of the US older adult population in the coming decades require us as HCPs, researchers, and educators to dedicate more resources to ethnoracially diverse populations. There are still a great many unknowns about Alzheimer disease and dementia, most especially among nonwhites. Research, clinical care, and education must focus on outreach to marginalized groups so we may better be able to diagnose and treat the fastest growing older adult populations in the US. A complex combination of educational, cultural, social, and environmental factors likely contribute to delayed diagnosis and care of these groups, as well as lack of access to medical care, research venues, and trust issues between minority groups and the medical establishment. We all have an obligation to acknowledge these disparities and elicit the support of our colleagues and workplaces to raise awareness and dedicate necessary resources to this growing concern.
Alzheimer disease and related dementias are a global health concern, affecting nearly 47 million people worldwide. Alzheimer disease and related dementias were among the top 10 causes of death worldwide in 2015 and are expected to increase by 10 million cases annually.1 Despite the ethnic diversity of the US, there are considerable gaps in the literature regarding dementia and how it is diagnosed and treated among many ethnic and racial groups.
In 2012, President Barack Obama signed a declaration with the intention of decreasing ethnoracial disparities in Alzheimer disease research and treatment by increasing clinical care, research, and services targeted to racial and ethnic minorities.2 Despite that declaration, in the US there are gaps in access to care for the geriatric population in general. The American Geriatrics Society estimates that the US has fewer than half the needed number of practicing geriatricians. In 2016, there was 1 geriatrician for every 1,924 Americans aged ≥ 65 years.3 Furthermore, health care providers (HCPs) are often not of the same ethnicity or adequately trained to assess and build relationships with ethnically and racially diverse populations.2 Given the projected growth in the numbers of individuals worldwide with dementia, we have a responsibility to continue to develop strategies to provide more inclusive care.
By 2060, minority populations aged ≥ 65 years are expected to represent 45% of the US population, up from 22% in 2014.4 The growth of racial and ethnic minority groups are expected to exceed the growth of the non-Hispanic white population in the next few decades. By 2060, it is estimated that the US population will increase by 75% for non-Hispanic whites, 172% for African Americans, 270% for Asian and Pacific Islanders, 274% for American Indian and Alaska Natives, and 391% for Hispanics.4
A growing body of evidence suggests that Alzheimer disease and related dementias may disproportionately afflict minority groups in the US, which will become quite significant in the years ahead. The Alzheimer’s Association estimates that the prevalence of Alzheimer disease and other dementias among those aged > 65 years, is about twice the rate in African Americans and about 1.5 times the rate in Hispanics when compared with non-Hispanic whites.5 While increases in the incidence of Alzheimer disease and related dementias in non-Hispanic whites is expected to plateau around 2050, its incidence in ethnic and racial minority groups will continue to grow, especially among Hispanics.4 This stark realization provides additional compelling reasons for the US to develop preventative interventions or treatment options that may help delay the onset of the disease and to improve the quality of life of those with the disease or caregiving for those afflicted with it. Culturally competent care of these individuals is paramount.
Diagnosis
Early and accurate diagnosis of individuals with dementia confers many benefits, including early treatment; clinical trial participation; management of comorbid conditions; training, education, and support for patients and families; and legal, financial, and end of life care planning.3 Beyond the logistical concerns (such as HCP shortages), one of the challenges of assessing minority groups is finding staff who are culturally competent or speak the language necessary to accurately communicate and interact with these subgroups. Hispanics and African Americans often receive delayed or inadequate health care services or are diagnosed in an emergency department or other nontraditional setting.5
Even those individuals seeking or receiving care in primary care settings are not always forthcoming about their cognitive status. Only 56% of respondents in a recent survey of patients who had experienced subjective cognitive decline reported that they had discussed it with their HCP.4 This reticence is thought to be influenced by multiple factors, including distrust of the medical establishment, religious or spiritual beliefs, cultural or family beliefs and expectations about geriatric care, and lack of understanding about normal aging vs cognitive disorders. Furthermore, the sensitivity and specificity of current diagnostic tests for dementia have been questioned for nonwhite populations given the clinical presentation of dementia can vary across ethnoracial groups.5
As Luria noted, cognitive assessment tools developed and validated for use with one culture frequently results in experimental failures and are not valid for use with other cultural groups.1 Cognitive testing results are influenced by educational and cultural factors, and this is one of the challenges in correctly diagnosing those of differing ethnoracial backgrounds. Individuals in racial and ethnic minorities may have limited formal education and/or high illiteracy rates and/or cultural nuances to problem solving, thinking, and memory that may not be reflected in current assessment tools.1
There is hope that testing bias could be altered or eliminated using neuroimaging or biomarkers. However, the Alzheimer’s Disease Neuroimaging Initiative study of patients in the US and Canada included < 5% African American or Hispanic participants in its total sample. Few studies have systematically examined ethnoracial differences in amyloid positron emission tomography, and none have been published to date in ethnoracially diverse groups that assess the more recently developed tau imaging agents.1
Diversity Among Caregivers
The research community must make greater efforts to improve recruitment of more diverse populations into clinical trials. Recent efforts by the National Institute on Aging in conjunction with the Alzheimer’s Association include developing a national strategy for clinical research recruitment and retention with an emphasis on local and diverse populations. This strategy should include various training modules, webinars, and similar educational opportunities for researchers and clinical HCPs, including HCPs from diverse ethnoracial backgrounds, to implement culturally appropriate research methodologies across these diverse groups. It is important that these educational materials be disseminated to caregivers in a way they can comprehend, as the impact on caregivers of those with Alzheimer disease and related dementias is considerable.
The US currently has 7 unpaid caregivers for every adult in the high-risk group of patients aged ≥ 65 years, but this will decline to a ratio of 4:1 by 2030.4 More than two-thirds of caregivers are non-Hispanic white, while 10% are African American, 8% are Hispanic, and 5% are Asian.3 About 34% of caregivers are themselves aged ≥ 65 years and are at risk for declines in their own health given the time and financial requirements of caring for someone else.3 In 2017, the 16.1 million family and other unpaid caregivers of people with dementia provided an estimated 18.4 billion hours of unpaid care, often resulting in considerable financial strain for these individuals. More than half of the caregivers report providing ≥ 21 hours of care per week; and 42% reported providing an average of 9 hours of care per day for people with dementia.
Caregivers report increased stress, sleep deprivation, depression and anxiety, and uncertainty in their ability to provide quality care to the individual with Alzheimer or a related dementia.3 The disproportionate prevalence of Alzheimer disease and other dementias in racially and ethnically diverse populations could further magnify already existing socioeconomic and other disparities and potentially lead to worsening of health outcomes in these groups.4 Given that minority populations tend to cluster geographically, community partnerships with local churches, senior centers, community centers, and other nontraditional settings may offer better opportunities for connecting with caregivers.
Conclusions
The growth and increasing diversity of the US older adult population in the coming decades require us as HCPs, researchers, and educators to dedicate more resources to ethnoracially diverse populations. There are still a great many unknowns about Alzheimer disease and dementia, most especially among nonwhites. Research, clinical care, and education must focus on outreach to marginalized groups so we may better be able to diagnose and treat the fastest growing older adult populations in the US. A complex combination of educational, cultural, social, and environmental factors likely contribute to delayed diagnosis and care of these groups, as well as lack of access to medical care, research venues, and trust issues between minority groups and the medical establishment. We all have an obligation to acknowledge these disparities and elicit the support of our colleagues and workplaces to raise awareness and dedicate necessary resources to this growing concern.
1. Babulal GM, Quiroz YT, Albensi BC, et al; International Society to Advance Alzheimer’s Research and Treatment, Alzheimer’s Association. Perspectives on ethnic and racial disparities in Alzheimer’s disease and related dementias: update and areas of immediate need. Alzheimers Dement. 2019;15(2):292-312.
2. Brewster P, Barnes L, Haan M, et al. Progress and future challenges in aging and diversity research in the United States. Alzheimers Dement. 2019;15(7):995-1003.
3. Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019;15(3):321-387.
4. Matthews KA, Xu W, Gaglioti AH, et al. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015-2060) in adults aged ≥65 years. Alzheimers Dement. 2019;15(1):17-24.
5. Chin AL, Negash S, Hamilton R. Diversity and disparity in dementia: the impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25(3):187-195.
1. Babulal GM, Quiroz YT, Albensi BC, et al; International Society to Advance Alzheimer’s Research and Treatment, Alzheimer’s Association. Perspectives on ethnic and racial disparities in Alzheimer’s disease and related dementias: update and areas of immediate need. Alzheimers Dement. 2019;15(2):292-312.
2. Brewster P, Barnes L, Haan M, et al. Progress and future challenges in aging and diversity research in the United States. Alzheimers Dement. 2019;15(7):995-1003.
3. Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019;15(3):321-387.
4. Matthews KA, Xu W, Gaglioti AH, et al. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015-2060) in adults aged ≥65 years. Alzheimers Dement. 2019;15(1):17-24.
5. Chin AL, Negash S, Hamilton R. Diversity and disparity in dementia: the impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25(3):187-195.
Defending the Home Planet
Like me, some of you may have been following the agonizing news about the unprecedented brushfires in Australia that have devastated human, animal, and vegetative life in that country so culturally akin to our own.1 For many people who believe the overwhelming majority of scientific reports on climate change, these apocalyptic fires are an empirical demonstration of the truth of the dire prophecies for the future of our planet. Scientists have demonstrated that although climate change may not have caused the worst fires in Australia’s history, they may have contributed to the conditions that enabled them to spread so far and wide and reach such a destructive intensity.2The heartbreaking pictures of singed koalas and displaced people and the helpless feeling that all I can do from here is donate money set me to thinking about the relationship between the military, health, and climate change, which is the subject of this column.
As I write this in mid-January of a new decade and glance at the weather headlines, I read about an earthquake in Puerto Rico and tornadoes in the southern US. This makes it quite plausible that our comfortable lifestyle and technological civilization could in the coming decades go the way of the dinosaurs, also victims of climate change.
Initially, my first thought about this relationship is a negative one—images of scorched earth policies that stretch back to ancient wars jump to mind. Reflection and research on the topic though suggest that the relationship may be more complicated and conflicted. Alas, I can only touch on a few of the themes in this brief format.
It may not be as obvious that climate change also threatens the military, which is the guardian of that civilization. In 2018, for example, Hurricane Michael caused nearly $5 billion in damages to Tyndall Air Force Base in Florida.3 A year later, the US Department of Defense (DoD) released a report on the effects of climate change as mandated by Congress.4 Even though some congressional critics expressed concern about the report’s lack of depth and detail,5 the report asserted that, “The effects of a changing climate are a national security issue with potential impacts to Department of Defense (DoD or the Department) missions, operational plans, and installations.”4
The US Department of Veterans Affairs (VA) is not immune either. Natural disasters have already disrupted the delivery of health care at its many aging facilities. Climate change was called the “engine”6 driving Hurricane Maria, which in 2017 slammed into Puerto Rico, including its VA medical center, and resulted in shortages of supplies, staff, and basic utilities.7 The facility and the island are still trying to rebuild. In response to weather-exposed vulnerability in VA infrastructure, Senator and presidential candidate Elizabeth Warren (D-MA) and Senator Brian Schatz (D-HI), the ranking member of the Subcommittee on Military Construction, sent a letter to VA leadership arguing that “Strengthening VA’s resilience to climate change is consistent with the agency’s mission to deliver timely, high-quality care and benefits to America’s veterans.”8
It has been reported that the current administration has countered initiatives to prepare for the challenges of providing health care to service members and veterans in a climate changed world.9 Sadly, but predictably, in the politicized federal health care arena, the safety of our service members and, in turn, the domestic and national security and peace that depend on them are caught in the partisan debate over global warming, though it is not likely Congress or federal agency leaders will abandon planning to safeguard service members who will see duty and combat in a radically altered ecology and veterans and who will need to have VA continue to be the reliable safety net despite an increasingly erratic environment.10
Climate change is a divisive political issue; there is a proud tradition of conservatism and self-reliance in military members, active duty and veteran alike. That was why I was surprised and impressed when I saw the results of a recent survey on climate change. In January 2019, 293 active-duty service members and veterans were surveyed.
Participants were selected to reflect the ethnic makeup, educational level, and political allegiance of the military population, which enhanced the validity of the findings.11Participants were asked to indicate whether they believed that the earth was warming secondary to human or natural processes; not growing warmer at all; or whether they were unsure. Similar to the general population, 46% agreed that climate change is anthropogenic.11 More than three-fourths believed it was likely climate change would adversely affect the places they worked, like military installations; 61% thought it likely that global warming could lead to armed conflict over resources. Seven in 10 respondents believed that climate is changing vs 46% who did not. Of respondents who believe climate change is real, 87% see it as a threat to military bases compared with 60% who do not accept the science that the earth is warming.11
This survey, though, is only a small study, and the military and VA are big tents under which a wide range of political persuasions and diverse beliefs co-exist. There are many readers of Federal Practitioner who will no doubt reject nearly every word I have written, in what I know is a controversial column. But it matters that the military and veteran constituency are thinking and speaking about the issue of climate change.11 Why? The answer takes us back to the disaster in Australia. When the fires and the devastation they wrought escalated beyond the powers of the civil authorities to handle, it was the military whose technical skill, coordinated readiness, and personal courage and dedication that was called on to rescue thousands of civilians from the inferno.12 So it will be in our country and around the world when disasters—manmade, natural, or both—threaten to engulf life in all its wondrous variety. Those who battle extreme weather will have unique health needs, and their valiant sacrifices deserve to have health care systems ready and able to treat them.
1. Thompson A. Australia’s bushfires have likely devastated wildlife–and the impact will only get worse. Scientific American. https://www.scientificamerican.com/article/australias-bushfires-have-likely-devastated-wildlife-and-the-impact-will-only-get-worse. Published January 8, 2020. Accessed January 16, 2020.
2. Gibbens S. Intense ‘firestorms’ forming from Australia’s deadly wildfires. https://www.nationalgeographic.com/science/2020/01/australian-wildfires-cause-firestorms. Published January 9, 2020. Accessed January 15, 2020.
3. Shapiro A. Tyndall Air Force Base still faces challenges in recovering from Hurricane Michael. https://www.npr.org/2019/05/31/728754872/tyndall-air-force-base-still-faces-challenges-in-recovering-from-hurricane-micha. Published May 31, 2019. Accessed January 16, 2020.
4. US Department of Defense, Office of the Undersecretary for Acquisition and Sustainment. Report on effects of a changing climate to the Department of Defense. https://www.documentcloud.org/documents/5689153-DoD-Final-Climate-Report.html. Published January 2019. Accessed January 16, 2020.
5. Maucione S. DoD justifies climate change report, says response was mission-centric. https://federalnewsnetwork.com/defense-main/2019/03/dod-justifies-climate-change-report-says-response-was-mission-centric. Published March 28, 2019. Accessed January 16, 2020.
6. Shane L 3rd. Puerto Rico’s VA hospital weathers Maria, but challenges loom. https://www.armytimes.com/veterans/2017/09/22/puerto-ricos-va-hospital-weathers-hurricane-maria-but-challenges-loom. Published September 22, 2017. Accessed January 16, 2020.
7. Hersher R. Climate change was the engine that powered Hurricane Maria’s devastating rains. https://www.npr.org/2019/04/17/714098828/climate-change-was-the-engine-that-powered-hurricane-marias-devastating-rains. Published April 17, 2019. Accessed January 16, 2020.
8. Senators Warren and Schatz request an update from the Department of Veterans Affairs on efforts to build resilience to climate change [press release]. https://www.warren.senate.gov/oversight/letters/senators-warren-and-schatz-request-an-update-from-the-department-of-veterans-affairs-on-efforts-to-build-resilience-to-climate-change. Published October 1, 2019. Accessed January 16, 2020.
9. Simkins JD. Navy quietly ends climate change task force, reversing Obama initiative. https://www.navytimes.com/off-duty/military-culture/2019/08/26/navy-quietly-ends-climate-change-task-force-reversing-obama-initiative. Published August 26, 2019. Accessed January 16, 2020.
10. Eilperin J, Dennis B, Ryan M. As White House questions climate change, U.S. military is planning for it. https://www.washingtonpost.com/national/health-science/as-white-house-questions-climate-change-us-military-is-planning-for-it/2019/04/08/78142546-57c0-11e9-814f-e2f46684196e_story.html. Published April 8, 2019. Accessed January 16, 2020.
11. Motta M, Spindel J, Ralston R. Veterans are concerned about climate change and that matters. http://theconversation.com/veterans-are-concerned-about-climate-change-and-that-matters-110685. Published March 8, 2019. Accessed January 16, 2020.
12. Albeck-Ripka L, Kwai I, Fuller T, Tarabay J. ‘It’s an atomic bomb’: Australia deploys military as fires spread. https://www.nytimes.com/2020/01/04/world/australia/fires-military.html. Updated January 5, 2020. Accessed January 18, 2020.
Like me, some of you may have been following the agonizing news about the unprecedented brushfires in Australia that have devastated human, animal, and vegetative life in that country so culturally akin to our own.1 For many people who believe the overwhelming majority of scientific reports on climate change, these apocalyptic fires are an empirical demonstration of the truth of the dire prophecies for the future of our planet. Scientists have demonstrated that although climate change may not have caused the worst fires in Australia’s history, they may have contributed to the conditions that enabled them to spread so far and wide and reach such a destructive intensity.2The heartbreaking pictures of singed koalas and displaced people and the helpless feeling that all I can do from here is donate money set me to thinking about the relationship between the military, health, and climate change, which is the subject of this column.
As I write this in mid-January of a new decade and glance at the weather headlines, I read about an earthquake in Puerto Rico and tornadoes in the southern US. This makes it quite plausible that our comfortable lifestyle and technological civilization could in the coming decades go the way of the dinosaurs, also victims of climate change.
Initially, my first thought about this relationship is a negative one—images of scorched earth policies that stretch back to ancient wars jump to mind. Reflection and research on the topic though suggest that the relationship may be more complicated and conflicted. Alas, I can only touch on a few of the themes in this brief format.
It may not be as obvious that climate change also threatens the military, which is the guardian of that civilization. In 2018, for example, Hurricane Michael caused nearly $5 billion in damages to Tyndall Air Force Base in Florida.3 A year later, the US Department of Defense (DoD) released a report on the effects of climate change as mandated by Congress.4 Even though some congressional critics expressed concern about the report’s lack of depth and detail,5 the report asserted that, “The effects of a changing climate are a national security issue with potential impacts to Department of Defense (DoD or the Department) missions, operational plans, and installations.”4
The US Department of Veterans Affairs (VA) is not immune either. Natural disasters have already disrupted the delivery of health care at its many aging facilities. Climate change was called the “engine”6 driving Hurricane Maria, which in 2017 slammed into Puerto Rico, including its VA medical center, and resulted in shortages of supplies, staff, and basic utilities.7 The facility and the island are still trying to rebuild. In response to weather-exposed vulnerability in VA infrastructure, Senator and presidential candidate Elizabeth Warren (D-MA) and Senator Brian Schatz (D-HI), the ranking member of the Subcommittee on Military Construction, sent a letter to VA leadership arguing that “Strengthening VA’s resilience to climate change is consistent with the agency’s mission to deliver timely, high-quality care and benefits to America’s veterans.”8
It has been reported that the current administration has countered initiatives to prepare for the challenges of providing health care to service members and veterans in a climate changed world.9 Sadly, but predictably, in the politicized federal health care arena, the safety of our service members and, in turn, the domestic and national security and peace that depend on them are caught in the partisan debate over global warming, though it is not likely Congress or federal agency leaders will abandon planning to safeguard service members who will see duty and combat in a radically altered ecology and veterans and who will need to have VA continue to be the reliable safety net despite an increasingly erratic environment.10
Climate change is a divisive political issue; there is a proud tradition of conservatism and self-reliance in military members, active duty and veteran alike. That was why I was surprised and impressed when I saw the results of a recent survey on climate change. In January 2019, 293 active-duty service members and veterans were surveyed.
Participants were selected to reflect the ethnic makeup, educational level, and political allegiance of the military population, which enhanced the validity of the findings.11Participants were asked to indicate whether they believed that the earth was warming secondary to human or natural processes; not growing warmer at all; or whether they were unsure. Similar to the general population, 46% agreed that climate change is anthropogenic.11 More than three-fourths believed it was likely climate change would adversely affect the places they worked, like military installations; 61% thought it likely that global warming could lead to armed conflict over resources. Seven in 10 respondents believed that climate is changing vs 46% who did not. Of respondents who believe climate change is real, 87% see it as a threat to military bases compared with 60% who do not accept the science that the earth is warming.11
This survey, though, is only a small study, and the military and VA are big tents under which a wide range of political persuasions and diverse beliefs co-exist. There are many readers of Federal Practitioner who will no doubt reject nearly every word I have written, in what I know is a controversial column. But it matters that the military and veteran constituency are thinking and speaking about the issue of climate change.11 Why? The answer takes us back to the disaster in Australia. When the fires and the devastation they wrought escalated beyond the powers of the civil authorities to handle, it was the military whose technical skill, coordinated readiness, and personal courage and dedication that was called on to rescue thousands of civilians from the inferno.12 So it will be in our country and around the world when disasters—manmade, natural, or both—threaten to engulf life in all its wondrous variety. Those who battle extreme weather will have unique health needs, and their valiant sacrifices deserve to have health care systems ready and able to treat them.
Like me, some of you may have been following the agonizing news about the unprecedented brushfires in Australia that have devastated human, animal, and vegetative life in that country so culturally akin to our own.1 For many people who believe the overwhelming majority of scientific reports on climate change, these apocalyptic fires are an empirical demonstration of the truth of the dire prophecies for the future of our planet. Scientists have demonstrated that although climate change may not have caused the worst fires in Australia’s history, they may have contributed to the conditions that enabled them to spread so far and wide and reach such a destructive intensity.2The heartbreaking pictures of singed koalas and displaced people and the helpless feeling that all I can do from here is donate money set me to thinking about the relationship between the military, health, and climate change, which is the subject of this column.
As I write this in mid-January of a new decade and glance at the weather headlines, I read about an earthquake in Puerto Rico and tornadoes in the southern US. This makes it quite plausible that our comfortable lifestyle and technological civilization could in the coming decades go the way of the dinosaurs, also victims of climate change.
Initially, my first thought about this relationship is a negative one—images of scorched earth policies that stretch back to ancient wars jump to mind. Reflection and research on the topic though suggest that the relationship may be more complicated and conflicted. Alas, I can only touch on a few of the themes in this brief format.
It may not be as obvious that climate change also threatens the military, which is the guardian of that civilization. In 2018, for example, Hurricane Michael caused nearly $5 billion in damages to Tyndall Air Force Base in Florida.3 A year later, the US Department of Defense (DoD) released a report on the effects of climate change as mandated by Congress.4 Even though some congressional critics expressed concern about the report’s lack of depth and detail,5 the report asserted that, “The effects of a changing climate are a national security issue with potential impacts to Department of Defense (DoD or the Department) missions, operational plans, and installations.”4
The US Department of Veterans Affairs (VA) is not immune either. Natural disasters have already disrupted the delivery of health care at its many aging facilities. Climate change was called the “engine”6 driving Hurricane Maria, which in 2017 slammed into Puerto Rico, including its VA medical center, and resulted in shortages of supplies, staff, and basic utilities.7 The facility and the island are still trying to rebuild. In response to weather-exposed vulnerability in VA infrastructure, Senator and presidential candidate Elizabeth Warren (D-MA) and Senator Brian Schatz (D-HI), the ranking member of the Subcommittee on Military Construction, sent a letter to VA leadership arguing that “Strengthening VA’s resilience to climate change is consistent with the agency’s mission to deliver timely, high-quality care and benefits to America’s veterans.”8
It has been reported that the current administration has countered initiatives to prepare for the challenges of providing health care to service members and veterans in a climate changed world.9 Sadly, but predictably, in the politicized federal health care arena, the safety of our service members and, in turn, the domestic and national security and peace that depend on them are caught in the partisan debate over global warming, though it is not likely Congress or federal agency leaders will abandon planning to safeguard service members who will see duty and combat in a radically altered ecology and veterans and who will need to have VA continue to be the reliable safety net despite an increasingly erratic environment.10
Climate change is a divisive political issue; there is a proud tradition of conservatism and self-reliance in military members, active duty and veteran alike. That was why I was surprised and impressed when I saw the results of a recent survey on climate change. In January 2019, 293 active-duty service members and veterans were surveyed.
Participants were selected to reflect the ethnic makeup, educational level, and political allegiance of the military population, which enhanced the validity of the findings.11Participants were asked to indicate whether they believed that the earth was warming secondary to human or natural processes; not growing warmer at all; or whether they were unsure. Similar to the general population, 46% agreed that climate change is anthropogenic.11 More than three-fourths believed it was likely climate change would adversely affect the places they worked, like military installations; 61% thought it likely that global warming could lead to armed conflict over resources. Seven in 10 respondents believed that climate is changing vs 46% who did not. Of respondents who believe climate change is real, 87% see it as a threat to military bases compared with 60% who do not accept the science that the earth is warming.11
This survey, though, is only a small study, and the military and VA are big tents under which a wide range of political persuasions and diverse beliefs co-exist. There are many readers of Federal Practitioner who will no doubt reject nearly every word I have written, in what I know is a controversial column. But it matters that the military and veteran constituency are thinking and speaking about the issue of climate change.11 Why? The answer takes us back to the disaster in Australia. When the fires and the devastation they wrought escalated beyond the powers of the civil authorities to handle, it was the military whose technical skill, coordinated readiness, and personal courage and dedication that was called on to rescue thousands of civilians from the inferno.12 So it will be in our country and around the world when disasters—manmade, natural, or both—threaten to engulf life in all its wondrous variety. Those who battle extreme weather will have unique health needs, and their valiant sacrifices deserve to have health care systems ready and able to treat them.
1. Thompson A. Australia’s bushfires have likely devastated wildlife–and the impact will only get worse. Scientific American. https://www.scientificamerican.com/article/australias-bushfires-have-likely-devastated-wildlife-and-the-impact-will-only-get-worse. Published January 8, 2020. Accessed January 16, 2020.
2. Gibbens S. Intense ‘firestorms’ forming from Australia’s deadly wildfires. https://www.nationalgeographic.com/science/2020/01/australian-wildfires-cause-firestorms. Published January 9, 2020. Accessed January 15, 2020.
3. Shapiro A. Tyndall Air Force Base still faces challenges in recovering from Hurricane Michael. https://www.npr.org/2019/05/31/728754872/tyndall-air-force-base-still-faces-challenges-in-recovering-from-hurricane-micha. Published May 31, 2019. Accessed January 16, 2020.
4. US Department of Defense, Office of the Undersecretary for Acquisition and Sustainment. Report on effects of a changing climate to the Department of Defense. https://www.documentcloud.org/documents/5689153-DoD-Final-Climate-Report.html. Published January 2019. Accessed January 16, 2020.
5. Maucione S. DoD justifies climate change report, says response was mission-centric. https://federalnewsnetwork.com/defense-main/2019/03/dod-justifies-climate-change-report-says-response-was-mission-centric. Published March 28, 2019. Accessed January 16, 2020.
6. Shane L 3rd. Puerto Rico’s VA hospital weathers Maria, but challenges loom. https://www.armytimes.com/veterans/2017/09/22/puerto-ricos-va-hospital-weathers-hurricane-maria-but-challenges-loom. Published September 22, 2017. Accessed January 16, 2020.
7. Hersher R. Climate change was the engine that powered Hurricane Maria’s devastating rains. https://www.npr.org/2019/04/17/714098828/climate-change-was-the-engine-that-powered-hurricane-marias-devastating-rains. Published April 17, 2019. Accessed January 16, 2020.
8. Senators Warren and Schatz request an update from the Department of Veterans Affairs on efforts to build resilience to climate change [press release]. https://www.warren.senate.gov/oversight/letters/senators-warren-and-schatz-request-an-update-from-the-department-of-veterans-affairs-on-efforts-to-build-resilience-to-climate-change. Published October 1, 2019. Accessed January 16, 2020.
9. Simkins JD. Navy quietly ends climate change task force, reversing Obama initiative. https://www.navytimes.com/off-duty/military-culture/2019/08/26/navy-quietly-ends-climate-change-task-force-reversing-obama-initiative. Published August 26, 2019. Accessed January 16, 2020.
10. Eilperin J, Dennis B, Ryan M. As White House questions climate change, U.S. military is planning for it. https://www.washingtonpost.com/national/health-science/as-white-house-questions-climate-change-us-military-is-planning-for-it/2019/04/08/78142546-57c0-11e9-814f-e2f46684196e_story.html. Published April 8, 2019. Accessed January 16, 2020.
11. Motta M, Spindel J, Ralston R. Veterans are concerned about climate change and that matters. http://theconversation.com/veterans-are-concerned-about-climate-change-and-that-matters-110685. Published March 8, 2019. Accessed January 16, 2020.
12. Albeck-Ripka L, Kwai I, Fuller T, Tarabay J. ‘It’s an atomic bomb’: Australia deploys military as fires spread. https://www.nytimes.com/2020/01/04/world/australia/fires-military.html. Updated January 5, 2020. Accessed January 18, 2020.
1. Thompson A. Australia’s bushfires have likely devastated wildlife–and the impact will only get worse. Scientific American. https://www.scientificamerican.com/article/australias-bushfires-have-likely-devastated-wildlife-and-the-impact-will-only-get-worse. Published January 8, 2020. Accessed January 16, 2020.
2. Gibbens S. Intense ‘firestorms’ forming from Australia’s deadly wildfires. https://www.nationalgeographic.com/science/2020/01/australian-wildfires-cause-firestorms. Published January 9, 2020. Accessed January 15, 2020.
3. Shapiro A. Tyndall Air Force Base still faces challenges in recovering from Hurricane Michael. https://www.npr.org/2019/05/31/728754872/tyndall-air-force-base-still-faces-challenges-in-recovering-from-hurricane-micha. Published May 31, 2019. Accessed January 16, 2020.
4. US Department of Defense, Office of the Undersecretary for Acquisition and Sustainment. Report on effects of a changing climate to the Department of Defense. https://www.documentcloud.org/documents/5689153-DoD-Final-Climate-Report.html. Published January 2019. Accessed January 16, 2020.
5. Maucione S. DoD justifies climate change report, says response was mission-centric. https://federalnewsnetwork.com/defense-main/2019/03/dod-justifies-climate-change-report-says-response-was-mission-centric. Published March 28, 2019. Accessed January 16, 2020.
6. Shane L 3rd. Puerto Rico’s VA hospital weathers Maria, but challenges loom. https://www.armytimes.com/veterans/2017/09/22/puerto-ricos-va-hospital-weathers-hurricane-maria-but-challenges-loom. Published September 22, 2017. Accessed January 16, 2020.
7. Hersher R. Climate change was the engine that powered Hurricane Maria’s devastating rains. https://www.npr.org/2019/04/17/714098828/climate-change-was-the-engine-that-powered-hurricane-marias-devastating-rains. Published April 17, 2019. Accessed January 16, 2020.
8. Senators Warren and Schatz request an update from the Department of Veterans Affairs on efforts to build resilience to climate change [press release]. https://www.warren.senate.gov/oversight/letters/senators-warren-and-schatz-request-an-update-from-the-department-of-veterans-affairs-on-efforts-to-build-resilience-to-climate-change. Published October 1, 2019. Accessed January 16, 2020.
9. Simkins JD. Navy quietly ends climate change task force, reversing Obama initiative. https://www.navytimes.com/off-duty/military-culture/2019/08/26/navy-quietly-ends-climate-change-task-force-reversing-obama-initiative. Published August 26, 2019. Accessed January 16, 2020.
10. Eilperin J, Dennis B, Ryan M. As White House questions climate change, U.S. military is planning for it. https://www.washingtonpost.com/national/health-science/as-white-house-questions-climate-change-us-military-is-planning-for-it/2019/04/08/78142546-57c0-11e9-814f-e2f46684196e_story.html. Published April 8, 2019. Accessed January 16, 2020.
11. Motta M, Spindel J, Ralston R. Veterans are concerned about climate change and that matters. http://theconversation.com/veterans-are-concerned-about-climate-change-and-that-matters-110685. Published March 8, 2019. Accessed January 16, 2020.
12. Albeck-Ripka L, Kwai I, Fuller T, Tarabay J. ‘It’s an atomic bomb’: Australia deploys military as fires spread. https://www.nytimes.com/2020/01/04/world/australia/fires-military.html. Updated January 5, 2020. Accessed January 18, 2020.

Stimulant Medication Prescribing Practices Within a VA Health Care System
Dispensing of prescription stimulant medications, such as methylphenidate or amphetamine salts, has been expanding at a rapid rate over the past 2 decades. An astounding 58 million stimulant medications were prescribed in 2014.1,2 Adults now exceed youths in the proportion of prescribed stimulant medications.1,3
Off-label use of prescription stimulant medications, such as for performance enhancement, fatigue management, weight loss, medication-assisted therapy for stimulant use disorders, and adjunctive treatment for certain depressive disorders, is reported to be ≥ 40% of total stimulant use and is much more common in adults.1 A 2017 study assessing risk of amphetamine use disorder and mortality among veterans prescribed stimulant medications within the Veterans Health Administration (VHA) reported off-label use in nearly 3 of every 5 incident users in 2012.4 Off-label use also is significantly more common when prescribed by nonpsychiatric physicians compared with that of psychiatrists.1
One study assessing stimulant prescribing from 2006 to 2009 found that nearly 60% of adults were prescribed stimulant medications by nonpsychiatrist physicians, and only 34% of those adults prescribed a stimulant by a nonpsychiatrist physician had a diagnosis of attention-deficit hyperactivity disorder (ADHD).5 Findings from managed care plans covering years from 2000 to 2004 were similar, concluding that 30% of the adult patients who were prescribed methylphenidate had at least 1 medical claim with a diagnosis of ADHD.6 Of the approximately 16 million adults prescribed stimulant medications in 2017, > 5 million of them reported stimulant misuse.3 Much attention has been focused on misuse of stimulant medications by youths and young adults, but new information suggests that increased monitoring is needed among the US adult population. Per the US Department of Veterans Affairs (VA) Academic Detailing Stimulant Dashboard, as of October 2018 the national average of veterans with a documented substance use disorder (SUD) who are also prescribed stimulant medications through the VHA exceeds 20%, < 50% have an annual urine drug screen (UDS), and > 10% are coprescribed opioids and benzodiazepines.The percentage of veterans prescribed stimulant medications in the presence of a SUD has increased over the past decade, with a reported 8.7% incidence in 2002 increasing to 14.3% in 2012.4
There are currently no protocols, prescribing restrictions, or required monitoring parameters in place for prescription stimulant use within the Lexington VA Health Care System (LVAHCS). The purpose of this study was to evaluate the prescribing practices at LVAHCS of stimulant medications and identify opportunities for improvement in the prescribing and monitoring of this drug class.
Methods
This study was a single-center quality improvement project evaluating the prescribing practices of stimulant medications within LVAHCS and exempt from institutional review board approval. Veterans were included in the study if they were prescribed amphetamine salts, dextroamphetamine, lisdexamphetamine, or methylphenidate between January 1, 2018 and June 30, 2018; however, the veterans’ entire stimulant use history was assessed. Exclusion criteria included duration of use of < 2 months or < 2 prescriptions filled during the study period. Data for veterans who met the prespecified inclusion and exclusion criteria were collected via chart review and Microsoft SQL Server Management Studio.
Collected data included age, gender, stimulant regimen (drug name, dose, frequency), indication and duration of use, prescriber name and specialty, prescribing origin of initial stimulant medication, and whether stimulant use predated military service. Monitoring of stimulant medications was assessed via UDS at least annually, query of the prescription drug monitoring program (PDMP) at least quarterly, and average time between follow-up appointments with stimulant prescriber.
Monitoring parameters were assessed from January 1, 2017 through June 30, 2018, as it was felt that the 6-month study period would be too narrow to accurately assess monitoring trends. Mental health diagnoses, ADHD diagnostic testing if applicable, documented SUD or stimulant misuse past or present, and concomitant central nervous system (CNS) depressant use also were collected. CNS depressants evaluated were those that have abuse potential or significant psychotropic effects and included benzodiazepines, antipsychotics, opioids, gabapentin/pregabalin, Z-hypnotics, and muscle relaxants.
Results
The majority of participants were male (168/200) with an average age of 43.3 years. Dextroamphetamine/amphetamine was the most used stimulant (48.5%), followed by methylphenidate (40%), and dextroamphetamine (10%). Lisdexamphetamine was the least used stimulant, likely due to its formulary-restricted status within this facility. An extended release (ER) formulation was utilized in 1 of 4 participants, with 1 of 20 participants prescribed a combination of immediate release (IR) and ER formulations. Duration of use ranged from 3 months to 14 years, with an average duration of 4 years (Table 1).
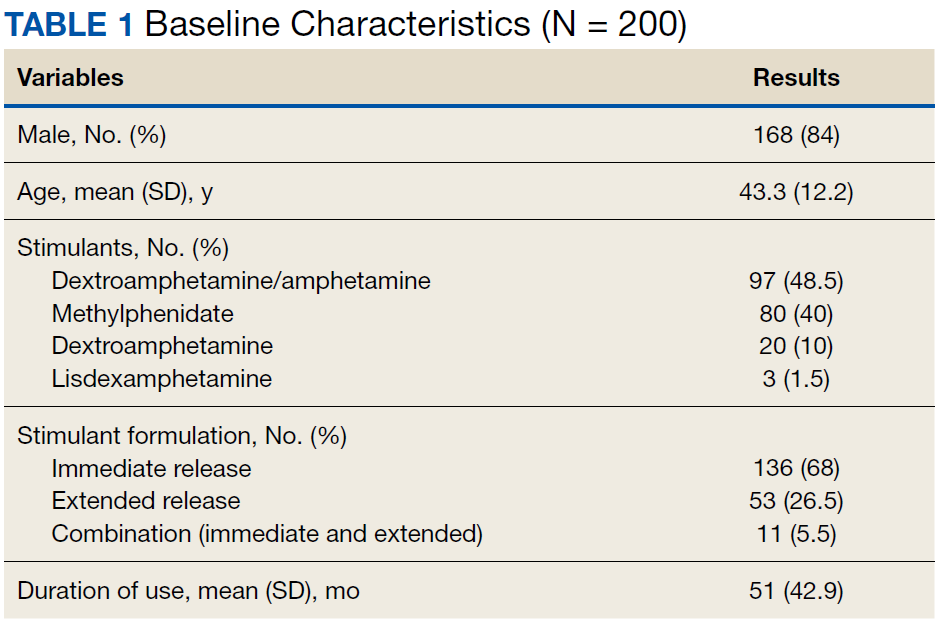
Nearly 40% of participants reported an origin of stimulant initiation outside of LVAHCS. Fourteen percent of participants were started on prescription stimulant medications while active-duty service members. Stimulant medications were initiated at another VA facility in 10.5% of instances, and 15% of participants reported being prescribed stimulant medications by a civilian prescriber prior to receiving them at LVAHCS. Seventy-four of 79 (93.6%) participants with an origin of stimulant prescription outside of LVAHCS reported a US Federal Food and Drug Administration (FDA)-approved indication for use.
Stimulant medications were used for FDA-approved indications (ADHD and narcolepsy) in 69.5% of participants. Note, this included patients who maintained an ADHD diagnosis in their medical record even if it was not substantiated with diagnostic testing. Of the participants reporting ADHD as an indication for stimulant use, diagnostic testing was conducted at LVAHCS to confirm an ADHD diagnosis in 58.6% (78/133) participants; 20.5% (16/78) of these diagnostic tests did not support the diagnosis of ADHD. All documented indications for use can be found in Table 2.
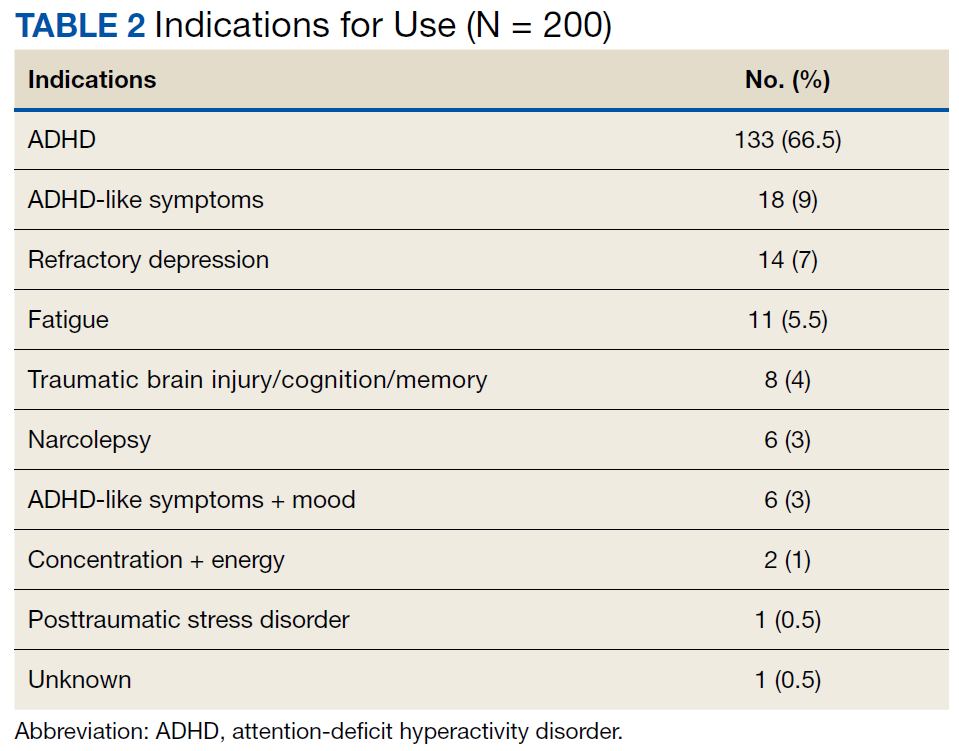
As expected, the most common indication was ADHD (66.5%), followed by ADHD-like symptoms (9%), refractory depression (7%), and fatigue (5.5%). Fourteen percent of participants had ≥ 1 change in indication for use, with some participants having up to 4 different documented indications while being prescribed stimulant medications. Twelve percent of participants were either denied stimulant initiation, or current stimulant medications were discontinued by one health provider and were restarted by another following a prescriber change. Aside from indication for stimulant use, 90% of participants had at least one additional mental health diagnosis. The rate of all mental health diagnoses documented in the medical record problem list can be found in Table 3.
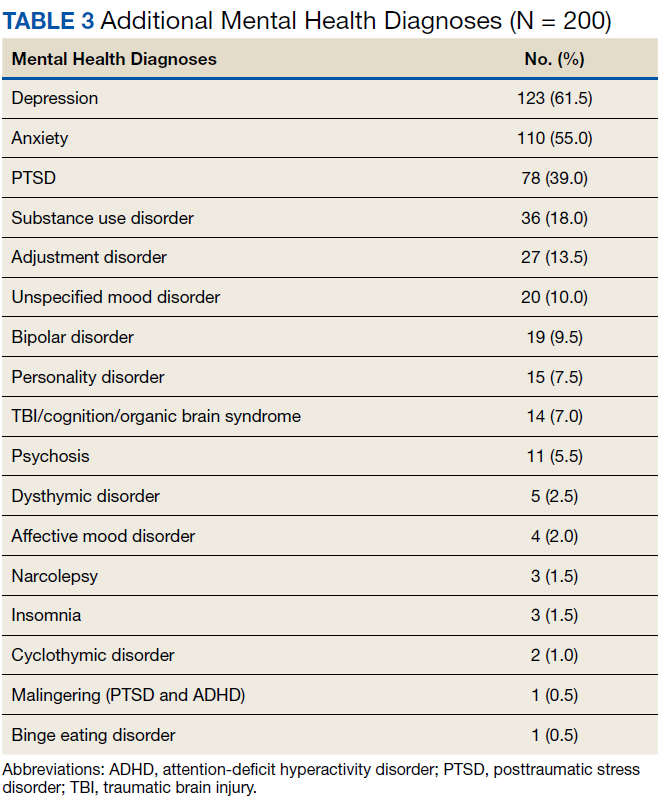
A UDS was collected at least annually in 37% of participants. A methylphenidate confirmatory screen was ordered to assess adherence in just 2 (2.5%) participants prescribed methylphenidate. While actively prescribed stimulant medications, PDMP was queried quarterly in 26% of participants. Time to follow-up with the prescriber ranged from 1 to 15 months, and 40% of participants had follow-up at least quarterly. Instance of SUD, either active or in remission, differed when searched via problem list (36/200) and prescriber documentation (63/200). The most common SUD was alcohol use disorder (13%), followed by cannabis use disorder (5%), polysubstance use disorder (5%), opioid use disorder (4.5%), stimulant use disorder (2.5%), and sedative use disorder (1%). Twenty-five participants currently prescribed stimulant medications had stimulant abuse/misuse documented in their medical record. Fifty-four percent of participants were prescribed at least 1 CNS depressant considered to have abuse potential or significant psychotropic effects. Opioids were most common (23%), followed by muscle relaxants (15.5%), benzodiazepines (15%), antipsychotics (13%), gabapentin/pregabalin (12%), and Z-hypnotics (12%).
Discussion
The source of the initial stimulant prescription was assessed. The majority of veterans had received medical care prior to receiving care at LVAHCS, whether on active duty, from another VA facility throughout the country, or by a private civilian prescriber. The origin of initial stimulant medication and indication for stimulant medication use were patient reported. Requiring medical records from civilian providers prior to continuing stimulant medication is prescriber-dependent and was not available for all participants.
As expected, the majority of participants
The reasons for discontinuation included a positive UDS result for cocaine, psychosis, broken narcotic contract, ADHD diagnosis not supported by psychological testing, chronic bipolar disorder secondary to stimulant use, diversion, stimulant misuse, and lack of indication for use. There also were a handful of veterans whose VA prescribers declined to initiate prescription stimulant medications for various reasons, so the veteran sought care from a civilian prescriber who prescribed stimulant medications, then returned to the VA for medication management, and stimulant medications were continued. Fourteen percent (28/200) of participants had multiple indications for use at some point during stimulant medication therapy. Eight of those were a reasonable change from ADHD to ADHD-like symptoms when diagnosis was not substantiated by testing. The cause of other changes in indication for use was not well documented and often unclear. One veteran had 4 different indications for use documented in the medical record, often changing with each change in prescriber. It appeared that the most recent prescriber was uncertain of the actual indication for use but did not want to discontinue the medication. This prescriber documented that the stimulant medication should continue for presumed ADHD/mood/fatigue/cognitive dysfunction, which were all of the indications documented by the veteran’s previous prescribers.
Reasons for Discontinuation
ADHD was the most prominent indication for use, although the indication was changed to ADHD-like symptoms in several veterans for whom diagnostic testing did not support the ADHD diagnosis. Seventy-eight of 133 veterans prescribed stimulant medications for ADHD received diagnostic testing via a psychologist at LVAHCS. For the 11 veterans who had testing after stimulant initiation, a stimulant-free period was required prior to testing to ensure an accurate diagnosis. For 21% of veterans, the ADHD diagnosis was unsubstantiated by formal testing; however, all of these veterans continued stimulant medication use. For 1 veteran, the psychologist performing the testing documented new diagnoses, including moderate to severe stimulant use disorder and malingering both for PTSD and ADHD. The rate of stimulant prescribing inconsistency, “prescriber-hopping,” and unsupported ADHD diagnosis results warrant a conversation about expectations for transitions of care regarding stimulant medications, not only from outside to inside LVAHCS, but from prescriber to prescriber within the facility.
In some cases, stimulant medications were discontinued by a prescriber secondary to a worsening of another mental health condition. More than half of the participants in this study had an anxiety disorder diagnosis. Whether or not anxiety predated stimulant use or whether the use of stimulant medications contributed to the diagnosis and thus the addition of an additional CNS depressant to treat anxiety may be an area of research for future consideration. Although bipolar disorder, anxiety disorders, psychosis, and SUD are not contraindications for use of stimulant medications, caution must be used in patients with these diagnoses. Prescribers must weigh risks vs benefits as well as perform close monitoring during use. Similarly, one might look further into stimulant medications prescribed for fatigue and assess the role of any simultaneously prescribed CNS depressants. Is the stimulant being used to treat the adverse effect (AE) of another medication? In 2 documented instances in this study, a psychologist conducted diagnostic testing who reported that the veteran did not meet the criteria for ADHD but that a stimulant may help counteract the iatrogenic effect of anticonvulsants. In both instances stimulant use continued.
Prescription Monitoring
Polysubstance use disorder (5%) was the third most common SUD recorded among study participants. The majority of those with polysubstance use disorder reported abuse/misuse of illicit or prescribed stimulants. Stimulant abuse/misuse was documented in 25 of 200 (12.5%) study participants. In several instances, abuse/misuse was detected by the LVAHCS delivery coordination pharmacist who tracks patterns of early fill requests and prescriptions reported lost/stolen. This pharmacist may request that the prescriber obtain PDMP query, UDS, or pill count if concerning patterns are noted. Lisdexamphetamine is a formulary-restricted medication at LVAHCS, but it was noted to be approved for use when prescribers requested an abuse-deterrent formulation. Investigators noticed a trend in veterans whose prescriptions exceeded the recommended maximum dosage also having stimulant abuse/misuse documented in their medical record. The highest documented total daily dose in this study was 120-mg amphetamine salts IR for ADHD, compared with the normal recommended dosing range of 5 to 40 mg/d for the same indication.
Various modalities were used to monitor participants but less than half of veterans had an annual UDS, quarterly PDMP query, and quarterly prescriber follow-up. PDMP queries and prescriber follow-up was assessed quarterly as would be reasonable given that private sector practitioners may issue multiple prescriptions authorizing the patient to receive up to a 90-day supply.7 Prescriber follow-up ranged from 1 to 15 months. A longer time to follow-up was seen more frequently in stimulant medications prescribed by primary care as compared with that of mental health.
Clinical Practice Protocol
Data from this study were collected with the intent to identify opportunities for improvement in the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a facility-specific clinical practice protocol (CPP) for stimulant prescribing. It may also be beneficial to formulate a chronic stimulant management agreement between patient and prescriber to provide informed consent and clear expectations prior to stimulant medication initiation.
A CPP could be used to establish stimulant prescribing rules within a facility, which may limit who can prescribe stimulant medications or include a review process and/or required documentation in the medical record when being prescribed outside of specified dosing range and indications for use designated in the CPP or other evidence-based guidelines. Transition of care was found to be an area of opportunity in this study, which could be mitigated with the requirement of a baseline assessment prior to stimulant initiation with the expectation that it be completed regardless of prior prescription stimulant medication use. There was a lack of consistent monitoring for participants in this study, which may be improved if required monitoring parameters and frequency were provided for prescribers. For example, monitoring of heart rate and blood pressure was not assessed in this study, but a CPP may include monitoring vital signs before and after each dose change and every 6 months, per recommendation from the National Institute for Health and Care Excellence ADHD Diagnosis and Management guideline published in 2018.8The CPP may list the responsibilities of all those involved in the prescribing of stimulant medications, such as mental health service leadership, prescribers, nursing staff, pharmacists, social workers, psychologists, and other mental health staff. For prescribers this may include a thorough baseline assessment and criteria for use that must be met prior to stimulant initiation, documentation that must be included in the medical record and required monitoring during stimulant treatment, and expectations for increased monitoring and/or termination of treatment with nonadherence, diversion, or abuse/misuse.
The responsibilities of pharmacists may include establishing criteria for use of nonformulary and restricted agents as well as completion of nonformulary/restricted requests, reviewing dosages that exceed the recommended FDA daily maximum, reviewing uncommon off-label uses of stimulant medications, review and document early fill requests, potential nonadherence, potential drug-seeking behavior, and communication of the following information to the primary prescriber. For other mental health staff this may include documenting any reported AEs of the medication, referring the patient to their prescriber or pharmacist for any medication questions or concerns, and assessment of effectiveness and/or worsening behavior during patient contact.
Limitations
One limitation of this study was the way that data were pulled from patient charts. For example, only 3/200 participants in this study had insomnia per diagnosis codes, whereas that number was substantially higher when chart review was used to assess active prescriptions for sleep aids or documented complaints of insomnia in prescriber progress notes. For this same reason, rates of SUDs must be interpreted with caution as well. SUD diagnosis, both current and in remission were taken into account during data collection. Per diagnosis codes, 36 (18%) veterans in this study had a history of SUD, but this number was higher (31.5%) during chart review. The majority of discrepancies were found when participants reported a history of SUD to the prescriber, but this information was not captured via the problem list or encounter codes. What some may consider a minor omission in documentation can have a large impact on patient care as it is unlikely that prescribers have adequate administrative time to complete a chart review in order to find a complete past medical history as was required of investigators in this study. For this reason, incomplete provider documentation and human error that can occur as a result of a retrospective chart review were also identified as study limitations.
Conclusion
Our data show that there is still substantial room for improvement in the prescribing and monitoring of stimulant medications. The rate of stimulant prescribing inconsistency, prescriber-hopping, and unsupported ADHD diagnosis resulting from formal diagnostic testing warrant a review in the processes for transition of care regarding stimulant medications, both within and outside of this facility. A lack of consistent monitoring was also identified in this study. One of the most appreciable areas of opportunity resulting from this study is the need for consistency in both the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a CPP for stimulant prescribing as well as a chronic stimulant management agreement to provide clear expectations for patients and prescribers prior to and during prescription stimulant use.
Acknowledgments
We thank Tori Wilhoit, PharmD candidate, and Dana Fischer, PharmD candidate, for their participation in data collection and Courtney Eatmon, PharmD, BCPP, for her general administrative support throughout this study.
1. Safer DJ. Recent trends in stimulant usage. J Atten Disord. 2016;20(6):471-477.
2. Christopher Jones; US Food and Drug Administration. The opioid epidemic overview and a look to the future. http://www.agencymeddirectors.wa.gov/Files/OpioidConference/2Jones_OPIOIDEPIDEMICOVERVIEW.pdf. Published June 12, 2015. Accessed January 16, 2020.
3. Compton WM, Han B, Blanco C, Johnson K, Jones CM. Prevalence and correlates of prescription stimulant use, misuse, use disorders, motivations for misuse among adults in the United States. Am J Psychiatry. 2018;175(8):741-755.
4. Westover AN, Nakonezney PA, Halm EA, Adinoff B. Risk of amphetamine use disorder and mortality among incident users of prescribed stimulant medications in the Veterans Administration. Addiction. 2018;113(5):857-867.
5. Olfson M, Blanco C, Wang S, Greenhill LL. Trends in office-based treatment of adults with stimulant medications in the United States. J Clin Psychiatry. 2013;74(1):43-50.
6. Olfson M, Marcus SC, Zhang HF, and Wan GJ. Continuity in methylphenidate treatment of adults with attention-deficit/hyperactivity disorder. J Manag Care Pharm. 2007;13(7): 570-577.
7. 21 CFR § 1306.12
8. National Collaborating Centre for Mental Health (UK). Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. NICE Clinical Guidelines, No. 87. Leicester, United Kingdom: British Psychological Society; 2018.
Dispensing of prescription stimulant medications, such as methylphenidate or amphetamine salts, has been expanding at a rapid rate over the past 2 decades. An astounding 58 million stimulant medications were prescribed in 2014.1,2 Adults now exceed youths in the proportion of prescribed stimulant medications.1,3
Off-label use of prescription stimulant medications, such as for performance enhancement, fatigue management, weight loss, medication-assisted therapy for stimulant use disorders, and adjunctive treatment for certain depressive disorders, is reported to be ≥ 40% of total stimulant use and is much more common in adults.1 A 2017 study assessing risk of amphetamine use disorder and mortality among veterans prescribed stimulant medications within the Veterans Health Administration (VHA) reported off-label use in nearly 3 of every 5 incident users in 2012.4 Off-label use also is significantly more common when prescribed by nonpsychiatric physicians compared with that of psychiatrists.1
One study assessing stimulant prescribing from 2006 to 2009 found that nearly 60% of adults were prescribed stimulant medications by nonpsychiatrist physicians, and only 34% of those adults prescribed a stimulant by a nonpsychiatrist physician had a diagnosis of attention-deficit hyperactivity disorder (ADHD).5 Findings from managed care plans covering years from 2000 to 2004 were similar, concluding that 30% of the adult patients who were prescribed methylphenidate had at least 1 medical claim with a diagnosis of ADHD.6 Of the approximately 16 million adults prescribed stimulant medications in 2017, > 5 million of them reported stimulant misuse.3 Much attention has been focused on misuse of stimulant medications by youths and young adults, but new information suggests that increased monitoring is needed among the US adult population. Per the US Department of Veterans Affairs (VA) Academic Detailing Stimulant Dashboard, as of October 2018 the national average of veterans with a documented substance use disorder (SUD) who are also prescribed stimulant medications through the VHA exceeds 20%, < 50% have an annual urine drug screen (UDS), and > 10% are coprescribed opioids and benzodiazepines.The percentage of veterans prescribed stimulant medications in the presence of a SUD has increased over the past decade, with a reported 8.7% incidence in 2002 increasing to 14.3% in 2012.4
There are currently no protocols, prescribing restrictions, or required monitoring parameters in place for prescription stimulant use within the Lexington VA Health Care System (LVAHCS). The purpose of this study was to evaluate the prescribing practices at LVAHCS of stimulant medications and identify opportunities for improvement in the prescribing and monitoring of this drug class.
Methods
This study was a single-center quality improvement project evaluating the prescribing practices of stimulant medications within LVAHCS and exempt from institutional review board approval. Veterans were included in the study if they were prescribed amphetamine salts, dextroamphetamine, lisdexamphetamine, or methylphenidate between January 1, 2018 and June 30, 2018; however, the veterans’ entire stimulant use history was assessed. Exclusion criteria included duration of use of < 2 months or < 2 prescriptions filled during the study period. Data for veterans who met the prespecified inclusion and exclusion criteria were collected via chart review and Microsoft SQL Server Management Studio.
Collected data included age, gender, stimulant regimen (drug name, dose, frequency), indication and duration of use, prescriber name and specialty, prescribing origin of initial stimulant medication, and whether stimulant use predated military service. Monitoring of stimulant medications was assessed via UDS at least annually, query of the prescription drug monitoring program (PDMP) at least quarterly, and average time between follow-up appointments with stimulant prescriber.
Monitoring parameters were assessed from January 1, 2017 through June 30, 2018, as it was felt that the 6-month study period would be too narrow to accurately assess monitoring trends. Mental health diagnoses, ADHD diagnostic testing if applicable, documented SUD or stimulant misuse past or present, and concomitant central nervous system (CNS) depressant use also were collected. CNS depressants evaluated were those that have abuse potential or significant psychotropic effects and included benzodiazepines, antipsychotics, opioids, gabapentin/pregabalin, Z-hypnotics, and muscle relaxants.
Results
The majority of participants were male (168/200) with an average age of 43.3 years. Dextroamphetamine/amphetamine was the most used stimulant (48.5%), followed by methylphenidate (40%), and dextroamphetamine (10%). Lisdexamphetamine was the least used stimulant, likely due to its formulary-restricted status within this facility. An extended release (ER) formulation was utilized in 1 of 4 participants, with 1 of 20 participants prescribed a combination of immediate release (IR) and ER formulations. Duration of use ranged from 3 months to 14 years, with an average duration of 4 years (Table 1).
Nearly 40% of participants reported an origin of stimulant initiation outside of LVAHCS. Fourteen percent of participants were started on prescription stimulant medications while active-duty service members. Stimulant medications were initiated at another VA facility in 10.5% of instances, and 15% of participants reported being prescribed stimulant medications by a civilian prescriber prior to receiving them at LVAHCS. Seventy-four of 79 (93.6%) participants with an origin of stimulant prescription outside of LVAHCS reported a US Federal Food and Drug Administration (FDA)-approved indication for use.
Stimulant medications were used for FDA-approved indications (ADHD and narcolepsy) in 69.5% of participants. Note, this included patients who maintained an ADHD diagnosis in their medical record even if it was not substantiated with diagnostic testing. Of the participants reporting ADHD as an indication for stimulant use, diagnostic testing was conducted at LVAHCS to confirm an ADHD diagnosis in 58.6% (78/133) participants; 20.5% (16/78) of these diagnostic tests did not support the diagnosis of ADHD. All documented indications for use can be found in Table 2.
As expected, the most common indication was ADHD (66.5%), followed by ADHD-like symptoms (9%), refractory depression (7%), and fatigue (5.5%). Fourteen percent of participants had ≥ 1 change in indication for use, with some participants having up to 4 different documented indications while being prescribed stimulant medications. Twelve percent of participants were either denied stimulant initiation, or current stimulant medications were discontinued by one health provider and were restarted by another following a prescriber change. Aside from indication for stimulant use, 90% of participants had at least one additional mental health diagnosis. The rate of all mental health diagnoses documented in the medical record problem list can be found in Table 3.
A UDS was collected at least annually in 37% of participants. A methylphenidate confirmatory screen was ordered to assess adherence in just 2 (2.5%) participants prescribed methylphenidate. While actively prescribed stimulant medications, PDMP was queried quarterly in 26% of participants. Time to follow-up with the prescriber ranged from 1 to 15 months, and 40% of participants had follow-up at least quarterly. Instance of SUD, either active or in remission, differed when searched via problem list (36/200) and prescriber documentation (63/200). The most common SUD was alcohol use disorder (13%), followed by cannabis use disorder (5%), polysubstance use disorder (5%), opioid use disorder (4.5%), stimulant use disorder (2.5%), and sedative use disorder (1%). Twenty-five participants currently prescribed stimulant medications had stimulant abuse/misuse documented in their medical record. Fifty-four percent of participants were prescribed at least 1 CNS depressant considered to have abuse potential or significant psychotropic effects. Opioids were most common (23%), followed by muscle relaxants (15.5%), benzodiazepines (15%), antipsychotics (13%), gabapentin/pregabalin (12%), and Z-hypnotics (12%).
Discussion
The source of the initial stimulant prescription was assessed. The majority of veterans had received medical care prior to receiving care at LVAHCS, whether on active duty, from another VA facility throughout the country, or by a private civilian prescriber. The origin of initial stimulant medication and indication for stimulant medication use were patient reported. Requiring medical records from civilian providers prior to continuing stimulant medication is prescriber-dependent and was not available for all participants.
As expected, the majority of participants
The reasons for discontinuation included a positive UDS result for cocaine, psychosis, broken narcotic contract, ADHD diagnosis not supported by psychological testing, chronic bipolar disorder secondary to stimulant use, diversion, stimulant misuse, and lack of indication for use. There also were a handful of veterans whose VA prescribers declined to initiate prescription stimulant medications for various reasons, so the veteran sought care from a civilian prescriber who prescribed stimulant medications, then returned to the VA for medication management, and stimulant medications were continued. Fourteen percent (28/200) of participants had multiple indications for use at some point during stimulant medication therapy. Eight of those were a reasonable change from ADHD to ADHD-like symptoms when diagnosis was not substantiated by testing. The cause of other changes in indication for use was not well documented and often unclear. One veteran had 4 different indications for use documented in the medical record, often changing with each change in prescriber. It appeared that the most recent prescriber was uncertain of the actual indication for use but did not want to discontinue the medication. This prescriber documented that the stimulant medication should continue for presumed ADHD/mood/fatigue/cognitive dysfunction, which were all of the indications documented by the veteran’s previous prescribers.
Reasons for Discontinuation
ADHD was the most prominent indication for use, although the indication was changed to ADHD-like symptoms in several veterans for whom diagnostic testing did not support the ADHD diagnosis. Seventy-eight of 133 veterans prescribed stimulant medications for ADHD received diagnostic testing via a psychologist at LVAHCS. For the 11 veterans who had testing after stimulant initiation, a stimulant-free period was required prior to testing to ensure an accurate diagnosis. For 21% of veterans, the ADHD diagnosis was unsubstantiated by formal testing; however, all of these veterans continued stimulant medication use. For 1 veteran, the psychologist performing the testing documented new diagnoses, including moderate to severe stimulant use disorder and malingering both for PTSD and ADHD. The rate of stimulant prescribing inconsistency, “prescriber-hopping,” and unsupported ADHD diagnosis results warrant a conversation about expectations for transitions of care regarding stimulant medications, not only from outside to inside LVAHCS, but from prescriber to prescriber within the facility.
In some cases, stimulant medications were discontinued by a prescriber secondary to a worsening of another mental health condition. More than half of the participants in this study had an anxiety disorder diagnosis. Whether or not anxiety predated stimulant use or whether the use of stimulant medications contributed to the diagnosis and thus the addition of an additional CNS depressant to treat anxiety may be an area of research for future consideration. Although bipolar disorder, anxiety disorders, psychosis, and SUD are not contraindications for use of stimulant medications, caution must be used in patients with these diagnoses. Prescribers must weigh risks vs benefits as well as perform close monitoring during use. Similarly, one might look further into stimulant medications prescribed for fatigue and assess the role of any simultaneously prescribed CNS depressants. Is the stimulant being used to treat the adverse effect (AE) of another medication? In 2 documented instances in this study, a psychologist conducted diagnostic testing who reported that the veteran did not meet the criteria for ADHD but that a stimulant may help counteract the iatrogenic effect of anticonvulsants. In both instances stimulant use continued.
Prescription Monitoring
Polysubstance use disorder (5%) was the third most common SUD recorded among study participants. The majority of those with polysubstance use disorder reported abuse/misuse of illicit or prescribed stimulants. Stimulant abuse/misuse was documented in 25 of 200 (12.5%) study participants. In several instances, abuse/misuse was detected by the LVAHCS delivery coordination pharmacist who tracks patterns of early fill requests and prescriptions reported lost/stolen. This pharmacist may request that the prescriber obtain PDMP query, UDS, or pill count if concerning patterns are noted. Lisdexamphetamine is a formulary-restricted medication at LVAHCS, but it was noted to be approved for use when prescribers requested an abuse-deterrent formulation. Investigators noticed a trend in veterans whose prescriptions exceeded the recommended maximum dosage also having stimulant abuse/misuse documented in their medical record. The highest documented total daily dose in this study was 120-mg amphetamine salts IR for ADHD, compared with the normal recommended dosing range of 5 to 40 mg/d for the same indication.
Various modalities were used to monitor participants but less than half of veterans had an annual UDS, quarterly PDMP query, and quarterly prescriber follow-up. PDMP queries and prescriber follow-up was assessed quarterly as would be reasonable given that private sector practitioners may issue multiple prescriptions authorizing the patient to receive up to a 90-day supply.7 Prescriber follow-up ranged from 1 to 15 months. A longer time to follow-up was seen more frequently in stimulant medications prescribed by primary care as compared with that of mental health.
Clinical Practice Protocol
Data from this study were collected with the intent to identify opportunities for improvement in the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a facility-specific clinical practice protocol (CPP) for stimulant prescribing. It may also be beneficial to formulate a chronic stimulant management agreement between patient and prescriber to provide informed consent and clear expectations prior to stimulant medication initiation.
A CPP could be used to establish stimulant prescribing rules within a facility, which may limit who can prescribe stimulant medications or include a review process and/or required documentation in the medical record when being prescribed outside of specified dosing range and indications for use designated in the CPP or other evidence-based guidelines. Transition of care was found to be an area of opportunity in this study, which could be mitigated with the requirement of a baseline assessment prior to stimulant initiation with the expectation that it be completed regardless of prior prescription stimulant medication use. There was a lack of consistent monitoring for participants in this study, which may be improved if required monitoring parameters and frequency were provided for prescribers. For example, monitoring of heart rate and blood pressure was not assessed in this study, but a CPP may include monitoring vital signs before and after each dose change and every 6 months, per recommendation from the National Institute for Health and Care Excellence ADHD Diagnosis and Management guideline published in 2018.8The CPP may list the responsibilities of all those involved in the prescribing of stimulant medications, such as mental health service leadership, prescribers, nursing staff, pharmacists, social workers, psychologists, and other mental health staff. For prescribers this may include a thorough baseline assessment and criteria for use that must be met prior to stimulant initiation, documentation that must be included in the medical record and required monitoring during stimulant treatment, and expectations for increased monitoring and/or termination of treatment with nonadherence, diversion, or abuse/misuse.
The responsibilities of pharmacists may include establishing criteria for use of nonformulary and restricted agents as well as completion of nonformulary/restricted requests, reviewing dosages that exceed the recommended FDA daily maximum, reviewing uncommon off-label uses of stimulant medications, review and document early fill requests, potential nonadherence, potential drug-seeking behavior, and communication of the following information to the primary prescriber. For other mental health staff this may include documenting any reported AEs of the medication, referring the patient to their prescriber or pharmacist for any medication questions or concerns, and assessment of effectiveness and/or worsening behavior during patient contact.
Limitations
One limitation of this study was the way that data were pulled from patient charts. For example, only 3/200 participants in this study had insomnia per diagnosis codes, whereas that number was substantially higher when chart review was used to assess active prescriptions for sleep aids or documented complaints of insomnia in prescriber progress notes. For this same reason, rates of SUDs must be interpreted with caution as well. SUD diagnosis, both current and in remission were taken into account during data collection. Per diagnosis codes, 36 (18%) veterans in this study had a history of SUD, but this number was higher (31.5%) during chart review. The majority of discrepancies were found when participants reported a history of SUD to the prescriber, but this information was not captured via the problem list or encounter codes. What some may consider a minor omission in documentation can have a large impact on patient care as it is unlikely that prescribers have adequate administrative time to complete a chart review in order to find a complete past medical history as was required of investigators in this study. For this reason, incomplete provider documentation and human error that can occur as a result of a retrospective chart review were also identified as study limitations.
Conclusion
Our data show that there is still substantial room for improvement in the prescribing and monitoring of stimulant medications. The rate of stimulant prescribing inconsistency, prescriber-hopping, and unsupported ADHD diagnosis resulting from formal diagnostic testing warrant a review in the processes for transition of care regarding stimulant medications, both within and outside of this facility. A lack of consistent monitoring was also identified in this study. One of the most appreciable areas of opportunity resulting from this study is the need for consistency in both the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a CPP for stimulant prescribing as well as a chronic stimulant management agreement to provide clear expectations for patients and prescribers prior to and during prescription stimulant use.
Acknowledgments
We thank Tori Wilhoit, PharmD candidate, and Dana Fischer, PharmD candidate, for their participation in data collection and Courtney Eatmon, PharmD, BCPP, for her general administrative support throughout this study.
Dispensing of prescription stimulant medications, such as methylphenidate or amphetamine salts, has been expanding at a rapid rate over the past 2 decades. An astounding 58 million stimulant medications were prescribed in 2014.1,2 Adults now exceed youths in the proportion of prescribed stimulant medications.1,3
Off-label use of prescription stimulant medications, such as for performance enhancement, fatigue management, weight loss, medication-assisted therapy for stimulant use disorders, and adjunctive treatment for certain depressive disorders, is reported to be ≥ 40% of total stimulant use and is much more common in adults.1 A 2017 study assessing risk of amphetamine use disorder and mortality among veterans prescribed stimulant medications within the Veterans Health Administration (VHA) reported off-label use in nearly 3 of every 5 incident users in 2012.4 Off-label use also is significantly more common when prescribed by nonpsychiatric physicians compared with that of psychiatrists.1
One study assessing stimulant prescribing from 2006 to 2009 found that nearly 60% of adults were prescribed stimulant medications by nonpsychiatrist physicians, and only 34% of those adults prescribed a stimulant by a nonpsychiatrist physician had a diagnosis of attention-deficit hyperactivity disorder (ADHD).5 Findings from managed care plans covering years from 2000 to 2004 were similar, concluding that 30% of the adult patients who were prescribed methylphenidate had at least 1 medical claim with a diagnosis of ADHD.6 Of the approximately 16 million adults prescribed stimulant medications in 2017, > 5 million of them reported stimulant misuse.3 Much attention has been focused on misuse of stimulant medications by youths and young adults, but new information suggests that increased monitoring is needed among the US adult population. Per the US Department of Veterans Affairs (VA) Academic Detailing Stimulant Dashboard, as of October 2018 the national average of veterans with a documented substance use disorder (SUD) who are also prescribed stimulant medications through the VHA exceeds 20%, < 50% have an annual urine drug screen (UDS), and > 10% are coprescribed opioids and benzodiazepines.The percentage of veterans prescribed stimulant medications in the presence of a SUD has increased over the past decade, with a reported 8.7% incidence in 2002 increasing to 14.3% in 2012.4
There are currently no protocols, prescribing restrictions, or required monitoring parameters in place for prescription stimulant use within the Lexington VA Health Care System (LVAHCS). The purpose of this study was to evaluate the prescribing practices at LVAHCS of stimulant medications and identify opportunities for improvement in the prescribing and monitoring of this drug class.
Methods
This study was a single-center quality improvement project evaluating the prescribing practices of stimulant medications within LVAHCS and exempt from institutional review board approval. Veterans were included in the study if they were prescribed amphetamine salts, dextroamphetamine, lisdexamphetamine, or methylphenidate between January 1, 2018 and June 30, 2018; however, the veterans’ entire stimulant use history was assessed. Exclusion criteria included duration of use of < 2 months or < 2 prescriptions filled during the study period. Data for veterans who met the prespecified inclusion and exclusion criteria were collected via chart review and Microsoft SQL Server Management Studio.
Collected data included age, gender, stimulant regimen (drug name, dose, frequency), indication and duration of use, prescriber name and specialty, prescribing origin of initial stimulant medication, and whether stimulant use predated military service. Monitoring of stimulant medications was assessed via UDS at least annually, query of the prescription drug monitoring program (PDMP) at least quarterly, and average time between follow-up appointments with stimulant prescriber.
Monitoring parameters were assessed from January 1, 2017 through June 30, 2018, as it was felt that the 6-month study period would be too narrow to accurately assess monitoring trends. Mental health diagnoses, ADHD diagnostic testing if applicable, documented SUD or stimulant misuse past or present, and concomitant central nervous system (CNS) depressant use also were collected. CNS depressants evaluated were those that have abuse potential or significant psychotropic effects and included benzodiazepines, antipsychotics, opioids, gabapentin/pregabalin, Z-hypnotics, and muscle relaxants.
Results
The majority of participants were male (168/200) with an average age of 43.3 years. Dextroamphetamine/amphetamine was the most used stimulant (48.5%), followed by methylphenidate (40%), and dextroamphetamine (10%). Lisdexamphetamine was the least used stimulant, likely due to its formulary-restricted status within this facility. An extended release (ER) formulation was utilized in 1 of 4 participants, with 1 of 20 participants prescribed a combination of immediate release (IR) and ER formulations. Duration of use ranged from 3 months to 14 years, with an average duration of 4 years (Table 1).
Nearly 40% of participants reported an origin of stimulant initiation outside of LVAHCS. Fourteen percent of participants were started on prescription stimulant medications while active-duty service members. Stimulant medications were initiated at another VA facility in 10.5% of instances, and 15% of participants reported being prescribed stimulant medications by a civilian prescriber prior to receiving them at LVAHCS. Seventy-four of 79 (93.6%) participants with an origin of stimulant prescription outside of LVAHCS reported a US Federal Food and Drug Administration (FDA)-approved indication for use.
Stimulant medications were used for FDA-approved indications (ADHD and narcolepsy) in 69.5% of participants. Note, this included patients who maintained an ADHD diagnosis in their medical record even if it was not substantiated with diagnostic testing. Of the participants reporting ADHD as an indication for stimulant use, diagnostic testing was conducted at LVAHCS to confirm an ADHD diagnosis in 58.6% (78/133) participants; 20.5% (16/78) of these diagnostic tests did not support the diagnosis of ADHD. All documented indications for use can be found in Table 2.
As expected, the most common indication was ADHD (66.5%), followed by ADHD-like symptoms (9%), refractory depression (7%), and fatigue (5.5%). Fourteen percent of participants had ≥ 1 change in indication for use, with some participants having up to 4 different documented indications while being prescribed stimulant medications. Twelve percent of participants were either denied stimulant initiation, or current stimulant medications were discontinued by one health provider and were restarted by another following a prescriber change. Aside from indication for stimulant use, 90% of participants had at least one additional mental health diagnosis. The rate of all mental health diagnoses documented in the medical record problem list can be found in Table 3.
A UDS was collected at least annually in 37% of participants. A methylphenidate confirmatory screen was ordered to assess adherence in just 2 (2.5%) participants prescribed methylphenidate. While actively prescribed stimulant medications, PDMP was queried quarterly in 26% of participants. Time to follow-up with the prescriber ranged from 1 to 15 months, and 40% of participants had follow-up at least quarterly. Instance of SUD, either active or in remission, differed when searched via problem list (36/200) and prescriber documentation (63/200). The most common SUD was alcohol use disorder (13%), followed by cannabis use disorder (5%), polysubstance use disorder (5%), opioid use disorder (4.5%), stimulant use disorder (2.5%), and sedative use disorder (1%). Twenty-five participants currently prescribed stimulant medications had stimulant abuse/misuse documented in their medical record. Fifty-four percent of participants were prescribed at least 1 CNS depressant considered to have abuse potential or significant psychotropic effects. Opioids were most common (23%), followed by muscle relaxants (15.5%), benzodiazepines (15%), antipsychotics (13%), gabapentin/pregabalin (12%), and Z-hypnotics (12%).
Discussion
The source of the initial stimulant prescription was assessed. The majority of veterans had received medical care prior to receiving care at LVAHCS, whether on active duty, from another VA facility throughout the country, or by a private civilian prescriber. The origin of initial stimulant medication and indication for stimulant medication use were patient reported. Requiring medical records from civilian providers prior to continuing stimulant medication is prescriber-dependent and was not available for all participants.
As expected, the majority of participants
The reasons for discontinuation included a positive UDS result for cocaine, psychosis, broken narcotic contract, ADHD diagnosis not supported by psychological testing, chronic bipolar disorder secondary to stimulant use, diversion, stimulant misuse, and lack of indication for use. There also were a handful of veterans whose VA prescribers declined to initiate prescription stimulant medications for various reasons, so the veteran sought care from a civilian prescriber who prescribed stimulant medications, then returned to the VA for medication management, and stimulant medications were continued. Fourteen percent (28/200) of participants had multiple indications for use at some point during stimulant medication therapy. Eight of those were a reasonable change from ADHD to ADHD-like symptoms when diagnosis was not substantiated by testing. The cause of other changes in indication for use was not well documented and often unclear. One veteran had 4 different indications for use documented in the medical record, often changing with each change in prescriber. It appeared that the most recent prescriber was uncertain of the actual indication for use but did not want to discontinue the medication. This prescriber documented that the stimulant medication should continue for presumed ADHD/mood/fatigue/cognitive dysfunction, which were all of the indications documented by the veteran’s previous prescribers.
Reasons for Discontinuation
ADHD was the most prominent indication for use, although the indication was changed to ADHD-like symptoms in several veterans for whom diagnostic testing did not support the ADHD diagnosis. Seventy-eight of 133 veterans prescribed stimulant medications for ADHD received diagnostic testing via a psychologist at LVAHCS. For the 11 veterans who had testing after stimulant initiation, a stimulant-free period was required prior to testing to ensure an accurate diagnosis. For 21% of veterans, the ADHD diagnosis was unsubstantiated by formal testing; however, all of these veterans continued stimulant medication use. For 1 veteran, the psychologist performing the testing documented new diagnoses, including moderate to severe stimulant use disorder and malingering both for PTSD and ADHD. The rate of stimulant prescribing inconsistency, “prescriber-hopping,” and unsupported ADHD diagnosis results warrant a conversation about expectations for transitions of care regarding stimulant medications, not only from outside to inside LVAHCS, but from prescriber to prescriber within the facility.
In some cases, stimulant medications were discontinued by a prescriber secondary to a worsening of another mental health condition. More than half of the participants in this study had an anxiety disorder diagnosis. Whether or not anxiety predated stimulant use or whether the use of stimulant medications contributed to the diagnosis and thus the addition of an additional CNS depressant to treat anxiety may be an area of research for future consideration. Although bipolar disorder, anxiety disorders, psychosis, and SUD are not contraindications for use of stimulant medications, caution must be used in patients with these diagnoses. Prescribers must weigh risks vs benefits as well as perform close monitoring during use. Similarly, one might look further into stimulant medications prescribed for fatigue and assess the role of any simultaneously prescribed CNS depressants. Is the stimulant being used to treat the adverse effect (AE) of another medication? In 2 documented instances in this study, a psychologist conducted diagnostic testing who reported that the veteran did not meet the criteria for ADHD but that a stimulant may help counteract the iatrogenic effect of anticonvulsants. In both instances stimulant use continued.
Prescription Monitoring
Polysubstance use disorder (5%) was the third most common SUD recorded among study participants. The majority of those with polysubstance use disorder reported abuse/misuse of illicit or prescribed stimulants. Stimulant abuse/misuse was documented in 25 of 200 (12.5%) study participants. In several instances, abuse/misuse was detected by the LVAHCS delivery coordination pharmacist who tracks patterns of early fill requests and prescriptions reported lost/stolen. This pharmacist may request that the prescriber obtain PDMP query, UDS, or pill count if concerning patterns are noted. Lisdexamphetamine is a formulary-restricted medication at LVAHCS, but it was noted to be approved for use when prescribers requested an abuse-deterrent formulation. Investigators noticed a trend in veterans whose prescriptions exceeded the recommended maximum dosage also having stimulant abuse/misuse documented in their medical record. The highest documented total daily dose in this study was 120-mg amphetamine salts IR for ADHD, compared with the normal recommended dosing range of 5 to 40 mg/d for the same indication.
Various modalities were used to monitor participants but less than half of veterans had an annual UDS, quarterly PDMP query, and quarterly prescriber follow-up. PDMP queries and prescriber follow-up was assessed quarterly as would be reasonable given that private sector practitioners may issue multiple prescriptions authorizing the patient to receive up to a 90-day supply.7 Prescriber follow-up ranged from 1 to 15 months. A longer time to follow-up was seen more frequently in stimulant medications prescribed by primary care as compared with that of mental health.
Clinical Practice Protocol
Data from this study were collected with the intent to identify opportunities for improvement in the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a facility-specific clinical practice protocol (CPP) for stimulant prescribing. It may also be beneficial to formulate a chronic stimulant management agreement between patient and prescriber to provide informed consent and clear expectations prior to stimulant medication initiation.
A CPP could be used to establish stimulant prescribing rules within a facility, which may limit who can prescribe stimulant medications or include a review process and/or required documentation in the medical record when being prescribed outside of specified dosing range and indications for use designated in the CPP or other evidence-based guidelines. Transition of care was found to be an area of opportunity in this study, which could be mitigated with the requirement of a baseline assessment prior to stimulant initiation with the expectation that it be completed regardless of prior prescription stimulant medication use. There was a lack of consistent monitoring for participants in this study, which may be improved if required monitoring parameters and frequency were provided for prescribers. For example, monitoring of heart rate and blood pressure was not assessed in this study, but a CPP may include monitoring vital signs before and after each dose change and every 6 months, per recommendation from the National Institute for Health and Care Excellence ADHD Diagnosis and Management guideline published in 2018.8The CPP may list the responsibilities of all those involved in the prescribing of stimulant medications, such as mental health service leadership, prescribers, nursing staff, pharmacists, social workers, psychologists, and other mental health staff. For prescribers this may include a thorough baseline assessment and criteria for use that must be met prior to stimulant initiation, documentation that must be included in the medical record and required monitoring during stimulant treatment, and expectations for increased monitoring and/or termination of treatment with nonadherence, diversion, or abuse/misuse.
The responsibilities of pharmacists may include establishing criteria for use of nonformulary and restricted agents as well as completion of nonformulary/restricted requests, reviewing dosages that exceed the recommended FDA daily maximum, reviewing uncommon off-label uses of stimulant medications, review and document early fill requests, potential nonadherence, potential drug-seeking behavior, and communication of the following information to the primary prescriber. For other mental health staff this may include documenting any reported AEs of the medication, referring the patient to their prescriber or pharmacist for any medication questions or concerns, and assessment of effectiveness and/or worsening behavior during patient contact.
Limitations
One limitation of this study was the way that data were pulled from patient charts. For example, only 3/200 participants in this study had insomnia per diagnosis codes, whereas that number was substantially higher when chart review was used to assess active prescriptions for sleep aids or documented complaints of insomnia in prescriber progress notes. For this same reason, rates of SUDs must be interpreted with caution as well. SUD diagnosis, both current and in remission were taken into account during data collection. Per diagnosis codes, 36 (18%) veterans in this study had a history of SUD, but this number was higher (31.5%) during chart review. The majority of discrepancies were found when participants reported a history of SUD to the prescriber, but this information was not captured via the problem list or encounter codes. What some may consider a minor omission in documentation can have a large impact on patient care as it is unlikely that prescribers have adequate administrative time to complete a chart review in order to find a complete past medical history as was required of investigators in this study. For this reason, incomplete provider documentation and human error that can occur as a result of a retrospective chart review were also identified as study limitations.
Conclusion
Our data show that there is still substantial room for improvement in the prescribing and monitoring of stimulant medications. The rate of stimulant prescribing inconsistency, prescriber-hopping, and unsupported ADHD diagnosis resulting from formal diagnostic testing warrant a review in the processes for transition of care regarding stimulant medications, both within and outside of this facility. A lack of consistent monitoring was also identified in this study. One of the most appreciable areas of opportunity resulting from this study is the need for consistency in both the prescribing and monitoring of stimulant medications. From the above results investigators concluded that this facility may benefit from implementation of a CPP for stimulant prescribing as well as a chronic stimulant management agreement to provide clear expectations for patients and prescribers prior to and during prescription stimulant use.
Acknowledgments
We thank Tori Wilhoit, PharmD candidate, and Dana Fischer, PharmD candidate, for their participation in data collection and Courtney Eatmon, PharmD, BCPP, for her general administrative support throughout this study.
1. Safer DJ. Recent trends in stimulant usage. J Atten Disord. 2016;20(6):471-477.
2. Christopher Jones; US Food and Drug Administration. The opioid epidemic overview and a look to the future. http://www.agencymeddirectors.wa.gov/Files/OpioidConference/2Jones_OPIOIDEPIDEMICOVERVIEW.pdf. Published June 12, 2015. Accessed January 16, 2020.
3. Compton WM, Han B, Blanco C, Johnson K, Jones CM. Prevalence and correlates of prescription stimulant use, misuse, use disorders, motivations for misuse among adults in the United States. Am J Psychiatry. 2018;175(8):741-755.
4. Westover AN, Nakonezney PA, Halm EA, Adinoff B. Risk of amphetamine use disorder and mortality among incident users of prescribed stimulant medications in the Veterans Administration. Addiction. 2018;113(5):857-867.
5. Olfson M, Blanco C, Wang S, Greenhill LL. Trends in office-based treatment of adults with stimulant medications in the United States. J Clin Psychiatry. 2013;74(1):43-50.
6. Olfson M, Marcus SC, Zhang HF, and Wan GJ. Continuity in methylphenidate treatment of adults with attention-deficit/hyperactivity disorder. J Manag Care Pharm. 2007;13(7): 570-577.
7. 21 CFR § 1306.12
8. National Collaborating Centre for Mental Health (UK). Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. NICE Clinical Guidelines, No. 87. Leicester, United Kingdom: British Psychological Society; 2018.
1. Safer DJ. Recent trends in stimulant usage. J Atten Disord. 2016;20(6):471-477.
2. Christopher Jones; US Food and Drug Administration. The opioid epidemic overview and a look to the future. http://www.agencymeddirectors.wa.gov/Files/OpioidConference/2Jones_OPIOIDEPIDEMICOVERVIEW.pdf. Published June 12, 2015. Accessed January 16, 2020.
3. Compton WM, Han B, Blanco C, Johnson K, Jones CM. Prevalence and correlates of prescription stimulant use, misuse, use disorders, motivations for misuse among adults in the United States. Am J Psychiatry. 2018;175(8):741-755.
4. Westover AN, Nakonezney PA, Halm EA, Adinoff B. Risk of amphetamine use disorder and mortality among incident users of prescribed stimulant medications in the Veterans Administration. Addiction. 2018;113(5):857-867.
5. Olfson M, Blanco C, Wang S, Greenhill LL. Trends in office-based treatment of adults with stimulant medications in the United States. J Clin Psychiatry. 2013;74(1):43-50.
6. Olfson M, Marcus SC, Zhang HF, and Wan GJ. Continuity in methylphenidate treatment of adults with attention-deficit/hyperactivity disorder. J Manag Care Pharm. 2007;13(7): 570-577.
7. 21 CFR § 1306.12
8. National Collaborating Centre for Mental Health (UK). Attention deficit hyperactivity disorder: diagnosis and management of ADHD in children, young people and adults. NICE Clinical Guidelines, No. 87. Leicester, United Kingdom: British Psychological Society; 2018.
The Group Practice Manager in the VHA: A View From the Field
The Veterans Health Administration (VHA) provides care for 9 million veterans at 1,255 health care sites linked to one of 170 local medical systems.1 Recognizing that providing timely care requires effective access management, the US Congress mandated training of VHA staff to manage and improve access to care but did not provide additional local funds for new positions.2 In response, the VHA created the group practice manager (GPM), a new position responsible for improving clinical practice management and unifying access improvement across leadership levels, professions, and services within each local medical system.
In May 2015, the VHA began hiring and training GPMs to spearhead management of access to services. The US Department of Veterans Affairs (VA) Office of Veteran Access to Care spearheaded GPM training, including face-to-face sessions, national calls, webinars, and educational materials. Five local medical systems were selected by the VA Office of Veteran Access to Care to implement the GPM role to allow for an early evaluation of the program that would inform a subsequent nationwide rollout. Implementation of the GPM role remained in the hands of local medical systems.
Longer wait times are shown to impact patient health.3,4 Open access scheduling and other patient-centered access management interventions have been shown to improve availability of primary care appointments.5 However, little empirical evidence exists regarding the managers who focus on clinic access interventions. While the nonpeer-reviewed literature includes references to such roles, including GPMs, the empirical literature has focused on external practice faciliators,6-8 “mid-level managers,”9 and clinic staff.10 We found no peer-reviewed articles on the needs and experiences of practice managers who are focused on improving access. The purpose of this study was to examine GPM prototype sites to both enhance subsequent nationwide implementation and to advance empirical literature on managing patient access within health care.
Methods
In 2015, the VA identified 5 prototype sites representing diverse geographic locations, size, and complexity for the implementation of the GPM role (Table 1). These sites self-identified as having clinical practice management experience. GPMs attended 4 training sessions between February and August 2015.
Data Collection
Participants from each prototype site included GPMs, national trainers, clinic leaders, and frontline staff. Table 2 includes the roles and sample size. Participants were recruited through purposive sampling followed by snowball sampling until thematic saturation was reached (the point at which subsequent data fail to produce new findings across sites and roles of interest).
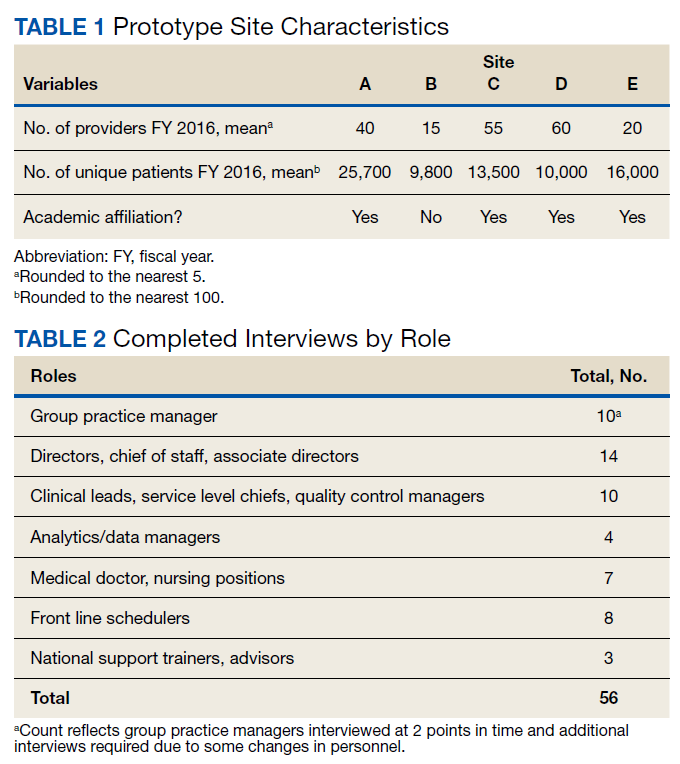
Guided by the Consolidated Framework for Implementation Research (CFIR), the research team developed semistructured interview guides tailored to participants’ roles to elicit rich descriptions regarding overall impressions, practice management strategies, goals, activities, relationship to clinic roles, data analytics usage, challenges, barriers, and facilitators.11 These guides included open-ended questions and structured prompts utilizing participant language for follow-up probes to minimize interviewing bias (eAppendix:
Data Analysis
Data were analyzed using iterative deductive and inductive content analysis.12 Deductive content analysis consisted of identifying quotes that fit within preidentified categories (ie, perceptions of national effort, organizational structure for GPM, challenges, facilitators, metrics and tools, and mobilizing access culture) developed by the interdisciplinary research team. Further content analysis entailed open-coding and iteratively revisiting and reconciling codes associated within each preidentified category as new codes emerged. The team analyzed the resulting codes to inductively and iteratively identify and stabilize themes regarding the GPM role: roles and tasks, GPM characteristics, issues, and challenges. Through this process we moved coded data to reconciled descriptions suited to addressing the purposes of this study. Dedoose 7.0.23 software was used for qualitative data management and analysis.
Results
The study identified participants’ overall impressions of the GPM initiative and key themes within 4 major domains regarding implementing the GPM role: roles and tasks (implementing clinic practice management, leading patient access, supporting data analytics, and enabling self and staff); GPM characteristics (familiarity with clinical services, knowledge of VHA systems, ability to analyze patient data, communication skills, and the ability to work with others); and issues, and challenges (technical, social, and structural).
Overall Impressions
Interviewees perceived the GPM initiative as a consolidation of existing distributed responsibilities into one role that directly reported to local top-level management with indirect reporting to national leaders. Many of the sites reported that they had designated or planned to designate a role resembling the GPM prior to the initiative. “There are staff who’ve been doing some of this work all along,” a GPM noted. “We just didn’t have them grouped together. They weren’t necessarily all working in the same type of service under the same type of structure.”
Whether the GPM position was new or not, participants referenced the importance and challenges of engaging the local facility in recognizing the agency associated with the GPM position. According to national support, the staff are trying to get the facility to understand “why the group practice manager is so important… we’ve got to embed that standard position in the system.”
While the GPM was recognized as the hub of access management, respondents recognized that transformation regarding access involved many players. “We have to create [an] orchestrated team inside each facility,” an advisor argued.
Respondents discussed how the initiative allows local facilities to appoint a specific person with a specific title and role who helps facilitate, organize, and legitimize an access focus at their sites. One GPM interviewee noted how the initiative helped refocus some of their previously less centralized efforts. “We’ve always looked at productivity; we’ve always looked at access; we’ve always looked at efficiency. I think the bigger difference is now there are individuals identified in the clinics, as practice managers as well…I interact with them. They interact with individual clinic staff, and it’s more of a group process than a single individual.”
The value of having tools available and being able to track and manage patient care as a specific example of the positive impact of this new role was noted by participants. A GPM noted that many health care providers will be happy to have tools to better manage their services and a process “that flows from a service level all the way up to executive management, where there is a common interest in making those things happen—I think that’s going to be a tremendous help.”
Participants expressed concern that the national GPM rollout would be a one-size-fits-all approach. These respondents emphasized the need to have the flexibility to customize their activities to meet their unique site and patient needs.
GPM Roles and Tasks
Participants described 4 primary roles that the GPM was expected to fill: implementing clinic practice management, leading patient access, supporting data analytics, and enabling self and staff. Some activities overlapped in that they served to support multiple role areas (Figure 1).
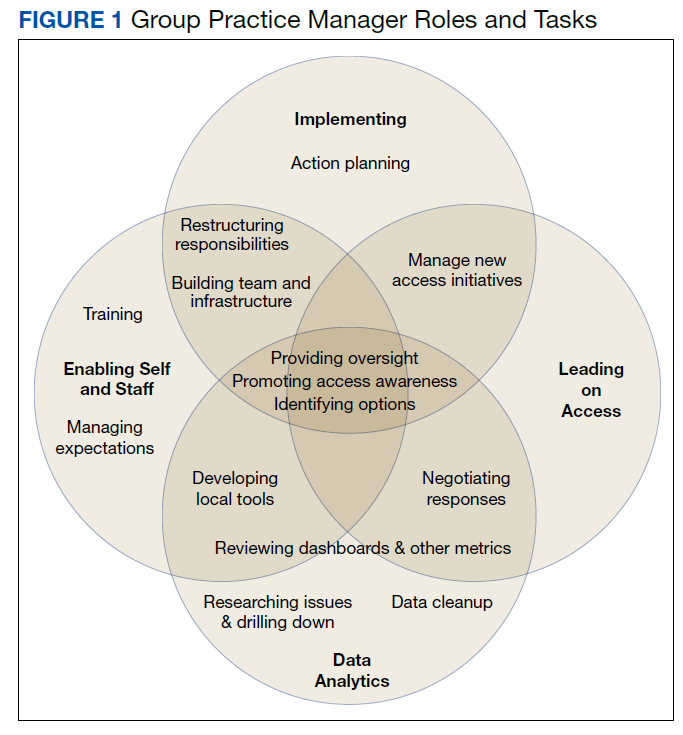
Implementing clinic practice management. In the early stages of the initiative, the GPM’s primary role was to prepare the facility to implement a standardized set of clinic practice management (CPM) team processes. Part of standardizing the CPM process was defining the scope and tasks of the GPM, which requires significant planning for the implementation. “My big job is to finalize what we think group practice management is going to look [like] here,” a GPM reported.
Each prototype site had latitude to interpret the GPM initiative in a way that would work in their context within given VHA boundaries and ongoing initiatives. To achieve the high-level vision and purpose, the GPM first had to develop action plans that accounted for the operating environment of the facility. According to one GPM, VA national officials are “constantly” asking for action plans, which required significant time by specific deadlines. “They want an action plan [and to] clean up all your consults, [and to] clean up all your recall reminders.”
Leading on improving access efforts. Participants saw the GPM as the central staff member responsible for providing oversight of any activities and people involved in improving access. “I ensure everybody is doing what they’re supposed to do,” one GPM reported. When the GPM sees areas that are not being addressed, the individual tries to develop a process or training to “close those gaps.”
GPMs promoted an awareness of their goals, changes in process, and new tools accompanying the initiative. However, other access initiatives were occurring simultaneously creating confusion for health care providers and patients; thus GPMs found they were managing a wide array of related initiatives.
GPMs have to negotiate with leaders across the VHA facility, many of whom operated at a higher leadership level and had different priorities, to address access problems.
“I’m a lieutenant as a GPM in a clinic, a GPM noted. “How is the lieutenant going to talk to a major or a colonel in the clinic and say your clinic has problems. How[‘s] that lieutenant...going to do that? With people skills!”
Managing expectations about the speed and to what extent a problem could be resolved was an important part of the GPM leadership role. “I see myself as managing expectations both up to the leadership and down to the frontline,” a GPM explained. “I find myself talking to leadership [about] our progress. But at the same time, we have to say, ‘not everything can be fixed overnight.’”
Providing leadership on access-related issues included developing a range of options for addressing patient access problems. One analytics manager recounted how the GPM role led to evaluating how physical space limited efficiency in clinic flow. The first step was identifying possible additional rooms to improve clinic flow. This required working with the space committee to “get someone to look at our overarching space and find someplace else for them to sit” to avoid adding to congestion in the clinic area.
Supporting data analytics.
GPMs routinely immersed themselves in the data to understand access issues. GPMs worked with clinic leaders to identify the underlying causes and various solutions. The GPMs maintained access through administrative scrubbing of the data and finding “smart ways to get patients scheduled,” a GPM explained. “I don’t think our facility would have taken care of as many veterans in the time frame as we did....We’ve cleared over 4,000 consults that were older than 90 days. We have cleared thousands and thousands of weekly reminders.”
GPMs expressed the need for aggregated (ie, dashboard) and standardized information to efficiently address access issues. “I would like to have some more standardization on what’s being reviewed; it seems to change frequently, and so [to] be able to track and trend and have something given to me to review,” one health care provider requested. On the other hand, participants also described a need for decision support tools that would lead to action aligned with best practices. “Instead of a dashboard or something that’s just measuring their performance, it’s more something that they can look at and take action” a national support staff advisor suggested.
Enabling self and staff. GPMs felt they were most effective if they enabled themselves and stakeholders through training and by cultivating relationships and team building. Figure 2 illustrates the various stakeholders GPMs reported engaging with. The GPMS should be building relationships, bridging relationships, developing trust, and then providing a higher level of hands-on management. However, “that doesn’t really happen right now, day to day,” one member of leadership reported.
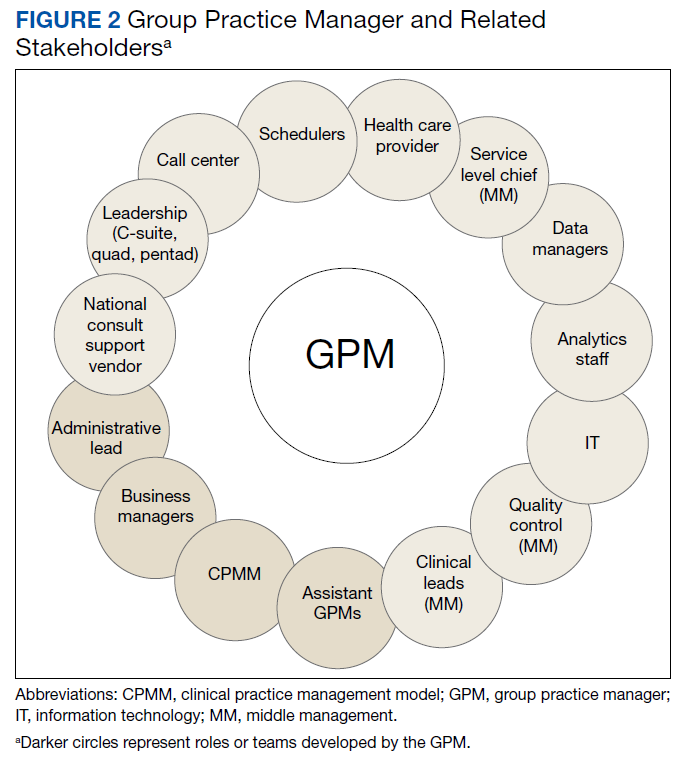
Key topics in GPM leadership training included both soft skills (change management, culture change, and negotiation skills) and crucial analytic/technical training (understanding each metric and dashboard available, data analytics, and supply/demand balancing techniques). The GPMs not only wanted to understand metrics as part of their training, but also want to know what to do about them.
An “operationalization” training approach (discerning the meaning of data, data-based decision making, and determining action from multiple options) inspired by real-life situations was preferred by participants. Other effective learning structures included job aids in the form of templated Gantt charts, process maps to guide GPMs through implementation of new processes, formalized peer learning (accumulated field insights shared during training courses), and informal peer sharing of direct experiences during calls.
GPMs also emphasized training for frontline clinical and support staff, including schedulers. VHA schedulers typically have less education and experience higher turnover rates than do other clinic staff, yet they carry out complex and critical tasks. Providing training and ensuring that any materials developed for training and education were appropriate to the level of education of schedulers was an important task for GPMs. “If they don’t understand all of the scheduling principles and potential,” one GPM explained, “we will not be maximizing the utilization of our parts.”
GPMs also provided informal education to clinicians. Participants noted GPMs have to avoid appearing to overstep their positions or presuming more knowledge and expertise than clinicians. They “have to be able to teach a physician without being overbearing, in a way a physician will accept it as advisement,” one program leader reported.
GPM knowledge, skills, and abilities. GPMs presented a complex range of knowledge, skills, and abilities, including clinical, administrative, analytics, and people skills. All interviewees reported that their prior education and experience did not sufficiently train them for the GPM role. GPMs identified a willingness to learn quickly as a critical characteristic. Many GPMs tended to have a formal education in health administration or business (eg, MBAs); others had administrative experience (eg, administrative assistance to executive leadership) or clinical training (eg, physician assistant). Detailed clinical knowledge was not expected, but clinical familiarity was helpful.
Some interviewees also mentioned previous experience and familiarity with the VHA system specifically as an advantage. This was especially true for VA outpatient flows, clinic flows, and understanding what an outpatient is in a VA context. Interviewees noted the importance of GPMs needing to be able to analyze patient demand metrics and underlying data in order to determine supply of providers and then to allocate adequate resources to complement providers. Forecasting skills were referenced as a key point. “They need to be able to be assured that they can recruit more providers if needed,” a national support staff advisor noted.
Given the importance of developing effective relationships, communication skills were mentioned by most participants and underscored as critical to establishing trust between GPMs and others as the initiative was being implemented. Interviewees indicated that relationship building was further enhanced when GPMs possessed the ability to “work with” rather than command clinicians and staff; navigate politics; and were respectful of other people’s knowledge, skills, abilities, and status. “They have to work with the nursing staff and teach them,” a leader described, “so that people understand that we are going to a different place to achieve our primary objectives and goals.”
Issues and Challenges
Participants identified several technical, social, and structural challenges and barriers to successfully implementing the GPM role.
Technical challenges. Recurring themes across all phases of data analytics were GPMs’ capability to challenge data use and use large volumes of information from multiple data sources (entering and accessing data; “drilling down” from summaries; generating reports; and analyzing and interpreting resulting metrics). Interviewees reported that information assessment and analytic support were not consistent. One GPM had a data analyst pulling reports needed to support clinical units while other GPMs trained staff to pull data. Even with support, some GPMs had issues due to limited information technology (IT) skills or access privileges leading to inefficiencies and delays. “Whenever I need anything from a programmer, I have to go through, you know, the IT gods in the sky,” one GPM remarked. “That usually takes a few months or more.”
Social challenges. Instituting the GPM role was a cultural change, and interviewees reported needing to address resistance to CPM model efforts. Resistance to change “is particularly hard in the VA just because it has a unique culture,” one leader noted. “There is a comfort in the legacy way of doing things.” The GPM initiative was introduced during a time when other national level initiatives were being implemented throughout the VHA. Fulfilling requests for information for these initiatives became the responsibility of the GPM and their team, which diverted attention from the mandate to improve access. Furthermore, GPMs were often considered the “change communicators” to clinics putting them in the role of “bad messenger,” which degraded trust and made it difficult to partner with clinicians.
Efforts to work through change management and build relationships included general program awareness presentation to internal stakeholders; including key stakeholders in GPM committees; pre-emptive conversations with unit chiefs; creating awareness of the GPM activities and progress through formal and informal update meetings; and identifying successes regarding access.
Structural challenges. The GPM role did not have direct supervision over clinical and administrative leaders, making it challenging to enact change. GPMs reported that “they do not always have authority over the area that they are being asked to manage,” which made their work difficult, requiring strong negotiation skills and political savvy to affect change. However, as the clinic staff and providers saw how the GPM could support and positively impact their practice, these challenges began to subside.
Discussion
This study provides empirical evidence regarding the implementation of a new access management strategy for health care systems focused on improving timeliness of care. First, the GPM position was seen as critical at each facility, as a single point person, to help local system leaders respond effectively to both national mandates and local context. Second, requiring the GPMs to report to the medical center director or chief of staff was important for integrating access perspectives across service lines and to facilitate a strong GPM role in strategic planning. Third, the intentional flexibility of the access management initiative, beyond the nationally specified aspects of the GPM role, was key for allowing individual sites to adapt to unique local challenges, resources, and population demands. Fourth, the initiative provided GPMs with opportunities to learn important skills and support the acquisition, utilization, and communication of a tremendous range of data toward responsive action.
According to our respondents, the GPM role demands functioning across a broad set of responsibilities; understanding the big picture as well as the complex underlying variables; engaging facility leaders, clinical and administrative staff; and prioritizing competing national and local demands. Consistent with previous findings, effective GPMs must possess a complex set of skills (interpersonal, analytic, and leadership) and the ability to create a supporting team.13
In practice, improving access at individual sites of care (VA medical centers and community-based outpatient clinics) poses significant challenges, especially in the early stages, even with the assistance of a GPM. For example, some respondents reported being overwhelmed by the volume of available data and dashboards, and responding to current requests for data analysis and dissemination sometimes impeded long range planning. Multiple national access-related initiatives and local pressures also generated excessive and potentially conflicting demands. Thus, while the creation of a GPM position seemed to be essential for the pilot sites to improve local access and timeliness to care, success also requires ongoing national and facility-wide communication, education, and support. Ongoing data analysis training and support will be critical to ensuring the sustainability of the position. Last, recruiting GPMs with the needed complex skill set may prove to be challenging, and it will be important to provide resources to identify, attract, and retain well-qualified GPMs.
Limitations and Future Work
This study was based on a small initial sample of pilot sites of varying sizes and, as such, may not reflect the experience of all VHA GPMs. In addition, the use of snowball sampling, while facilitating identification of key stakeholders, may introduce bias in participant sampling. Nonetheless, the results from this study provide findings that informed the national VHA GPM initiative and can inform further studies of practice management roles outside of the VA.
Further study of the VHA GPM implementation and similar roles in other health care systems is needed. As the GPM position is fully implemented across the VHA, large scale evaluation is needed to gain a more representative picture and allow for comparison of the GPM role at various types of facilities (eg, size, rurality, complexity, ranking based on access performance metrics).
Conclusion
Improving access to care is a central goal for health care systems. The incorporation of the GPM role is an innovative approach to improve access management strategies. Early study of prototype sites provided VHA leadership with valuable insights used to influence further rollout of this initiative. Based on our findings, national and local support are important to ongoing success. National access mandates, training, and resources should focus on ensuring sufficient GPM authority, enabling GPMs to use data, and ensuring GPMs engage with frontline clinical and administrative staff to improve veteran access to care.
1. US Department of Veterans Affairs. Veterans Health Administration. https://www.va.gov/health. Updated October 25, 2019. Accessed January 8, 2020.
2. Veterans Access, Choice, and Accountability Act of 2014. 38 CFR § 17.1500 (2014).
3. Fahmy N, Aprikian A, Al-Otaibi M, et al. Impact of treatment delay in patients with bladder cancer managed with partial cystectomy in Quebec: a population-based study. Can Urol Assoc J. 2009;3(2):131-135.
4. Hill CJ, Joonas K. The impact of unacceptable wait time on health care patients’ attitudes and actions. Health Mark Q. 2005;23(2):69-87.
5. Ansell D, Crispo JAG, Simard B, Bjerre LM. Interventions to reduce wait times for primary care appointments: a systematic review. BMC Health Serv Res. 2017;17(1):295.
6. Kotecha J, Han H, Green M, Russell G, Martin MI, Birtwhistle R. The role of the practice facilitators in Ontario primary healthcare quality improvement. BMC Fam Pract. 2015;16:93.
7. Taylor EF, Machta RM, Meyers DS, Genevro J, Peikes DN. Enhancing the primary care team to provide redesigned care: the roles of practice facilitators and care managers. Ann Fam Med. 2013;11(1):80-83.
8. Liddy C, Laferriere D, Baskerville B, Dahrouge S, Knox L, Hogg W. An overview of practice facilitation programs in Canada: current perspectives and future directions. Healthc Policy. 2013;8(3):58-67.
9. Birken SA, Lee SY, Weiner BJ, Chin MH, Schaefer CT. Improving the effectiveness of health care innovation implementation: middle managers as change agents. Med Care Res Rev. 2013;70(1):29-45.
10. Ahluwalia S, Offredy M. A qualitative study of the impact of the implementation of advanced access in primary healthcare on the working lives of general practice staff. BMC Fam Pract. 2005;6:39.
11. Damschroder LJ, Aron DC, Keith RE, Kirsh SR, Alexander JA, Lowery JC. Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4:50.
12. Elo S, Kyngäs H. The qualitative content analysis process. J Adv Nurs. 2008;62(1):107-115.
13. Stefl ME. Common competencies for all healthcare managers: the Healthcare Leadership Alliance model. J Healthc Manag. 2008;53(6):360-374.
The Veterans Health Administration (VHA) provides care for 9 million veterans at 1,255 health care sites linked to one of 170 local medical systems.1 Recognizing that providing timely care requires effective access management, the US Congress mandated training of VHA staff to manage and improve access to care but did not provide additional local funds for new positions.2 In response, the VHA created the group practice manager (GPM), a new position responsible for improving clinical practice management and unifying access improvement across leadership levels, professions, and services within each local medical system.
In May 2015, the VHA began hiring and training GPMs to spearhead management of access to services. The US Department of Veterans Affairs (VA) Office of Veteran Access to Care spearheaded GPM training, including face-to-face sessions, national calls, webinars, and educational materials. Five local medical systems were selected by the VA Office of Veteran Access to Care to implement the GPM role to allow for an early evaluation of the program that would inform a subsequent nationwide rollout. Implementation of the GPM role remained in the hands of local medical systems.
Longer wait times are shown to impact patient health.3,4 Open access scheduling and other patient-centered access management interventions have been shown to improve availability of primary care appointments.5 However, little empirical evidence exists regarding the managers who focus on clinic access interventions. While the nonpeer-reviewed literature includes references to such roles, including GPMs, the empirical literature has focused on external practice faciliators,6-8 “mid-level managers,”9 and clinic staff.10 We found no peer-reviewed articles on the needs and experiences of practice managers who are focused on improving access. The purpose of this study was to examine GPM prototype sites to both enhance subsequent nationwide implementation and to advance empirical literature on managing patient access within health care.
Methods
In 2015, the VA identified 5 prototype sites representing diverse geographic locations, size, and complexity for the implementation of the GPM role (Table 1). These sites self-identified as having clinical practice management experience. GPMs attended 4 training sessions between February and August 2015.
Data Collection
Participants from each prototype site included GPMs, national trainers, clinic leaders, and frontline staff. Table 2 includes the roles and sample size. Participants were recruited through purposive sampling followed by snowball sampling until thematic saturation was reached (the point at which subsequent data fail to produce new findings across sites and roles of interest).
Guided by the Consolidated Framework for Implementation Research (CFIR), the research team developed semistructured interview guides tailored to participants’ roles to elicit rich descriptions regarding overall impressions, practice management strategies, goals, activities, relationship to clinic roles, data analytics usage, challenges, barriers, and facilitators.11 These guides included open-ended questions and structured prompts utilizing participant language for follow-up probes to minimize interviewing bias (eAppendix:
Data Analysis
Data were analyzed using iterative deductive and inductive content analysis.12 Deductive content analysis consisted of identifying quotes that fit within preidentified categories (ie, perceptions of national effort, organizational structure for GPM, challenges, facilitators, metrics and tools, and mobilizing access culture) developed by the interdisciplinary research team. Further content analysis entailed open-coding and iteratively revisiting and reconciling codes associated within each preidentified category as new codes emerged. The team analyzed the resulting codes to inductively and iteratively identify and stabilize themes regarding the GPM role: roles and tasks, GPM characteristics, issues, and challenges. Through this process we moved coded data to reconciled descriptions suited to addressing the purposes of this study. Dedoose 7.0.23 software was used for qualitative data management and analysis.
Results
The study identified participants’ overall impressions of the GPM initiative and key themes within 4 major domains regarding implementing the GPM role: roles and tasks (implementing clinic practice management, leading patient access, supporting data analytics, and enabling self and staff); GPM characteristics (familiarity with clinical services, knowledge of VHA systems, ability to analyze patient data, communication skills, and the ability to work with others); and issues, and challenges (technical, social, and structural).
Overall Impressions
Interviewees perceived the GPM initiative as a consolidation of existing distributed responsibilities into one role that directly reported to local top-level management with indirect reporting to national leaders. Many of the sites reported that they had designated or planned to designate a role resembling the GPM prior to the initiative. “There are staff who’ve been doing some of this work all along,” a GPM noted. “We just didn’t have them grouped together. They weren’t necessarily all working in the same type of service under the same type of structure.”
Whether the GPM position was new or not, participants referenced the importance and challenges of engaging the local facility in recognizing the agency associated with the GPM position. According to national support, the staff are trying to get the facility to understand “why the group practice manager is so important… we’ve got to embed that standard position in the system.”
While the GPM was recognized as the hub of access management, respondents recognized that transformation regarding access involved many players. “We have to create [an] orchestrated team inside each facility,” an advisor argued.
Respondents discussed how the initiative allows local facilities to appoint a specific person with a specific title and role who helps facilitate, organize, and legitimize an access focus at their sites. One GPM interviewee noted how the initiative helped refocus some of their previously less centralized efforts. “We’ve always looked at productivity; we’ve always looked at access; we’ve always looked at efficiency. I think the bigger difference is now there are individuals identified in the clinics, as practice managers as well…I interact with them. They interact with individual clinic staff, and it’s more of a group process than a single individual.”
The value of having tools available and being able to track and manage patient care as a specific example of the positive impact of this new role was noted by participants. A GPM noted that many health care providers will be happy to have tools to better manage their services and a process “that flows from a service level all the way up to executive management, where there is a common interest in making those things happen—I think that’s going to be a tremendous help.”
Participants expressed concern that the national GPM rollout would be a one-size-fits-all approach. These respondents emphasized the need to have the flexibility to customize their activities to meet their unique site and patient needs.
GPM Roles and Tasks
Participants described 4 primary roles that the GPM was expected to fill: implementing clinic practice management, leading patient access, supporting data analytics, and enabling self and staff. Some activities overlapped in that they served to support multiple role areas (Figure 1).
Implementing clinic practice management. In the early stages of the initiative, the GPM’s primary role was to prepare the facility to implement a standardized set of clinic practice management (CPM) team processes. Part of standardizing the CPM process was defining the scope and tasks of the GPM, which requires significant planning for the implementation. “My big job is to finalize what we think group practice management is going to look [like] here,” a GPM reported.
Each prototype site had latitude to interpret the GPM initiative in a way that would work in their context within given VHA boundaries and ongoing initiatives. To achieve the high-level vision and purpose, the GPM first had to develop action plans that accounted for the operating environment of the facility. According to one GPM, VA national officials are “constantly” asking for action plans, which required significant time by specific deadlines. “They want an action plan [and to] clean up all your consults, [and to] clean up all your recall reminders.”
Leading on improving access efforts. Participants saw the GPM as the central staff member responsible for providing oversight of any activities and people involved in improving access. “I ensure everybody is doing what they’re supposed to do,” one GPM reported. When the GPM sees areas that are not being addressed, the individual tries to develop a process or training to “close those gaps.”
GPMs promoted an awareness of their goals, changes in process, and new tools accompanying the initiative. However, other access initiatives were occurring simultaneously creating confusion for health care providers and patients; thus GPMs found they were managing a wide array of related initiatives.
GPMs have to negotiate with leaders across the VHA facility, many of whom operated at a higher leadership level and had different priorities, to address access problems.
“I’m a lieutenant as a GPM in a clinic, a GPM noted. “How is the lieutenant going to talk to a major or a colonel in the clinic and say your clinic has problems. How[‘s] that lieutenant...going to do that? With people skills!”
Managing expectations about the speed and to what extent a problem could be resolved was an important part of the GPM leadership role. “I see myself as managing expectations both up to the leadership and down to the frontline,” a GPM explained. “I find myself talking to leadership [about] our progress. But at the same time, we have to say, ‘not everything can be fixed overnight.’”
Providing leadership on access-related issues included developing a range of options for addressing patient access problems. One analytics manager recounted how the GPM role led to evaluating how physical space limited efficiency in clinic flow. The first step was identifying possible additional rooms to improve clinic flow. This required working with the space committee to “get someone to look at our overarching space and find someplace else for them to sit” to avoid adding to congestion in the clinic area.
Supporting data analytics.
GPMs routinely immersed themselves in the data to understand access issues. GPMs worked with clinic leaders to identify the underlying causes and various solutions. The GPMs maintained access through administrative scrubbing of the data and finding “smart ways to get patients scheduled,” a GPM explained. “I don’t think our facility would have taken care of as many veterans in the time frame as we did....We’ve cleared over 4,000 consults that were older than 90 days. We have cleared thousands and thousands of weekly reminders.”
GPMs expressed the need for aggregated (ie, dashboard) and standardized information to efficiently address access issues. “I would like to have some more standardization on what’s being reviewed; it seems to change frequently, and so [to] be able to track and trend and have something given to me to review,” one health care provider requested. On the other hand, participants also described a need for decision support tools that would lead to action aligned with best practices. “Instead of a dashboard or something that’s just measuring their performance, it’s more something that they can look at and take action” a national support staff advisor suggested.
Enabling self and staff. GPMs felt they were most effective if they enabled themselves and stakeholders through training and by cultivating relationships and team building. Figure 2 illustrates the various stakeholders GPMs reported engaging with. The GPMS should be building relationships, bridging relationships, developing trust, and then providing a higher level of hands-on management. However, “that doesn’t really happen right now, day to day,” one member of leadership reported.
Key topics in GPM leadership training included both soft skills (change management, culture change, and negotiation skills) and crucial analytic/technical training (understanding each metric and dashboard available, data analytics, and supply/demand balancing techniques). The GPMs not only wanted to understand metrics as part of their training, but also want to know what to do about them.
An “operationalization” training approach (discerning the meaning of data, data-based decision making, and determining action from multiple options) inspired by real-life situations was preferred by participants. Other effective learning structures included job aids in the form of templated Gantt charts, process maps to guide GPMs through implementation of new processes, formalized peer learning (accumulated field insights shared during training courses), and informal peer sharing of direct experiences during calls.
GPMs also emphasized training for frontline clinical and support staff, including schedulers. VHA schedulers typically have less education and experience higher turnover rates than do other clinic staff, yet they carry out complex and critical tasks. Providing training and ensuring that any materials developed for training and education were appropriate to the level of education of schedulers was an important task for GPMs. “If they don’t understand all of the scheduling principles and potential,” one GPM explained, “we will not be maximizing the utilization of our parts.”
GPMs also provided informal education to clinicians. Participants noted GPMs have to avoid appearing to overstep their positions or presuming more knowledge and expertise than clinicians. They “have to be able to teach a physician without being overbearing, in a way a physician will accept it as advisement,” one program leader reported.
GPM knowledge, skills, and abilities. GPMs presented a complex range of knowledge, skills, and abilities, including clinical, administrative, analytics, and people skills. All interviewees reported that their prior education and experience did not sufficiently train them for the GPM role. GPMs identified a willingness to learn quickly as a critical characteristic. Many GPMs tended to have a formal education in health administration or business (eg, MBAs); others had administrative experience (eg, administrative assistance to executive leadership) or clinical training (eg, physician assistant). Detailed clinical knowledge was not expected, but clinical familiarity was helpful.
Some interviewees also mentioned previous experience and familiarity with the VHA system specifically as an advantage. This was especially true for VA outpatient flows, clinic flows, and understanding what an outpatient is in a VA context. Interviewees noted the importance of GPMs needing to be able to analyze patient demand metrics and underlying data in order to determine supply of providers and then to allocate adequate resources to complement providers. Forecasting skills were referenced as a key point. “They need to be able to be assured that they can recruit more providers if needed,” a national support staff advisor noted.
Given the importance of developing effective relationships, communication skills were mentioned by most participants and underscored as critical to establishing trust between GPMs and others as the initiative was being implemented. Interviewees indicated that relationship building was further enhanced when GPMs possessed the ability to “work with” rather than command clinicians and staff; navigate politics; and were respectful of other people’s knowledge, skills, abilities, and status. “They have to work with the nursing staff and teach them,” a leader described, “so that people understand that we are going to a different place to achieve our primary objectives and goals.”
Issues and Challenges
Participants identified several technical, social, and structural challenges and barriers to successfully implementing the GPM role.
Technical challenges. Recurring themes across all phases of data analytics were GPMs’ capability to challenge data use and use large volumes of information from multiple data sources (entering and accessing data; “drilling down” from summaries; generating reports; and analyzing and interpreting resulting metrics). Interviewees reported that information assessment and analytic support were not consistent. One GPM had a data analyst pulling reports needed to support clinical units while other GPMs trained staff to pull data. Even with support, some GPMs had issues due to limited information technology (IT) skills or access privileges leading to inefficiencies and delays. “Whenever I need anything from a programmer, I have to go through, you know, the IT gods in the sky,” one GPM remarked. “That usually takes a few months or more.”
Social challenges. Instituting the GPM role was a cultural change, and interviewees reported needing to address resistance to CPM model efforts. Resistance to change “is particularly hard in the VA just because it has a unique culture,” one leader noted. “There is a comfort in the legacy way of doing things.” The GPM initiative was introduced during a time when other national level initiatives were being implemented throughout the VHA. Fulfilling requests for information for these initiatives became the responsibility of the GPM and their team, which diverted attention from the mandate to improve access. Furthermore, GPMs were often considered the “change communicators” to clinics putting them in the role of “bad messenger,” which degraded trust and made it difficult to partner with clinicians.
Efforts to work through change management and build relationships included general program awareness presentation to internal stakeholders; including key stakeholders in GPM committees; pre-emptive conversations with unit chiefs; creating awareness of the GPM activities and progress through formal and informal update meetings; and identifying successes regarding access.
Structural challenges. The GPM role did not have direct supervision over clinical and administrative leaders, making it challenging to enact change. GPMs reported that “they do not always have authority over the area that they are being asked to manage,” which made their work difficult, requiring strong negotiation skills and political savvy to affect change. However, as the clinic staff and providers saw how the GPM could support and positively impact their practice, these challenges began to subside.
Discussion
This study provides empirical evidence regarding the implementation of a new access management strategy for health care systems focused on improving timeliness of care. First, the GPM position was seen as critical at each facility, as a single point person, to help local system leaders respond effectively to both national mandates and local context. Second, requiring the GPMs to report to the medical center director or chief of staff was important for integrating access perspectives across service lines and to facilitate a strong GPM role in strategic planning. Third, the intentional flexibility of the access management initiative, beyond the nationally specified aspects of the GPM role, was key for allowing individual sites to adapt to unique local challenges, resources, and population demands. Fourth, the initiative provided GPMs with opportunities to learn important skills and support the acquisition, utilization, and communication of a tremendous range of data toward responsive action.
According to our respondents, the GPM role demands functioning across a broad set of responsibilities; understanding the big picture as well as the complex underlying variables; engaging facility leaders, clinical and administrative staff; and prioritizing competing national and local demands. Consistent with previous findings, effective GPMs must possess a complex set of skills (interpersonal, analytic, and leadership) and the ability to create a supporting team.13
In practice, improving access at individual sites of care (VA medical centers and community-based outpatient clinics) poses significant challenges, especially in the early stages, even with the assistance of a GPM. For example, some respondents reported being overwhelmed by the volume of available data and dashboards, and responding to current requests for data analysis and dissemination sometimes impeded long range planning. Multiple national access-related initiatives and local pressures also generated excessive and potentially conflicting demands. Thus, while the creation of a GPM position seemed to be essential for the pilot sites to improve local access and timeliness to care, success also requires ongoing national and facility-wide communication, education, and support. Ongoing data analysis training and support will be critical to ensuring the sustainability of the position. Last, recruiting GPMs with the needed complex skill set may prove to be challenging, and it will be important to provide resources to identify, attract, and retain well-qualified GPMs.
Limitations and Future Work
This study was based on a small initial sample of pilot sites of varying sizes and, as such, may not reflect the experience of all VHA GPMs. In addition, the use of snowball sampling, while facilitating identification of key stakeholders, may introduce bias in participant sampling. Nonetheless, the results from this study provide findings that informed the national VHA GPM initiative and can inform further studies of practice management roles outside of the VA.
Further study of the VHA GPM implementation and similar roles in other health care systems is needed. As the GPM position is fully implemented across the VHA, large scale evaluation is needed to gain a more representative picture and allow for comparison of the GPM role at various types of facilities (eg, size, rurality, complexity, ranking based on access performance metrics).
Conclusion
Improving access to care is a central goal for health care systems. The incorporation of the GPM role is an innovative approach to improve access management strategies. Early study of prototype sites provided VHA leadership with valuable insights used to influence further rollout of this initiative. Based on our findings, national and local support are important to ongoing success. National access mandates, training, and resources should focus on ensuring sufficient GPM authority, enabling GPMs to use data, and ensuring GPMs engage with frontline clinical and administrative staff to improve veteran access to care.
The Veterans Health Administration (VHA) provides care for 9 million veterans at 1,255 health care sites linked to one of 170 local medical systems.1 Recognizing that providing timely care requires effective access management, the US Congress mandated training of VHA staff to manage and improve access to care but did not provide additional local funds for new positions.2 In response, the VHA created the group practice manager (GPM), a new position responsible for improving clinical practice management and unifying access improvement across leadership levels, professions, and services within each local medical system.
In May 2015, the VHA began hiring and training GPMs to spearhead management of access to services. The US Department of Veterans Affairs (VA) Office of Veteran Access to Care spearheaded GPM training, including face-to-face sessions, national calls, webinars, and educational materials. Five local medical systems were selected by the VA Office of Veteran Access to Care to implement the GPM role to allow for an early evaluation of the program that would inform a subsequent nationwide rollout. Implementation of the GPM role remained in the hands of local medical systems.
Longer wait times are shown to impact patient health.3,4 Open access scheduling and other patient-centered access management interventions have been shown to improve availability of primary care appointments.5 However, little empirical evidence exists regarding the managers who focus on clinic access interventions. While the nonpeer-reviewed literature includes references to such roles, including GPMs, the empirical literature has focused on external practice faciliators,6-8 “mid-level managers,”9 and clinic staff.10 We found no peer-reviewed articles on the needs and experiences of practice managers who are focused on improving access. The purpose of this study was to examine GPM prototype sites to both enhance subsequent nationwide implementation and to advance empirical literature on managing patient access within health care.
Methods
In 2015, the VA identified 5 prototype sites representing diverse geographic locations, size, and complexity for the implementation of the GPM role (Table 1). These sites self-identified as having clinical practice management experience. GPMs attended 4 training sessions between February and August 2015.
Data Collection
Participants from each prototype site included GPMs, national trainers, clinic leaders, and frontline staff. Table 2 includes the roles and sample size. Participants were recruited through purposive sampling followed by snowball sampling until thematic saturation was reached (the point at which subsequent data fail to produce new findings across sites and roles of interest).
Guided by the Consolidated Framework for Implementation Research (CFIR), the research team developed semistructured interview guides tailored to participants’ roles to elicit rich descriptions regarding overall impressions, practice management strategies, goals, activities, relationship to clinic roles, data analytics usage, challenges, barriers, and facilitators.11 These guides included open-ended questions and structured prompts utilizing participant language for follow-up probes to minimize interviewing bias (eAppendix:
Data Analysis
Data were analyzed using iterative deductive and inductive content analysis.12 Deductive content analysis consisted of identifying quotes that fit within preidentified categories (ie, perceptions of national effort, organizational structure for GPM, challenges, facilitators, metrics and tools, and mobilizing access culture) developed by the interdisciplinary research team. Further content analysis entailed open-coding and iteratively revisiting and reconciling codes associated within each preidentified category as new codes emerged. The team analyzed the resulting codes to inductively and iteratively identify and stabilize themes regarding the GPM role: roles and tasks, GPM characteristics, issues, and challenges. Through this process we moved coded data to reconciled descriptions suited to addressing the purposes of this study. Dedoose 7.0.23 software was used for qualitative data management and analysis.
Results
The study identified participants’ overall impressions of the GPM initiative and key themes within 4 major domains regarding implementing the GPM role: roles and tasks (implementing clinic practice management, leading patient access, supporting data analytics, and enabling self and staff); GPM characteristics (familiarity with clinical services, knowledge of VHA systems, ability to analyze patient data, communication skills, and the ability to work with others); and issues, and challenges (technical, social, and structural).
Overall Impressions
Interviewees perceived the GPM initiative as a consolidation of existing distributed responsibilities into one role that directly reported to local top-level management with indirect reporting to national leaders. Many of the sites reported that they had designated or planned to designate a role resembling the GPM prior to the initiative. “There are staff who’ve been doing some of this work all along,” a GPM noted. “We just didn’t have them grouped together. They weren’t necessarily all working in the same type of service under the same type of structure.”
Whether the GPM position was new or not, participants referenced the importance and challenges of engaging the local facility in recognizing the agency associated with the GPM position. According to national support, the staff are trying to get the facility to understand “why the group practice manager is so important… we’ve got to embed that standard position in the system.”
While the GPM was recognized as the hub of access management, respondents recognized that transformation regarding access involved many players. “We have to create [an] orchestrated team inside each facility,” an advisor argued.
Respondents discussed how the initiative allows local facilities to appoint a specific person with a specific title and role who helps facilitate, organize, and legitimize an access focus at their sites. One GPM interviewee noted how the initiative helped refocus some of their previously less centralized efforts. “We’ve always looked at productivity; we’ve always looked at access; we’ve always looked at efficiency. I think the bigger difference is now there are individuals identified in the clinics, as practice managers as well…I interact with them. They interact with individual clinic staff, and it’s more of a group process than a single individual.”
The value of having tools available and being able to track and manage patient care as a specific example of the positive impact of this new role was noted by participants. A GPM noted that many health care providers will be happy to have tools to better manage their services and a process “that flows from a service level all the way up to executive management, where there is a common interest in making those things happen—I think that’s going to be a tremendous help.”
Participants expressed concern that the national GPM rollout would be a one-size-fits-all approach. These respondents emphasized the need to have the flexibility to customize their activities to meet their unique site and patient needs.
GPM Roles and Tasks
Participants described 4 primary roles that the GPM was expected to fill: implementing clinic practice management, leading patient access, supporting data analytics, and enabling self and staff. Some activities overlapped in that they served to support multiple role areas (Figure 1).
Implementing clinic practice management. In the early stages of the initiative, the GPM’s primary role was to prepare the facility to implement a standardized set of clinic practice management (CPM) team processes. Part of standardizing the CPM process was defining the scope and tasks of the GPM, which requires significant planning for the implementation. “My big job is to finalize what we think group practice management is going to look [like] here,” a GPM reported.
Each prototype site had latitude to interpret the GPM initiative in a way that would work in their context within given VHA boundaries and ongoing initiatives. To achieve the high-level vision and purpose, the GPM first had to develop action plans that accounted for the operating environment of the facility. According to one GPM, VA national officials are “constantly” asking for action plans, which required significant time by specific deadlines. “They want an action plan [and to] clean up all your consults, [and to] clean up all your recall reminders.”
Leading on improving access efforts. Participants saw the GPM as the central staff member responsible for providing oversight of any activities and people involved in improving access. “I ensure everybody is doing what they’re supposed to do,” one GPM reported. When the GPM sees areas that are not being addressed, the individual tries to develop a process or training to “close those gaps.”
GPMs promoted an awareness of their goals, changes in process, and new tools accompanying the initiative. However, other access initiatives were occurring simultaneously creating confusion for health care providers and patients; thus GPMs found they were managing a wide array of related initiatives.
GPMs have to negotiate with leaders across the VHA facility, many of whom operated at a higher leadership level and had different priorities, to address access problems.
“I’m a lieutenant as a GPM in a clinic, a GPM noted. “How is the lieutenant going to talk to a major or a colonel in the clinic and say your clinic has problems. How[‘s] that lieutenant...going to do that? With people skills!”
Managing expectations about the speed and to what extent a problem could be resolved was an important part of the GPM leadership role. “I see myself as managing expectations both up to the leadership and down to the frontline,” a GPM explained. “I find myself talking to leadership [about] our progress. But at the same time, we have to say, ‘not everything can be fixed overnight.’”
Providing leadership on access-related issues included developing a range of options for addressing patient access problems. One analytics manager recounted how the GPM role led to evaluating how physical space limited efficiency in clinic flow. The first step was identifying possible additional rooms to improve clinic flow. This required working with the space committee to “get someone to look at our overarching space and find someplace else for them to sit” to avoid adding to congestion in the clinic area.
Supporting data analytics.
GPMs routinely immersed themselves in the data to understand access issues. GPMs worked with clinic leaders to identify the underlying causes and various solutions. The GPMs maintained access through administrative scrubbing of the data and finding “smart ways to get patients scheduled,” a GPM explained. “I don’t think our facility would have taken care of as many veterans in the time frame as we did....We’ve cleared over 4,000 consults that were older than 90 days. We have cleared thousands and thousands of weekly reminders.”
GPMs expressed the need for aggregated (ie, dashboard) and standardized information to efficiently address access issues. “I would like to have some more standardization on what’s being reviewed; it seems to change frequently, and so [to] be able to track and trend and have something given to me to review,” one health care provider requested. On the other hand, participants also described a need for decision support tools that would lead to action aligned with best practices. “Instead of a dashboard or something that’s just measuring their performance, it’s more something that they can look at and take action” a national support staff advisor suggested.
Enabling self and staff. GPMs felt they were most effective if they enabled themselves and stakeholders through training and by cultivating relationships and team building. Figure 2 illustrates the various stakeholders GPMs reported engaging with. The GPMS should be building relationships, bridging relationships, developing trust, and then providing a higher level of hands-on management. However, “that doesn’t really happen right now, day to day,” one member of leadership reported.
Key topics in GPM leadership training included both soft skills (change management, culture change, and negotiation skills) and crucial analytic/technical training (understanding each metric and dashboard available, data analytics, and supply/demand balancing techniques). The GPMs not only wanted to understand metrics as part of their training, but also want to know what to do about them.
An “operationalization” training approach (discerning the meaning of data, data-based decision making, and determining action from multiple options) inspired by real-life situations was preferred by participants. Other effective learning structures included job aids in the form of templated Gantt charts, process maps to guide GPMs through implementation of new processes, formalized peer learning (accumulated field insights shared during training courses), and informal peer sharing of direct experiences during calls.
GPMs also emphasized training for frontline clinical and support staff, including schedulers. VHA schedulers typically have less education and experience higher turnover rates than do other clinic staff, yet they carry out complex and critical tasks. Providing training and ensuring that any materials developed for training and education were appropriate to the level of education of schedulers was an important task for GPMs. “If they don’t understand all of the scheduling principles and potential,” one GPM explained, “we will not be maximizing the utilization of our parts.”
GPMs also provided informal education to clinicians. Participants noted GPMs have to avoid appearing to overstep their positions or presuming more knowledge and expertise than clinicians. They “have to be able to teach a physician without being overbearing, in a way a physician will accept it as advisement,” one program leader reported.
GPM knowledge, skills, and abilities. GPMs presented a complex range of knowledge, skills, and abilities, including clinical, administrative, analytics, and people skills. All interviewees reported that their prior education and experience did not sufficiently train them for the GPM role. GPMs identified a willingness to learn quickly as a critical characteristic. Many GPMs tended to have a formal education in health administration or business (eg, MBAs); others had administrative experience (eg, administrative assistance to executive leadership) or clinical training (eg, physician assistant). Detailed clinical knowledge was not expected, but clinical familiarity was helpful.
Some interviewees also mentioned previous experience and familiarity with the VHA system specifically as an advantage. This was especially true for VA outpatient flows, clinic flows, and understanding what an outpatient is in a VA context. Interviewees noted the importance of GPMs needing to be able to analyze patient demand metrics and underlying data in order to determine supply of providers and then to allocate adequate resources to complement providers. Forecasting skills were referenced as a key point. “They need to be able to be assured that they can recruit more providers if needed,” a national support staff advisor noted.
Given the importance of developing effective relationships, communication skills were mentioned by most participants and underscored as critical to establishing trust between GPMs and others as the initiative was being implemented. Interviewees indicated that relationship building was further enhanced when GPMs possessed the ability to “work with” rather than command clinicians and staff; navigate politics; and were respectful of other people’s knowledge, skills, abilities, and status. “They have to work with the nursing staff and teach them,” a leader described, “so that people understand that we are going to a different place to achieve our primary objectives and goals.”
Issues and Challenges
Participants identified several technical, social, and structural challenges and barriers to successfully implementing the GPM role.
Technical challenges. Recurring themes across all phases of data analytics were GPMs’ capability to challenge data use and use large volumes of information from multiple data sources (entering and accessing data; “drilling down” from summaries; generating reports; and analyzing and interpreting resulting metrics). Interviewees reported that information assessment and analytic support were not consistent. One GPM had a data analyst pulling reports needed to support clinical units while other GPMs trained staff to pull data. Even with support, some GPMs had issues due to limited information technology (IT) skills or access privileges leading to inefficiencies and delays. “Whenever I need anything from a programmer, I have to go through, you know, the IT gods in the sky,” one GPM remarked. “That usually takes a few months or more.”
Social challenges. Instituting the GPM role was a cultural change, and interviewees reported needing to address resistance to CPM model efforts. Resistance to change “is particularly hard in the VA just because it has a unique culture,” one leader noted. “There is a comfort in the legacy way of doing things.” The GPM initiative was introduced during a time when other national level initiatives were being implemented throughout the VHA. Fulfilling requests for information for these initiatives became the responsibility of the GPM and their team, which diverted attention from the mandate to improve access. Furthermore, GPMs were often considered the “change communicators” to clinics putting them in the role of “bad messenger,” which degraded trust and made it difficult to partner with clinicians.
Efforts to work through change management and build relationships included general program awareness presentation to internal stakeholders; including key stakeholders in GPM committees; pre-emptive conversations with unit chiefs; creating awareness of the GPM activities and progress through formal and informal update meetings; and identifying successes regarding access.
Structural challenges. The GPM role did not have direct supervision over clinical and administrative leaders, making it challenging to enact change. GPMs reported that “they do not always have authority over the area that they are being asked to manage,” which made their work difficult, requiring strong negotiation skills and political savvy to affect change. However, as the clinic staff and providers saw how the GPM could support and positively impact their practice, these challenges began to subside.
Discussion
This study provides empirical evidence regarding the implementation of a new access management strategy for health care systems focused on improving timeliness of care. First, the GPM position was seen as critical at each facility, as a single point person, to help local system leaders respond effectively to both national mandates and local context. Second, requiring the GPMs to report to the medical center director or chief of staff was important for integrating access perspectives across service lines and to facilitate a strong GPM role in strategic planning. Third, the intentional flexibility of the access management initiative, beyond the nationally specified aspects of the GPM role, was key for allowing individual sites to adapt to unique local challenges, resources, and population demands. Fourth, the initiative provided GPMs with opportunities to learn important skills and support the acquisition, utilization, and communication of a tremendous range of data toward responsive action.
According to our respondents, the GPM role demands functioning across a broad set of responsibilities; understanding the big picture as well as the complex underlying variables; engaging facility leaders, clinical and administrative staff; and prioritizing competing national and local demands. Consistent with previous findings, effective GPMs must possess a complex set of skills (interpersonal, analytic, and leadership) and the ability to create a supporting team.13
In practice, improving access at individual sites of care (VA medical centers and community-based outpatient clinics) poses significant challenges, especially in the early stages, even with the assistance of a GPM. For example, some respondents reported being overwhelmed by the volume of available data and dashboards, and responding to current requests for data analysis and dissemination sometimes impeded long range planning. Multiple national access-related initiatives and local pressures also generated excessive and potentially conflicting demands. Thus, while the creation of a GPM position seemed to be essential for the pilot sites to improve local access and timeliness to care, success also requires ongoing national and facility-wide communication, education, and support. Ongoing data analysis training and support will be critical to ensuring the sustainability of the position. Last, recruiting GPMs with the needed complex skill set may prove to be challenging, and it will be important to provide resources to identify, attract, and retain well-qualified GPMs.
Limitations and Future Work
This study was based on a small initial sample of pilot sites of varying sizes and, as such, may not reflect the experience of all VHA GPMs. In addition, the use of snowball sampling, while facilitating identification of key stakeholders, may introduce bias in participant sampling. Nonetheless, the results from this study provide findings that informed the national VHA GPM initiative and can inform further studies of practice management roles outside of the VA.
Further study of the VHA GPM implementation and similar roles in other health care systems is needed. As the GPM position is fully implemented across the VHA, large scale evaluation is needed to gain a more representative picture and allow for comparison of the GPM role at various types of facilities (eg, size, rurality, complexity, ranking based on access performance metrics).
Conclusion
Improving access to care is a central goal for health care systems. The incorporation of the GPM role is an innovative approach to improve access management strategies. Early study of prototype sites provided VHA leadership with valuable insights used to influence further rollout of this initiative. Based on our findings, national and local support are important to ongoing success. National access mandates, training, and resources should focus on ensuring sufficient GPM authority, enabling GPMs to use data, and ensuring GPMs engage with frontline clinical and administrative staff to improve veteran access to care.
1. US Department of Veterans Affairs. Veterans Health Administration. https://www.va.gov/health. Updated October 25, 2019. Accessed January 8, 2020.
2. Veterans Access, Choice, and Accountability Act of 2014. 38 CFR § 17.1500 (2014).
3. Fahmy N, Aprikian A, Al-Otaibi M, et al. Impact of treatment delay in patients with bladder cancer managed with partial cystectomy in Quebec: a population-based study. Can Urol Assoc J. 2009;3(2):131-135.
4. Hill CJ, Joonas K. The impact of unacceptable wait time on health care patients’ attitudes and actions. Health Mark Q. 2005;23(2):69-87.
5. Ansell D, Crispo JAG, Simard B, Bjerre LM. Interventions to reduce wait times for primary care appointments: a systematic review. BMC Health Serv Res. 2017;17(1):295.
6. Kotecha J, Han H, Green M, Russell G, Martin MI, Birtwhistle R. The role of the practice facilitators in Ontario primary healthcare quality improvement. BMC Fam Pract. 2015;16:93.
7. Taylor EF, Machta RM, Meyers DS, Genevro J, Peikes DN. Enhancing the primary care team to provide redesigned care: the roles of practice facilitators and care managers. Ann Fam Med. 2013;11(1):80-83.
8. Liddy C, Laferriere D, Baskerville B, Dahrouge S, Knox L, Hogg W. An overview of practice facilitation programs in Canada: current perspectives and future directions. Healthc Policy. 2013;8(3):58-67.
9. Birken SA, Lee SY, Weiner BJ, Chin MH, Schaefer CT. Improving the effectiveness of health care innovation implementation: middle managers as change agents. Med Care Res Rev. 2013;70(1):29-45.
10. Ahluwalia S, Offredy M. A qualitative study of the impact of the implementation of advanced access in primary healthcare on the working lives of general practice staff. BMC Fam Pract. 2005;6:39.
11. Damschroder LJ, Aron DC, Keith RE, Kirsh SR, Alexander JA, Lowery JC. Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4:50.
12. Elo S, Kyngäs H. The qualitative content analysis process. J Adv Nurs. 2008;62(1):107-115.
13. Stefl ME. Common competencies for all healthcare managers: the Healthcare Leadership Alliance model. J Healthc Manag. 2008;53(6):360-374.
1. US Department of Veterans Affairs. Veterans Health Administration. https://www.va.gov/health. Updated October 25, 2019. Accessed January 8, 2020.
2. Veterans Access, Choice, and Accountability Act of 2014. 38 CFR § 17.1500 (2014).
3. Fahmy N, Aprikian A, Al-Otaibi M, et al. Impact of treatment delay in patients with bladder cancer managed with partial cystectomy in Quebec: a population-based study. Can Urol Assoc J. 2009;3(2):131-135.
4. Hill CJ, Joonas K. The impact of unacceptable wait time on health care patients’ attitudes and actions. Health Mark Q. 2005;23(2):69-87.
5. Ansell D, Crispo JAG, Simard B, Bjerre LM. Interventions to reduce wait times for primary care appointments: a systematic review. BMC Health Serv Res. 2017;17(1):295.
6. Kotecha J, Han H, Green M, Russell G, Martin MI, Birtwhistle R. The role of the practice facilitators in Ontario primary healthcare quality improvement. BMC Fam Pract. 2015;16:93.
7. Taylor EF, Machta RM, Meyers DS, Genevro J, Peikes DN. Enhancing the primary care team to provide redesigned care: the roles of practice facilitators and care managers. Ann Fam Med. 2013;11(1):80-83.
8. Liddy C, Laferriere D, Baskerville B, Dahrouge S, Knox L, Hogg W. An overview of practice facilitation programs in Canada: current perspectives and future directions. Healthc Policy. 2013;8(3):58-67.
9. Birken SA, Lee SY, Weiner BJ, Chin MH, Schaefer CT. Improving the effectiveness of health care innovation implementation: middle managers as change agents. Med Care Res Rev. 2013;70(1):29-45.
10. Ahluwalia S, Offredy M. A qualitative study of the impact of the implementation of advanced access in primary healthcare on the working lives of general practice staff. BMC Fam Pract. 2005;6:39.
11. Damschroder LJ, Aron DC, Keith RE, Kirsh SR, Alexander JA, Lowery JC. Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4:50.
12. Elo S, Kyngäs H. The qualitative content analysis process. J Adv Nurs. 2008;62(1):107-115.
13. Stefl ME. Common competencies for all healthcare managers: the Healthcare Leadership Alliance model. J Healthc Manag. 2008;53(6):360-374.
Prazosin Outcomes in Older Veterans With Posttraumatic Stress Disorder
Posttraumatic stress disorder (PTSD) is a common psychiatric condition in the veteran population and is associated with significant sleep disturbances and trauma-related nightmares.1 PTSD can present with intrusive symptoms, such as recurrent memories or dreams, which are associated with traumatic events.2 Clinical studies have described an increase in central nervous system (CNS) noradrenergic activity in PTSD; specifically, noradrenergic outflow and/or postsynaptic adrenoreceptor responsiveness is increased.3,4 Targeting a reduction in noradrenergic activity via antagonism of noradrenergic receptors has been a therapeutic treatment strategy in PTSD.
Prazosin crosses the blood-brain barrier and works to antagonize α-1 adrenoreceptors to decrease noradrenergic outflow.5 It has been shown in multiple trials to effectively reduce nightmares and improve sleep quality in the veteran population.6-12 However, a recent negative trial contributed to a downgraded recommendation for prazosin in the treatment of PTSD-related nightmares in the joint PTSD guideline from the US Department of Veterans Affairs (VA) and US Department of Defense (DoD).13,14
The diagnosis of PTSD in veterans aged ≥ 65 years has been increasing due to improved recognition.15 As a result, prazosin may be considered more frequently as a treatment option for those patients who report PTSD-related nightmares. It is important to recognize that the normal physiologic process of aging is associated with increased noradrenergic outflow, which may change the pharmacodynamics of prazosin in geriatric patients.12,16 This may necessitate increased doses to adequately antagonize the α-1 adenoreceptor.17 High doses of prazosin may increase the risk of hypotension in older patients.12 This increased risk is especially concerning for patients who already receive multiple medications or have comorbid conditions that impact blood pressure (BP).
The existing literature has few studies that have reported on outcomes with prazosin use in older veterans.11,12 The few existing reports provide clinically valuable descriptions of tolerability and efficacy with prazosin. For example, Peskind and colleagues showed prazosin to be an effective agent in the treatment of PTSD-related nightmares.12 However, in older veterans prazosin dosing > 4 mg has not been described or reported in the literature.
There appears to be a lack of clinical guidance with regards to dosing of prazosin in older patients. The goal of the current study was to assess the outcomes of older veterans with PTSD under pharmacist management of prazosin at our outpatient Prazosin Titration Clinic (PTC) in order to contribute to the minimal, yet valuable, existing clinical literature.
Methods
This study was approved by the University of Iowa Institutional Review Board and Iowa City Veterans Affairs Health Care System (ICVAHCS) research and development committee. The study was a retrospective chart review of older patients with consultations referred to the ICVAHCS PTC. To be eligible for inclusion, veterans with a PTSD diagnosis must have been evaluated at an initial consult appointment with a mental health clinical pharmacy specialist (MH CPS) from February 1, 2016 to August 31, 2018, and had at least 1 follow-up appointment. Follow-up visits were conducted either by telephone or in a face-to-face clinic visit.
Prazosin Titration Clinics
VA health care systems use pharmacists to manage veterans prescribed prazosin through PTC consultations. PTCs provide a process for close follow-up and assessment of PTSD-related outcomes. Due to the frequency of follow-up, this service may be beneficial for older veterans with more complex comorbidities and medication regimens. Any veteran with PTSD-related nightmares may be referred to the PTC for a consultation by any health care provider. Once referred to the clinic, MH CPSs assume responsibility for the prazosin prescription, including dose adjustments. For example, if a veteran reported no issues with tolerability but continued to have frequent and distressing nightmares, the dose may be increased, typically by 1-mg to 2-mg increments. Once the veteran reaches a stable and tolerable dose of prazosin, they are discharged from the PTC, and the referring health care provider resumes responsibility for the prazosin prescription.

Clinically Measured Outcomes
Nightmare frequency and intensity were measured using the Recurrent Distressing Dreams item B2 of the Clinician Administered PTSD Scale (CAPS) (Table 1). The PTSD Checklist (PCL-5), Insomnia Severity Index (ISI), and total sleep hours were used to determine the effect of prazosin on symptom severity (Table 2). The PCL-5 is a 20-item self-report used to monitor and quantify symptom level and change over time. It evaluates the frequency over the past month that a patient was bothered by any of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) PTSD criterion.2 Scores range from 0 (not at all) to 4 (extreme), with a maximum score of 80. The ISI is a 7-item self-report of sleep symptoms, with a total score of 28, where increasing scores indicate increasing severity of insomnia (Table 3).

Clinically measured outcome scales were performed and assessed by MH CPSs. CAPS frequency and intensity were measured at each clinic visit. PCL-5 and ISI scores were assessed at baseline and at the endpoint of study or discharge from clinic (Table 4). Patients who continued in the PTC after the end of the study date or who were lost to follow-up did not complete these measures at time of discharge.
![]()
Data Analysis
The primary outcome was change in CAPS nightmare frequency and intensity from time of initial clinic visit to time of discharge or end of study. The secondary outcomes included change in PCL-5, ISI, and sleep hours. Other secondary outcomes included measures of tolerability: BP changes, adverse effects (AEs) reported, and outcome of prazosin therapy when AEs were reported. Change in PTSD symptoms, PCL-5, and ISI were assessed using the Wilcoxon signed rank tests. Findings were considered to be statistically significant at P ≤ .05. Other variables were reported descriptively.
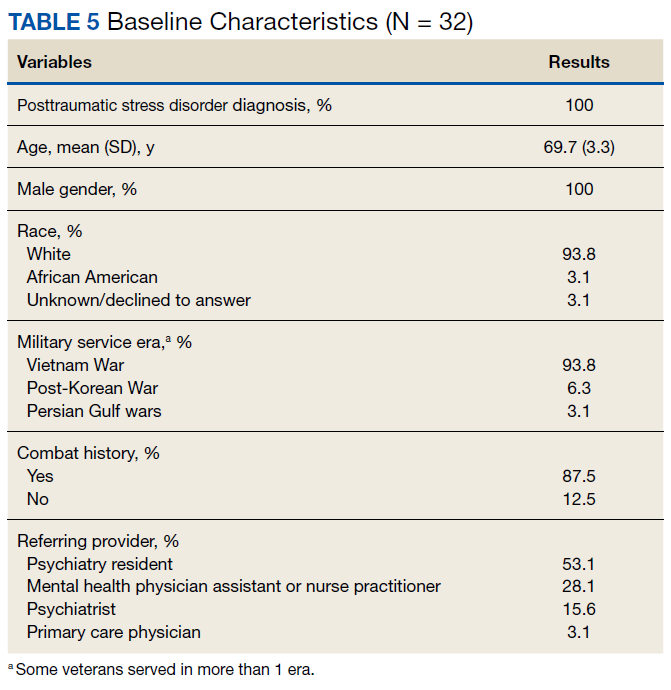
Results
Thirty-two veterans, aged ≥ 65 years, with clinical diagnosis of PTSD at the time of referral to the PTC were reviewed (Table 5). All patients were male and 93.8% were white. Thirty were Vietnam era veterans, 1 served in the Persian Gulf era, and 2 served in the post-Korean War era. Twenty-eight veterans had a combat history. Severe PTSD symptoms were reported as indicated by baseline PCL-5 scores, and moderate severity insomnia symptoms as indicated by baseline ISI scores.
All veterans had at least 1 comorbid medical condition, and the majority had multiple medical comorbidities. All were taking multiple medical and psychiatric medications. More than 80% of veterans were taking antihypertensive agents at baseline (Table 6). Twenty-two of the 32 veterans were prescribed a VA/DoD PTSD guideline-recommended antidepressant.
Primary Outcomes
The baseline, final, and changes in the primary outcomes are included in the Figure. Treatment with prazosin was associated with significant improvement in median scores from baseline to endpoint for CAPS nightmare frequency (-2, P = .0001), CAPS nightmare intensity (-2, P = .001), and total CAPS item score (-4, P < .001).
Secondary Outcomes
Of the 32 patients included in the study, PCL-5 was obtained from 20 veterans and ISI from 17 veterans at discharge from clinic. Thirty veterans reported final sleep hours, 2 veterans were unable to quantify average sleep hours per night at their final visit. PTSD symptom severity showed significant median change from baseline to endpoint of management in PTC for PCL-5 (-20.5, P = .0002) and ISI (-6.5, P = .002). Total sleep hours also showed significant improvement from baseline to endpoint (1.5, P = .003) (Table 7).
Prazosin Dosing
Maximum prazosin total daily doses were evaluated from the study baseline to the endpoint (Table 8). The mean (SD) maximum total daily dose of prazosin reached was 5.6 (5.1) mg (median, 3.5 mg; range, 1-17 mg). The mean (SD) total daily dose of prazosin at endpoint of study was 5.1 (5.3) mg (median, 2.5 mg; range, 0-17 mg). The average (SD) change of prazosin dose from baseline to endpoint was 3.5 (4.6) mg (median, 2 mg; range, -2 to 15 mg).
Tolerability
The average (SD) baseline systolic BP (SBP) was 135.8 (20.5) mm Hg and diastolic BP (DBP) was 77.2 (11.0) mm Hg. The average SBP and DBP at study endpoint were 131.8 (16.6) mm Hg and 75.9 (13.7) mm Hg, respectively. Endpoint BP values were missing for 6 patients.
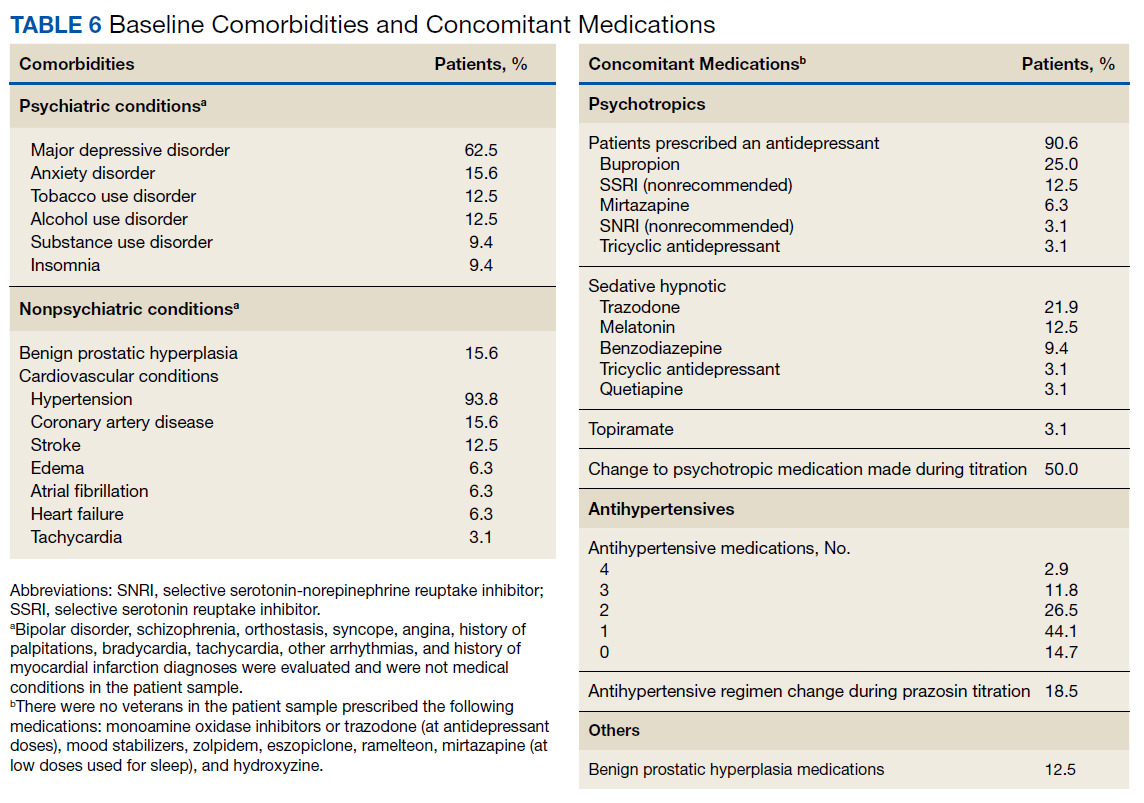
Nine of 32 veterans reported AEs during PTC management of prazosin. Dizziness was the most common AE reported. Other AEs noted included orthostatic hypotension, headache, and falls. Of 12 reported AEs, 8 were related to dizziness, 5 of which were transient or tolerable. One veteran had a dose reduction of prazosin due to dizziness, and 3 veterans discontinued prazosin due to orthostasis. Several veterans had changes made to their antihypertensive medication regimen during prazosin titration, including dose reductions and/or decreased number of medications. If indicated, the MH CPS collaborated with the antihypertensive prescriber to make dosing adjustments. Two veterans reported a fall during prazosin titration; 1 veteran had other mobility-related factors thought to precipitate to their fall, and neither veterans were injured because of the falls.
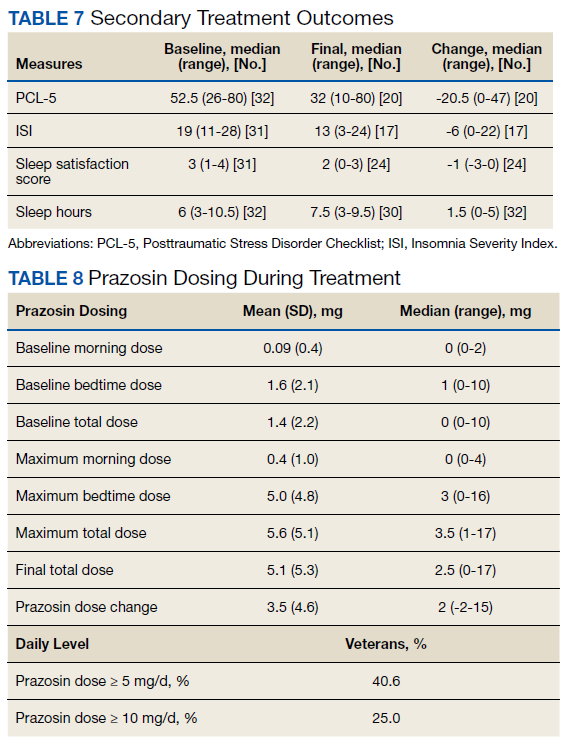
Twenty-eight veterans (87.5%) treated in the PTC continued prazosin therapy after discharge. Six months postdischarge, 70% of veterans had maintained prazosin therapy. Two veterans required a dose increase postdischarge from PTC, and 1 veteran required a dose reduction. About one-third of veterans included in this study continued in the PTC beyond the end of the study period. Common reasons for clinic discharge were symptom resolution (37.5%), adverse reactions (12.5%), lost to follow-up (6.3%), or nonadherence (3.1%).
Discussion
The existing literature reports few outcomes for older veterans prescribed prazosin for PTSD. One report included a 75-year-old otherwise-healthy veteran, who received 2-mg prazosin at bedtime. At this dose, he reported good tolerability and response, as indicated by a reduction in his CAPS nightmare severity score.11 An open-label trial assessed prazosin in 9 geriatric men with chronic PTSD and found low-dose prazosin (average [SD] maximum prazosin dose reported was 2.3 [0.7] mg, range 2-4 mg per day) greatly reduced nightmares and overall PTSD severity in 8 of 9 subjects.12 Despite the veterans in that study having multiple medical comorbid conditions and taking concomitant medications, prazosin was reported to be well tolerated, and changes in BP were determined to be clinically insignificant.12 A recent study of middle-aged veterans (average [SD] age 52 [14] years) reported prazosin did not significantly alleviate PTSD-related nightmares.13 However, we observed prazosin therapy significantly reduced nightmares and sleep disturbances, and significantly improved PTSD severity in our older veteran population.
To our knowledge, the current study is the largest retrospective study that evaluates prazosin therapy for the treatment of PTSD-related nightmares in older veterans. The findings of this study are similar to a previous study in older veterans as well as studies of prazosin in younger and middle-aged adult veterans, with the average age ranging from 30 to 56 years.6-12 Like the previously reported studies, prazosin also was well tolerated in our sample of veterans with multiple comorbidities and concomitant medications. Changes in BP were not clinically significant.
Studies have demonstrated increased noradrenergic activity as a component of the normal aging process.16,17 This may require utilizing caution during prazosin dose titration and frequent patient assessment, due to the concern for risk of hypotension in older patients and in particular those who may require increased doses to achieve efficacy. In our study, favorable outcomes were achieved at an average (SD) total daily dose of 5.1 (5.3) mg (median, 2.5 mg; range 0-17 mg). A previous report showed efficacy of prazosin around an average (SD) maximum dose of 2.3 (0.7) mg, which is lower than the doses reported in the current study.12 In addition, 13 veterans (40.6%) from our sample reached doses of ≥ 5 mg per day, and 8 veterans (25.0%) reached doses of ≥ 10 mg per day.
The doses reached in this study were reflective of a management approach using assessment of patient-reported symptoms at weekly to biweekly follow-up visits. The individualized management approach applied in the PTC by MH CPSs aids in uncovering the most efficacious and tolerated dose of prazosin for each veteran. Evaluation of symptom change during treatment in PTC was facilitated use of objective rating scales, which helped measure nightmare frequency and intensity, sleep satisfaction, and global PTSD severity. Given the variability in dosing of prazosin reported in the literature, further studies may be warranted to provide more definitive clinical guidance as far as dosing prazosin in older patients.
The study by Peskind and colleaguesrationalized that lower doses of prazosin may be used in older patients given pharmacokinetic effects of aging, age-associated changes in PTSD pathophysiology, and effects and interactions of concomitant medications.12 However, our study found that prazosin could be well tolerated at higher doses. The rate of discontinuation due to intolerable AEs was low. AEs reported were consistent with the established AE profile of prazosin, with dizziness, orthostasis, and headache most commonly reported. Similar to the Peskind and colleagues study, BP had a tendency to decrease in this current study; however, the change was not clinically significant.12 That study also reported transient dizziness with prazosin titration, which was shown to be tolerable in the majority of our veterans reporting dizziness.12 Other common AEs with prazosin, such as rash, priapism, sedation, syncope, other cardiac AEs, and sleep disturbance were not reported in our study population.
MH CPS-managed PTCs are one venue that may allow veterans to achieve favorable outcomes through frequent follow-up. As prazosin dosing is specific to each individual patient, frequent follow-up visits are helpful in determining optimal doses that maximize efficacy while minimizing intolerable AEs. The majority of veterans treated in our PTC continued use of prazosin 6 months postdischarge, while 3 veterans required a postdischarge dose change.
The 2017 VA/DoD PTSD guidelines recommend individual, trauma-focused psychotherapy over pharmacologic therapy for the primary treatment of PTSD.14 About half of the veterans in the current study participated in either group or individual psychotherapy during enrollment in the PTC. A systematic review of psychotherapy in older veterans reported mixed results, with 4 studies indicating positive effects of therapy, while the other 3 studies reported no benefit or mixed effects for PTSD symptoms. The review concluded that fewer older adults experience complete remission of symptoms with psychotherapy alone.18 A previous study of older veterans described improvement in PTSD-related symptoms with prazosin without concurrent psychotherapy.12
Limitations and Strengths
While this study is the largest study to evaluate outcomes of prazosin in older patients with PTSD, there are several important limitations. The study population was small and all were male. The results of this study may not be applicable to women. Another limitation was several missing values in our data set, as some secondary outcomes were not collected via telephone follow-up visits. This could potentially contribute a measurement bias in the reported secondary outcomes results, specifically for the PCL-5 and ISI. Additionally, some veterans in this study may have reported symptomatic improvement based on the additional supportive intervention that clinical pharmacists were able to offer, as well as concomitant participation in psychotherapy. This may be reflected in the study results. This study did not have a true placebo group, as we may find a reduction in symptoms with placebo.
Strengths of this study include multiple data points for assessment of prazosin tolerability and a pre- and poststudy design, which allowed for the veterans to serve as their own control. Another strength of this study is that data were complete for primary outcome measures, including the CAPS Recurrent and Distressing Dreams Item, where prazosin showed significant benefit in reduction of PTSD-related nightmares. While the results of this study are reassuring, further randomized, double-blind, placebo-controlled trials are likely needed in order to establish efficacy and tolerability of prazosin in older veterans for PTSD related nightmares.
Conclusion
These results demonstrate prazosin therapy in older veterans can significantly improve PTSD-related nightmares and PTSD severity. Prazosin was well tolerated in this population at doses higher than previously reported in other studies. This study shows that prazosin therapy can be effectively managed and tolerated in older veterans with complex medical and psychiatric comorbidities to provide favorable patient outcomes.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Iowa City VA Health Care System and by the Health Services Research and Development Service, US Department of Veterans Affairs.
1. Ross RJ, Ball WA, Sullivan KA, Caroff SN. Sleep disturbance as the hallmark of posttraumatic stress disorder. Am J Psychiatry. 1989;146(6):697-707.
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington VA: American Psychiatric Association; 2013.
3. Southwick SM, Krystal JH, Morgan CA, et al. Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry. 1993;50(4):266-274.
4. Geracioti TD Jr, Baker DG, Ekhator NN, et al. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry. 2001;158(8):1227-1230.
5. Friedman MJ. Posttraumatic and Acute Stress Disorders. 6th ed. New York: Springer Publishing; 2015.
6. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
7. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61(8):928-934.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. Germain A, Richardson R, Moul DE, et al. Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res. 2012;72(2):89-96.
10. Taylor HR, Freeman MK, Cates ME. Prazosin for treatment of nightmares related to posttraumatic stress disorder. Am J Health Syst Pharm. 2008;65(8):716-722.
11. Raskind MA, Dobie DJ, Kanter ED, Petrie EC, Thompson CE, Peskind ER. The alpha1-adrenergic antagonist prazosin ameliorates combat trauma nightmares in veterans with posttraumatic stress disorder: a report of 4 cases. J Clin Psychiatry. 2000;61(2):129-133.
12. Peskind ER, Bonner LT, Hoff DJ, Raskind MA. Prazosin reduces trauma-related nightmares in older men with chronic posttraumatic stress disorder. J Geriatr Psychiatry Neurol. 2003;16(3):165-171.
13. Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
14. The Management of Posttraumatic Stress Disorder Work Group. VA/DoD clinical practice guideline for the management of posttraumatic stress disorder and acute stress disorder. Version 3.0–2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/VADoDPTSDCPGFinal.pdf. Published June 2017. Accessed January 7, 2020.
15. Nichols BL, Czirr R. 24/Post-traumatic stress disorder: hidden syndrome in elders. Clin Gerontol. 1986;5(3-4):417-433.
16. Supiano MA, Linares OA, Smith MJ, Halter JB. Age-related differences in norepinephrine kinetics: effect of posture and sodium-restricted diet. Am J Physiol. 1990;259(3, pt 1):E422-E431.
17. Raskind MA, Peskind ER, Holmes C, Goldstein DS. Patterns of cerebrospinal fluid catechols support increased central noradrenergic responsiveness in aging and Alzheimer’s disease. Biol Psychiatry. 1999;46(6):756-765.
18. Dinnen S, Simiola V, Cook JM. Post-traumatic stress disorder in older adults: a systematic review of the psychotherapy treatment literature. Aging Ment Health. 2015;19(2):144-150.
Posttraumatic stress disorder (PTSD) is a common psychiatric condition in the veteran population and is associated with significant sleep disturbances and trauma-related nightmares.1 PTSD can present with intrusive symptoms, such as recurrent memories or dreams, which are associated with traumatic events.2 Clinical studies have described an increase in central nervous system (CNS) noradrenergic activity in PTSD; specifically, noradrenergic outflow and/or postsynaptic adrenoreceptor responsiveness is increased.3,4 Targeting a reduction in noradrenergic activity via antagonism of noradrenergic receptors has been a therapeutic treatment strategy in PTSD.
Prazosin crosses the blood-brain barrier and works to antagonize α-1 adrenoreceptors to decrease noradrenergic outflow.5 It has been shown in multiple trials to effectively reduce nightmares and improve sleep quality in the veteran population.6-12 However, a recent negative trial contributed to a downgraded recommendation for prazosin in the treatment of PTSD-related nightmares in the joint PTSD guideline from the US Department of Veterans Affairs (VA) and US Department of Defense (DoD).13,14
The diagnosis of PTSD in veterans aged ≥ 65 years has been increasing due to improved recognition.15 As a result, prazosin may be considered more frequently as a treatment option for those patients who report PTSD-related nightmares. It is important to recognize that the normal physiologic process of aging is associated with increased noradrenergic outflow, which may change the pharmacodynamics of prazosin in geriatric patients.12,16 This may necessitate increased doses to adequately antagonize the α-1 adenoreceptor.17 High doses of prazosin may increase the risk of hypotension in older patients.12 This increased risk is especially concerning for patients who already receive multiple medications or have comorbid conditions that impact blood pressure (BP).
The existing literature has few studies that have reported on outcomes with prazosin use in older veterans.11,12 The few existing reports provide clinically valuable descriptions of tolerability and efficacy with prazosin. For example, Peskind and colleagues showed prazosin to be an effective agent in the treatment of PTSD-related nightmares.12 However, in older veterans prazosin dosing > 4 mg has not been described or reported in the literature.
There appears to be a lack of clinical guidance with regards to dosing of prazosin in older patients. The goal of the current study was to assess the outcomes of older veterans with PTSD under pharmacist management of prazosin at our outpatient Prazosin Titration Clinic (PTC) in order to contribute to the minimal, yet valuable, existing clinical literature.
Methods
This study was approved by the University of Iowa Institutional Review Board and Iowa City Veterans Affairs Health Care System (ICVAHCS) research and development committee. The study was a retrospective chart review of older patients with consultations referred to the ICVAHCS PTC. To be eligible for inclusion, veterans with a PTSD diagnosis must have been evaluated at an initial consult appointment with a mental health clinical pharmacy specialist (MH CPS) from February 1, 2016 to August 31, 2018, and had at least 1 follow-up appointment. Follow-up visits were conducted either by telephone or in a face-to-face clinic visit.
Prazosin Titration Clinics
VA health care systems use pharmacists to manage veterans prescribed prazosin through PTC consultations. PTCs provide a process for close follow-up and assessment of PTSD-related outcomes. Due to the frequency of follow-up, this service may be beneficial for older veterans with more complex comorbidities and medication regimens. Any veteran with PTSD-related nightmares may be referred to the PTC for a consultation by any health care provider. Once referred to the clinic, MH CPSs assume responsibility for the prazosin prescription, including dose adjustments. For example, if a veteran reported no issues with tolerability but continued to have frequent and distressing nightmares, the dose may be increased, typically by 1-mg to 2-mg increments. Once the veteran reaches a stable and tolerable dose of prazosin, they are discharged from the PTC, and the referring health care provider resumes responsibility for the prazosin prescription.
Clinically Measured Outcomes
Nightmare frequency and intensity were measured using the Recurrent Distressing Dreams item B2 of the Clinician Administered PTSD Scale (CAPS) (Table 1). The PTSD Checklist (PCL-5), Insomnia Severity Index (ISI), and total sleep hours were used to determine the effect of prazosin on symptom severity (Table 2). The PCL-5 is a 20-item self-report used to monitor and quantify symptom level and change over time. It evaluates the frequency over the past month that a patient was bothered by any of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) PTSD criterion.2 Scores range from 0 (not at all) to 4 (extreme), with a maximum score of 80. The ISI is a 7-item self-report of sleep symptoms, with a total score of 28, where increasing scores indicate increasing severity of insomnia (Table 3).
Clinically measured outcome scales were performed and assessed by MH CPSs. CAPS frequency and intensity were measured at each clinic visit. PCL-5 and ISI scores were assessed at baseline and at the endpoint of study or discharge from clinic (Table 4). Patients who continued in the PTC after the end of the study date or who were lost to follow-up did not complete these measures at time of discharge.
![]()
Data Analysis
The primary outcome was change in CAPS nightmare frequency and intensity from time of initial clinic visit to time of discharge or end of study. The secondary outcomes included change in PCL-5, ISI, and sleep hours. Other secondary outcomes included measures of tolerability: BP changes, adverse effects (AEs) reported, and outcome of prazosin therapy when AEs were reported. Change in PTSD symptoms, PCL-5, and ISI were assessed using the Wilcoxon signed rank tests. Findings were considered to be statistically significant at P ≤ .05. Other variables were reported descriptively.
Results
Thirty-two veterans, aged ≥ 65 years, with clinical diagnosis of PTSD at the time of referral to the PTC were reviewed (Table 5). All patients were male and 93.8% were white. Thirty were Vietnam era veterans, 1 served in the Persian Gulf era, and 2 served in the post-Korean War era. Twenty-eight veterans had a combat history. Severe PTSD symptoms were reported as indicated by baseline PCL-5 scores, and moderate severity insomnia symptoms as indicated by baseline ISI scores.
All veterans had at least 1 comorbid medical condition, and the majority had multiple medical comorbidities. All were taking multiple medical and psychiatric medications. More than 80% of veterans were taking antihypertensive agents at baseline (Table 6). Twenty-two of the 32 veterans were prescribed a VA/DoD PTSD guideline-recommended antidepressant.
Primary Outcomes
The baseline, final, and changes in the primary outcomes are included in the Figure. Treatment with prazosin was associated with significant improvement in median scores from baseline to endpoint for CAPS nightmare frequency (-2, P = .0001), CAPS nightmare intensity (-2, P = .001), and total CAPS item score (-4, P < .001).
Secondary Outcomes
Of the 32 patients included in the study, PCL-5 was obtained from 20 veterans and ISI from 17 veterans at discharge from clinic. Thirty veterans reported final sleep hours, 2 veterans were unable to quantify average sleep hours per night at their final visit. PTSD symptom severity showed significant median change from baseline to endpoint of management in PTC for PCL-5 (-20.5, P = .0002) and ISI (-6.5, P = .002). Total sleep hours also showed significant improvement from baseline to endpoint (1.5, P = .003) (Table 7).
Prazosin Dosing
Maximum prazosin total daily doses were evaluated from the study baseline to the endpoint (Table 8). The mean (SD) maximum total daily dose of prazosin reached was 5.6 (5.1) mg (median, 3.5 mg; range, 1-17 mg). The mean (SD) total daily dose of prazosin at endpoint of study was 5.1 (5.3) mg (median, 2.5 mg; range, 0-17 mg). The average (SD) change of prazosin dose from baseline to endpoint was 3.5 (4.6) mg (median, 2 mg; range, -2 to 15 mg).
Tolerability
The average (SD) baseline systolic BP (SBP) was 135.8 (20.5) mm Hg and diastolic BP (DBP) was 77.2 (11.0) mm Hg. The average SBP and DBP at study endpoint were 131.8 (16.6) mm Hg and 75.9 (13.7) mm Hg, respectively. Endpoint BP values were missing for 6 patients.
Nine of 32 veterans reported AEs during PTC management of prazosin. Dizziness was the most common AE reported. Other AEs noted included orthostatic hypotension, headache, and falls. Of 12 reported AEs, 8 were related to dizziness, 5 of which were transient or tolerable. One veteran had a dose reduction of prazosin due to dizziness, and 3 veterans discontinued prazosin due to orthostasis. Several veterans had changes made to their antihypertensive medication regimen during prazosin titration, including dose reductions and/or decreased number of medications. If indicated, the MH CPS collaborated with the antihypertensive prescriber to make dosing adjustments. Two veterans reported a fall during prazosin titration; 1 veteran had other mobility-related factors thought to precipitate to their fall, and neither veterans were injured because of the falls.
Twenty-eight veterans (87.5%) treated in the PTC continued prazosin therapy after discharge. Six months postdischarge, 70% of veterans had maintained prazosin therapy. Two veterans required a dose increase postdischarge from PTC, and 1 veteran required a dose reduction. About one-third of veterans included in this study continued in the PTC beyond the end of the study period. Common reasons for clinic discharge were symptom resolution (37.5%), adverse reactions (12.5%), lost to follow-up (6.3%), or nonadherence (3.1%).
Discussion
The existing literature reports few outcomes for older veterans prescribed prazosin for PTSD. One report included a 75-year-old otherwise-healthy veteran, who received 2-mg prazosin at bedtime. At this dose, he reported good tolerability and response, as indicated by a reduction in his CAPS nightmare severity score.11 An open-label trial assessed prazosin in 9 geriatric men with chronic PTSD and found low-dose prazosin (average [SD] maximum prazosin dose reported was 2.3 [0.7] mg, range 2-4 mg per day) greatly reduced nightmares and overall PTSD severity in 8 of 9 subjects.12 Despite the veterans in that study having multiple medical comorbid conditions and taking concomitant medications, prazosin was reported to be well tolerated, and changes in BP were determined to be clinically insignificant.12 A recent study of middle-aged veterans (average [SD] age 52 [14] years) reported prazosin did not significantly alleviate PTSD-related nightmares.13 However, we observed prazosin therapy significantly reduced nightmares and sleep disturbances, and significantly improved PTSD severity in our older veteran population.
To our knowledge, the current study is the largest retrospective study that evaluates prazosin therapy for the treatment of PTSD-related nightmares in older veterans. The findings of this study are similar to a previous study in older veterans as well as studies of prazosin in younger and middle-aged adult veterans, with the average age ranging from 30 to 56 years.6-12 Like the previously reported studies, prazosin also was well tolerated in our sample of veterans with multiple comorbidities and concomitant medications. Changes in BP were not clinically significant.
Studies have demonstrated increased noradrenergic activity as a component of the normal aging process.16,17 This may require utilizing caution during prazosin dose titration and frequent patient assessment, due to the concern for risk of hypotension in older patients and in particular those who may require increased doses to achieve efficacy. In our study, favorable outcomes were achieved at an average (SD) total daily dose of 5.1 (5.3) mg (median, 2.5 mg; range 0-17 mg). A previous report showed efficacy of prazosin around an average (SD) maximum dose of 2.3 (0.7) mg, which is lower than the doses reported in the current study.12 In addition, 13 veterans (40.6%) from our sample reached doses of ≥ 5 mg per day, and 8 veterans (25.0%) reached doses of ≥ 10 mg per day.
The doses reached in this study were reflective of a management approach using assessment of patient-reported symptoms at weekly to biweekly follow-up visits. The individualized management approach applied in the PTC by MH CPSs aids in uncovering the most efficacious and tolerated dose of prazosin for each veteran. Evaluation of symptom change during treatment in PTC was facilitated use of objective rating scales, which helped measure nightmare frequency and intensity, sleep satisfaction, and global PTSD severity. Given the variability in dosing of prazosin reported in the literature, further studies may be warranted to provide more definitive clinical guidance as far as dosing prazosin in older patients.
The study by Peskind and colleaguesrationalized that lower doses of prazosin may be used in older patients given pharmacokinetic effects of aging, age-associated changes in PTSD pathophysiology, and effects and interactions of concomitant medications.12 However, our study found that prazosin could be well tolerated at higher doses. The rate of discontinuation due to intolerable AEs was low. AEs reported were consistent with the established AE profile of prazosin, with dizziness, orthostasis, and headache most commonly reported. Similar to the Peskind and colleagues study, BP had a tendency to decrease in this current study; however, the change was not clinically significant.12 That study also reported transient dizziness with prazosin titration, which was shown to be tolerable in the majority of our veterans reporting dizziness.12 Other common AEs with prazosin, such as rash, priapism, sedation, syncope, other cardiac AEs, and sleep disturbance were not reported in our study population.
MH CPS-managed PTCs are one venue that may allow veterans to achieve favorable outcomes through frequent follow-up. As prazosin dosing is specific to each individual patient, frequent follow-up visits are helpful in determining optimal doses that maximize efficacy while minimizing intolerable AEs. The majority of veterans treated in our PTC continued use of prazosin 6 months postdischarge, while 3 veterans required a postdischarge dose change.
The 2017 VA/DoD PTSD guidelines recommend individual, trauma-focused psychotherapy over pharmacologic therapy for the primary treatment of PTSD.14 About half of the veterans in the current study participated in either group or individual psychotherapy during enrollment in the PTC. A systematic review of psychotherapy in older veterans reported mixed results, with 4 studies indicating positive effects of therapy, while the other 3 studies reported no benefit or mixed effects for PTSD symptoms. The review concluded that fewer older adults experience complete remission of symptoms with psychotherapy alone.18 A previous study of older veterans described improvement in PTSD-related symptoms with prazosin without concurrent psychotherapy.12
Limitations and Strengths
While this study is the largest study to evaluate outcomes of prazosin in older patients with PTSD, there are several important limitations. The study population was small and all were male. The results of this study may not be applicable to women. Another limitation was several missing values in our data set, as some secondary outcomes were not collected via telephone follow-up visits. This could potentially contribute a measurement bias in the reported secondary outcomes results, specifically for the PCL-5 and ISI. Additionally, some veterans in this study may have reported symptomatic improvement based on the additional supportive intervention that clinical pharmacists were able to offer, as well as concomitant participation in psychotherapy. This may be reflected in the study results. This study did not have a true placebo group, as we may find a reduction in symptoms with placebo.
Strengths of this study include multiple data points for assessment of prazosin tolerability and a pre- and poststudy design, which allowed for the veterans to serve as their own control. Another strength of this study is that data were complete for primary outcome measures, including the CAPS Recurrent and Distressing Dreams Item, where prazosin showed significant benefit in reduction of PTSD-related nightmares. While the results of this study are reassuring, further randomized, double-blind, placebo-controlled trials are likely needed in order to establish efficacy and tolerability of prazosin in older veterans for PTSD related nightmares.
Conclusion
These results demonstrate prazosin therapy in older veterans can significantly improve PTSD-related nightmares and PTSD severity. Prazosin was well tolerated in this population at doses higher than previously reported in other studies. This study shows that prazosin therapy can be effectively managed and tolerated in older veterans with complex medical and psychiatric comorbidities to provide favorable patient outcomes.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Iowa City VA Health Care System and by the Health Services Research and Development Service, US Department of Veterans Affairs.
Posttraumatic stress disorder (PTSD) is a common psychiatric condition in the veteran population and is associated with significant sleep disturbances and trauma-related nightmares.1 PTSD can present with intrusive symptoms, such as recurrent memories or dreams, which are associated with traumatic events.2 Clinical studies have described an increase in central nervous system (CNS) noradrenergic activity in PTSD; specifically, noradrenergic outflow and/or postsynaptic adrenoreceptor responsiveness is increased.3,4 Targeting a reduction in noradrenergic activity via antagonism of noradrenergic receptors has been a therapeutic treatment strategy in PTSD.
Prazosin crosses the blood-brain barrier and works to antagonize α-1 adrenoreceptors to decrease noradrenergic outflow.5 It has been shown in multiple trials to effectively reduce nightmares and improve sleep quality in the veteran population.6-12 However, a recent negative trial contributed to a downgraded recommendation for prazosin in the treatment of PTSD-related nightmares in the joint PTSD guideline from the US Department of Veterans Affairs (VA) and US Department of Defense (DoD).13,14
The diagnosis of PTSD in veterans aged ≥ 65 years has been increasing due to improved recognition.15 As a result, prazosin may be considered more frequently as a treatment option for those patients who report PTSD-related nightmares. It is important to recognize that the normal physiologic process of aging is associated with increased noradrenergic outflow, which may change the pharmacodynamics of prazosin in geriatric patients.12,16 This may necessitate increased doses to adequately antagonize the α-1 adenoreceptor.17 High doses of prazosin may increase the risk of hypotension in older patients.12 This increased risk is especially concerning for patients who already receive multiple medications or have comorbid conditions that impact blood pressure (BP).
The existing literature has few studies that have reported on outcomes with prazosin use in older veterans.11,12 The few existing reports provide clinically valuable descriptions of tolerability and efficacy with prazosin. For example, Peskind and colleagues showed prazosin to be an effective agent in the treatment of PTSD-related nightmares.12 However, in older veterans prazosin dosing > 4 mg has not been described or reported in the literature.
There appears to be a lack of clinical guidance with regards to dosing of prazosin in older patients. The goal of the current study was to assess the outcomes of older veterans with PTSD under pharmacist management of prazosin at our outpatient Prazosin Titration Clinic (PTC) in order to contribute to the minimal, yet valuable, existing clinical literature.
Methods
This study was approved by the University of Iowa Institutional Review Board and Iowa City Veterans Affairs Health Care System (ICVAHCS) research and development committee. The study was a retrospective chart review of older patients with consultations referred to the ICVAHCS PTC. To be eligible for inclusion, veterans with a PTSD diagnosis must have been evaluated at an initial consult appointment with a mental health clinical pharmacy specialist (MH CPS) from February 1, 2016 to August 31, 2018, and had at least 1 follow-up appointment. Follow-up visits were conducted either by telephone or in a face-to-face clinic visit.
Prazosin Titration Clinics
VA health care systems use pharmacists to manage veterans prescribed prazosin through PTC consultations. PTCs provide a process for close follow-up and assessment of PTSD-related outcomes. Due to the frequency of follow-up, this service may be beneficial for older veterans with more complex comorbidities and medication regimens. Any veteran with PTSD-related nightmares may be referred to the PTC for a consultation by any health care provider. Once referred to the clinic, MH CPSs assume responsibility for the prazosin prescription, including dose adjustments. For example, if a veteran reported no issues with tolerability but continued to have frequent and distressing nightmares, the dose may be increased, typically by 1-mg to 2-mg increments. Once the veteran reaches a stable and tolerable dose of prazosin, they are discharged from the PTC, and the referring health care provider resumes responsibility for the prazosin prescription.
Clinically Measured Outcomes
Nightmare frequency and intensity were measured using the Recurrent Distressing Dreams item B2 of the Clinician Administered PTSD Scale (CAPS) (Table 1). The PTSD Checklist (PCL-5), Insomnia Severity Index (ISI), and total sleep hours were used to determine the effect of prazosin on symptom severity (Table 2). The PCL-5 is a 20-item self-report used to monitor and quantify symptom level and change over time. It evaluates the frequency over the past month that a patient was bothered by any of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) PTSD criterion.2 Scores range from 0 (not at all) to 4 (extreme), with a maximum score of 80. The ISI is a 7-item self-report of sleep symptoms, with a total score of 28, where increasing scores indicate increasing severity of insomnia (Table 3).
Clinically measured outcome scales were performed and assessed by MH CPSs. CAPS frequency and intensity were measured at each clinic visit. PCL-5 and ISI scores were assessed at baseline and at the endpoint of study or discharge from clinic (Table 4). Patients who continued in the PTC after the end of the study date or who were lost to follow-up did not complete these measures at time of discharge.
![]()
Data Analysis
The primary outcome was change in CAPS nightmare frequency and intensity from time of initial clinic visit to time of discharge or end of study. The secondary outcomes included change in PCL-5, ISI, and sleep hours. Other secondary outcomes included measures of tolerability: BP changes, adverse effects (AEs) reported, and outcome of prazosin therapy when AEs were reported. Change in PTSD symptoms, PCL-5, and ISI were assessed using the Wilcoxon signed rank tests. Findings were considered to be statistically significant at P ≤ .05. Other variables were reported descriptively.
Results
Thirty-two veterans, aged ≥ 65 years, with clinical diagnosis of PTSD at the time of referral to the PTC were reviewed (Table 5). All patients were male and 93.8% were white. Thirty were Vietnam era veterans, 1 served in the Persian Gulf era, and 2 served in the post-Korean War era. Twenty-eight veterans had a combat history. Severe PTSD symptoms were reported as indicated by baseline PCL-5 scores, and moderate severity insomnia symptoms as indicated by baseline ISI scores.
All veterans had at least 1 comorbid medical condition, and the majority had multiple medical comorbidities. All were taking multiple medical and psychiatric medications. More than 80% of veterans were taking antihypertensive agents at baseline (Table 6). Twenty-two of the 32 veterans were prescribed a VA/DoD PTSD guideline-recommended antidepressant.
Primary Outcomes
The baseline, final, and changes in the primary outcomes are included in the Figure. Treatment with prazosin was associated with significant improvement in median scores from baseline to endpoint for CAPS nightmare frequency (-2, P = .0001), CAPS nightmare intensity (-2, P = .001), and total CAPS item score (-4, P < .001).
Secondary Outcomes
Of the 32 patients included in the study, PCL-5 was obtained from 20 veterans and ISI from 17 veterans at discharge from clinic. Thirty veterans reported final sleep hours, 2 veterans were unable to quantify average sleep hours per night at their final visit. PTSD symptom severity showed significant median change from baseline to endpoint of management in PTC for PCL-5 (-20.5, P = .0002) and ISI (-6.5, P = .002). Total sleep hours also showed significant improvement from baseline to endpoint (1.5, P = .003) (Table 7).
Prazosin Dosing
Maximum prazosin total daily doses were evaluated from the study baseline to the endpoint (Table 8). The mean (SD) maximum total daily dose of prazosin reached was 5.6 (5.1) mg (median, 3.5 mg; range, 1-17 mg). The mean (SD) total daily dose of prazosin at endpoint of study was 5.1 (5.3) mg (median, 2.5 mg; range, 0-17 mg). The average (SD) change of prazosin dose from baseline to endpoint was 3.5 (4.6) mg (median, 2 mg; range, -2 to 15 mg).
Tolerability
The average (SD) baseline systolic BP (SBP) was 135.8 (20.5) mm Hg and diastolic BP (DBP) was 77.2 (11.0) mm Hg. The average SBP and DBP at study endpoint were 131.8 (16.6) mm Hg and 75.9 (13.7) mm Hg, respectively. Endpoint BP values were missing for 6 patients.
Nine of 32 veterans reported AEs during PTC management of prazosin. Dizziness was the most common AE reported. Other AEs noted included orthostatic hypotension, headache, and falls. Of 12 reported AEs, 8 were related to dizziness, 5 of which were transient or tolerable. One veteran had a dose reduction of prazosin due to dizziness, and 3 veterans discontinued prazosin due to orthostasis. Several veterans had changes made to their antihypertensive medication regimen during prazosin titration, including dose reductions and/or decreased number of medications. If indicated, the MH CPS collaborated with the antihypertensive prescriber to make dosing adjustments. Two veterans reported a fall during prazosin titration; 1 veteran had other mobility-related factors thought to precipitate to their fall, and neither veterans were injured because of the falls.
Twenty-eight veterans (87.5%) treated in the PTC continued prazosin therapy after discharge. Six months postdischarge, 70% of veterans had maintained prazosin therapy. Two veterans required a dose increase postdischarge from PTC, and 1 veteran required a dose reduction. About one-third of veterans included in this study continued in the PTC beyond the end of the study period. Common reasons for clinic discharge were symptom resolution (37.5%), adverse reactions (12.5%), lost to follow-up (6.3%), or nonadherence (3.1%).
Discussion
The existing literature reports few outcomes for older veterans prescribed prazosin for PTSD. One report included a 75-year-old otherwise-healthy veteran, who received 2-mg prazosin at bedtime. At this dose, he reported good tolerability and response, as indicated by a reduction in his CAPS nightmare severity score.11 An open-label trial assessed prazosin in 9 geriatric men with chronic PTSD and found low-dose prazosin (average [SD] maximum prazosin dose reported was 2.3 [0.7] mg, range 2-4 mg per day) greatly reduced nightmares and overall PTSD severity in 8 of 9 subjects.12 Despite the veterans in that study having multiple medical comorbid conditions and taking concomitant medications, prazosin was reported to be well tolerated, and changes in BP were determined to be clinically insignificant.12 A recent study of middle-aged veterans (average [SD] age 52 [14] years) reported prazosin did not significantly alleviate PTSD-related nightmares.13 However, we observed prazosin therapy significantly reduced nightmares and sleep disturbances, and significantly improved PTSD severity in our older veteran population.
To our knowledge, the current study is the largest retrospective study that evaluates prazosin therapy for the treatment of PTSD-related nightmares in older veterans. The findings of this study are similar to a previous study in older veterans as well as studies of prazosin in younger and middle-aged adult veterans, with the average age ranging from 30 to 56 years.6-12 Like the previously reported studies, prazosin also was well tolerated in our sample of veterans with multiple comorbidities and concomitant medications. Changes in BP were not clinically significant.
Studies have demonstrated increased noradrenergic activity as a component of the normal aging process.16,17 This may require utilizing caution during prazosin dose titration and frequent patient assessment, due to the concern for risk of hypotension in older patients and in particular those who may require increased doses to achieve efficacy. In our study, favorable outcomes were achieved at an average (SD) total daily dose of 5.1 (5.3) mg (median, 2.5 mg; range 0-17 mg). A previous report showed efficacy of prazosin around an average (SD) maximum dose of 2.3 (0.7) mg, which is lower than the doses reported in the current study.12 In addition, 13 veterans (40.6%) from our sample reached doses of ≥ 5 mg per day, and 8 veterans (25.0%) reached doses of ≥ 10 mg per day.
The doses reached in this study were reflective of a management approach using assessment of patient-reported symptoms at weekly to biweekly follow-up visits. The individualized management approach applied in the PTC by MH CPSs aids in uncovering the most efficacious and tolerated dose of prazosin for each veteran. Evaluation of symptom change during treatment in PTC was facilitated use of objective rating scales, which helped measure nightmare frequency and intensity, sleep satisfaction, and global PTSD severity. Given the variability in dosing of prazosin reported in the literature, further studies may be warranted to provide more definitive clinical guidance as far as dosing prazosin in older patients.
The study by Peskind and colleaguesrationalized that lower doses of prazosin may be used in older patients given pharmacokinetic effects of aging, age-associated changes in PTSD pathophysiology, and effects and interactions of concomitant medications.12 However, our study found that prazosin could be well tolerated at higher doses. The rate of discontinuation due to intolerable AEs was low. AEs reported were consistent with the established AE profile of prazosin, with dizziness, orthostasis, and headache most commonly reported. Similar to the Peskind and colleagues study, BP had a tendency to decrease in this current study; however, the change was not clinically significant.12 That study also reported transient dizziness with prazosin titration, which was shown to be tolerable in the majority of our veterans reporting dizziness.12 Other common AEs with prazosin, such as rash, priapism, sedation, syncope, other cardiac AEs, and sleep disturbance were not reported in our study population.
MH CPS-managed PTCs are one venue that may allow veterans to achieve favorable outcomes through frequent follow-up. As prazosin dosing is specific to each individual patient, frequent follow-up visits are helpful in determining optimal doses that maximize efficacy while minimizing intolerable AEs. The majority of veterans treated in our PTC continued use of prazosin 6 months postdischarge, while 3 veterans required a postdischarge dose change.
The 2017 VA/DoD PTSD guidelines recommend individual, trauma-focused psychotherapy over pharmacologic therapy for the primary treatment of PTSD.14 About half of the veterans in the current study participated in either group or individual psychotherapy during enrollment in the PTC. A systematic review of psychotherapy in older veterans reported mixed results, with 4 studies indicating positive effects of therapy, while the other 3 studies reported no benefit or mixed effects for PTSD symptoms. The review concluded that fewer older adults experience complete remission of symptoms with psychotherapy alone.18 A previous study of older veterans described improvement in PTSD-related symptoms with prazosin without concurrent psychotherapy.12
Limitations and Strengths
While this study is the largest study to evaluate outcomes of prazosin in older patients with PTSD, there are several important limitations. The study population was small and all were male. The results of this study may not be applicable to women. Another limitation was several missing values in our data set, as some secondary outcomes were not collected via telephone follow-up visits. This could potentially contribute a measurement bias in the reported secondary outcomes results, specifically for the PCL-5 and ISI. Additionally, some veterans in this study may have reported symptomatic improvement based on the additional supportive intervention that clinical pharmacists were able to offer, as well as concomitant participation in psychotherapy. This may be reflected in the study results. This study did not have a true placebo group, as we may find a reduction in symptoms with placebo.
Strengths of this study include multiple data points for assessment of prazosin tolerability and a pre- and poststudy design, which allowed for the veterans to serve as their own control. Another strength of this study is that data were complete for primary outcome measures, including the CAPS Recurrent and Distressing Dreams Item, where prazosin showed significant benefit in reduction of PTSD-related nightmares. While the results of this study are reassuring, further randomized, double-blind, placebo-controlled trials are likely needed in order to establish efficacy and tolerability of prazosin in older veterans for PTSD related nightmares.
Conclusion
These results demonstrate prazosin therapy in older veterans can significantly improve PTSD-related nightmares and PTSD severity. Prazosin was well tolerated in this population at doses higher than previously reported in other studies. This study shows that prazosin therapy can be effectively managed and tolerated in older veterans with complex medical and psychiatric comorbidities to provide favorable patient outcomes.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Iowa City VA Health Care System and by the Health Services Research and Development Service, US Department of Veterans Affairs.
1. Ross RJ, Ball WA, Sullivan KA, Caroff SN. Sleep disturbance as the hallmark of posttraumatic stress disorder. Am J Psychiatry. 1989;146(6):697-707.
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington VA: American Psychiatric Association; 2013.
3. Southwick SM, Krystal JH, Morgan CA, et al. Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry. 1993;50(4):266-274.
4. Geracioti TD Jr, Baker DG, Ekhator NN, et al. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry. 2001;158(8):1227-1230.
5. Friedman MJ. Posttraumatic and Acute Stress Disorders. 6th ed. New York: Springer Publishing; 2015.
6. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
7. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61(8):928-934.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. Germain A, Richardson R, Moul DE, et al. Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res. 2012;72(2):89-96.
10. Taylor HR, Freeman MK, Cates ME. Prazosin for treatment of nightmares related to posttraumatic stress disorder. Am J Health Syst Pharm. 2008;65(8):716-722.
11. Raskind MA, Dobie DJ, Kanter ED, Petrie EC, Thompson CE, Peskind ER. The alpha1-adrenergic antagonist prazosin ameliorates combat trauma nightmares in veterans with posttraumatic stress disorder: a report of 4 cases. J Clin Psychiatry. 2000;61(2):129-133.
12. Peskind ER, Bonner LT, Hoff DJ, Raskind MA. Prazosin reduces trauma-related nightmares in older men with chronic posttraumatic stress disorder. J Geriatr Psychiatry Neurol. 2003;16(3):165-171.
13. Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
14. The Management of Posttraumatic Stress Disorder Work Group. VA/DoD clinical practice guideline for the management of posttraumatic stress disorder and acute stress disorder. Version 3.0–2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/VADoDPTSDCPGFinal.pdf. Published June 2017. Accessed January 7, 2020.
15. Nichols BL, Czirr R. 24/Post-traumatic stress disorder: hidden syndrome in elders. Clin Gerontol. 1986;5(3-4):417-433.
16. Supiano MA, Linares OA, Smith MJ, Halter JB. Age-related differences in norepinephrine kinetics: effect of posture and sodium-restricted diet. Am J Physiol. 1990;259(3, pt 1):E422-E431.
17. Raskind MA, Peskind ER, Holmes C, Goldstein DS. Patterns of cerebrospinal fluid catechols support increased central noradrenergic responsiveness in aging and Alzheimer’s disease. Biol Psychiatry. 1999;46(6):756-765.
18. Dinnen S, Simiola V, Cook JM. Post-traumatic stress disorder in older adults: a systematic review of the psychotherapy treatment literature. Aging Ment Health. 2015;19(2):144-150.
1. Ross RJ, Ball WA, Sullivan KA, Caroff SN. Sleep disturbance as the hallmark of posttraumatic stress disorder. Am J Psychiatry. 1989;146(6):697-707.
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington VA: American Psychiatric Association; 2013.
3. Southwick SM, Krystal JH, Morgan CA, et al. Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry. 1993;50(4):266-274.
4. Geracioti TD Jr, Baker DG, Ekhator NN, et al. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry. 2001;158(8):1227-1230.
5. Friedman MJ. Posttraumatic and Acute Stress Disorders. 6th ed. New York: Springer Publishing; 2015.
6. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
7. Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61(8):928-934.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. Germain A, Richardson R, Moul DE, et al. Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res. 2012;72(2):89-96.
10. Taylor HR, Freeman MK, Cates ME. Prazosin for treatment of nightmares related to posttraumatic stress disorder. Am J Health Syst Pharm. 2008;65(8):716-722.
11. Raskind MA, Dobie DJ, Kanter ED, Petrie EC, Thompson CE, Peskind ER. The alpha1-adrenergic antagonist prazosin ameliorates combat trauma nightmares in veterans with posttraumatic stress disorder: a report of 4 cases. J Clin Psychiatry. 2000;61(2):129-133.
12. Peskind ER, Bonner LT, Hoff DJ, Raskind MA. Prazosin reduces trauma-related nightmares in older men with chronic posttraumatic stress disorder. J Geriatr Psychiatry Neurol. 2003;16(3):165-171.
13. Raskind MA, Peskind ER, Chow B, et al. Trial of prazosin for post-traumatic stress disorder in military veterans. N Engl J Med. 2018;378(6):507-517.
14. The Management of Posttraumatic Stress Disorder Work Group. VA/DoD clinical practice guideline for the management of posttraumatic stress disorder and acute stress disorder. Version 3.0–2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/VADoDPTSDCPGFinal.pdf. Published June 2017. Accessed January 7, 2020.
15. Nichols BL, Czirr R. 24/Post-traumatic stress disorder: hidden syndrome in elders. Clin Gerontol. 1986;5(3-4):417-433.
16. Supiano MA, Linares OA, Smith MJ, Halter JB. Age-related differences in norepinephrine kinetics: effect of posture and sodium-restricted diet. Am J Physiol. 1990;259(3, pt 1):E422-E431.
17. Raskind MA, Peskind ER, Holmes C, Goldstein DS. Patterns of cerebrospinal fluid catechols support increased central noradrenergic responsiveness in aging and Alzheimer’s disease. Biol Psychiatry. 1999;46(6):756-765.
18. Dinnen S, Simiola V, Cook JM. Post-traumatic stress disorder in older adults: a systematic review of the psychotherapy treatment literature. Aging Ment Health. 2015;19(2):144-150.
A Case-Based Review of Iron Overload With an Emphasis on Porphyria Cutanea Tarda, Hepatitis C, C282Y Heterozygosity, and Coronary Artery Disease
Sporadic porphyria cutanea tarda (PCT) is the most common cause of porphyria worldwide.1,2 Unlike other forms of porphyria, PCT usually is an acquired disease precipitated by extrinsic risk factors that commonly include excessive alcohol consumption, smoking, and chronic hepatitis C virus (HCV) infection. Additional risk factors include myeloproliferative disorders, exposure to polyhalogenated compounds, estrogen therapy, diseases of iron overload like hereditary hemochromatosis (HH), and potentially, HIV infection.1-3
In this case report, we present a patient with an iron overload (due in part to an HFE gene mutation) and concomitant PCT,
Case Presentation
Mr. M is a 59-year-old white male of Irish background with a medical history that includes coronary artery disease. He is status post ST-elevation myocardial infarction and percutaneous coronary intervention with placement of 2 drug-eluting stents. Additional medical issues include PCT and HCV infection with cirrhosis. He is an active smoker.
The patient has a long history of developing blisters with minor trauma, such as rubbing against his mattress/bed sheets or bumping into doors. These blisters primarily occur on his upper extremities, but also can occur on his face after shaving. Mr. M was diagnosed with HCV infection in 1979 while on active military duty. At that time, he had an acute HCV infection and jaundice that required a prolonged hospitalization. He reported no IV drug use and that many others on his military base had similar manifestations. He drinks 1 to 2 beers daily, but reports no binge drinking.
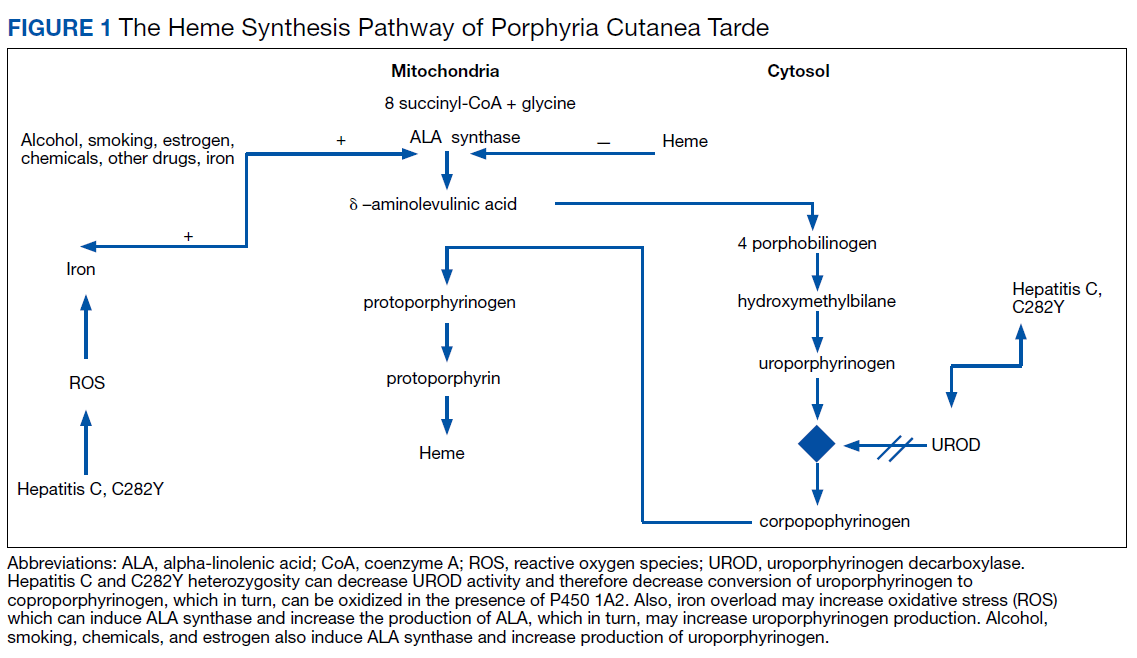
His laboratory studies were notable for ferritin, 2,069 ng/mL; serum iron, 317 mcg/dL; total iron binding capacity, 320 mcg/dL; transferrin, 239 mg/dl; liver function test alanine aminotransferase, 151 U/L; aspartate aminotransferase, 159 U/L; total bilirubin, 1.73 mg/dL; albumin, 3.6 g/dL; alkaline phosphatase, 119 U/L; INR, 1.1; and transferrin saturation, 99%. Mr. M’s HCV viral load was 28,700 IU/L with genotype 1b. Hemochromatosis genetic studies were notable for a heterozygous C282Y gene mutation and negative for H63D and S65C mutations. He repeatedly declined completing a 24-hour urine study of porphyrins. Ultrasonography was consistent with cirrhosis and splenomegaly. The patient was treatment naïve for HCV. He declined multiple offers for treatment of his HCV, citing financial considerations.
Porphyria Cutanea Tarda
The pathogenesis of PCT is related to the intrahepatic deficiency of uroporphyrinogen decarboxylase (UROD), an enzyme in the heme biosynthetic pathway (Figure 1). Decreased activity of UROD leads to accumulation of uroporphyrinogen and its derivatives, which most likely are oxidized in presence of cytochrome P450 1A2. Up to 80% of PCT cases are sporadic, in which the deficiency of UROD is acquired by exogenous risk factors as mentioned above. However, the remaining 20% of PCT cases are due to an autosomal dominant mutation of UROD that causes the partial deficiency (up to 50%) of UROD. In these cases, additional risk factors are needed to decrease UROD activity to < 75% for symptoms to occur.
Clinical Manifestation
Patients with PCT typically develop blisters, skin fragility, and peeling with sun exposure or minor trauma. They also may experience delayed wound healing in sun-exposed skin.3 The photosensitivity of PCT is believed to be related to the saturation of highly carboxylated uroporphyrins in the liver, which are then released into the circulation. Sun exposure then activates these products facilitating an immune reaction and subsequent skin damage.2 In chronic cases, fibrotic reactions and scaring occur which can be mistaken for scleroderma. Other skin manifestations include hyperpigmentation, hypertrichosis, alopecia due to scaring and purplish heliotrope suffusion of periorbital areas.
Patients can develop cirrhosis due to accumulation of porphyria in the hepatocytes and subsequent parenchymal damage. Hepatocellular carcinoma surveillance is recommended for patients with PCT, although its incidence is rare in those patients.
Diagnosis and Treatment
PCT is mainly a clinical diagnosis. Physicians should consider PCT in patients with photosensitivity and blisters after minor trauma (Figure 2). The urine of a patient with PCT is often pink or red when exposed to air or light due to its high concentration of porphyrin products. Mild elevation of liver enzymes and fatty liver on ultrasound are also noted. Evidence of iron overload is seen in most cases. Screening for risk factors like HCV, HIV, hepatitis B virus, and HH is recommended. Confirmation of PCT typically requires measurement of the porphyria level in a 24-hour urine collection.
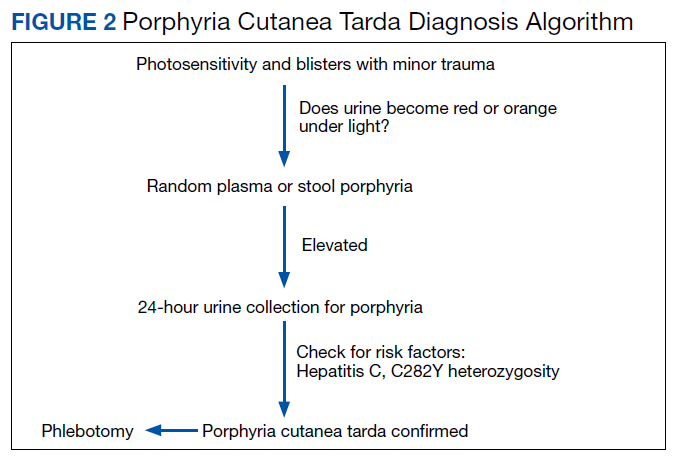
Avoiding sun exposure is fundamental in decreasing the development of skin lesions and scaring. Additionally, patients should be advised about the adverse effects of alcohol, smoking, and estrogen therapy on PCT. Treatment of PCT is frequently focused on iron overload and subsequent increased porphyrin oxidation.1,2 Iron can increase reactive oxygen species (ROS), which, in turn, increases the rate of oxidation of uroporphyrinogens. Excess iron also decreases the activity of UROD and increases δ-aminolevulinic acid (ALA) production (the precursor of uroporphyrinogen). Phlebotomy to treat iron overload should be done to a target ferritin level of 20 ng/mL. Clinical manifestations, including skin lesions, typically will normalize before the laboratory findings. Therapeutic remission is expected after 6 to 7 phlebotomy attempts, while clinical improvement can occur after 2 to 3 phlebotomies.
In addition to phlebotomy, 4-aminoquinoline medications (chloroquine and hydroxychloroquine) can be used effectively to treat PCT. Hydroxychloroquine is generally preferred due to its better safety profile. Although the exact mechanism of action of 4-aminoquinolines is not clear, it has been suggested that they bind to porphyrins and form water-soluble products, which are then excreted in the urine. Again, clinical remission occurs much sooner than chemical remission, (3 months vs 12 months). A 4-aminoquinoline should not be used in patients with severe liver disease, renal insufficiency, pregnancy, or G6PD deficiency. When used, they should be used in lower than typical doses due to the rapid removal of accumulated porphyrin from the hepatocytes potentially causing necrosis and acute hepatitis.
Iron chelation also is effective, but it is slower in achieving remission and more expensive than phlebotomy. Treatment of PCT should be individualized. For example, 4-aminoquinolines are contraindicated for patients with end-stage renal disease (ESRD), while phlebotomy could present a problem for patients with preexisting anemia. In this instance, removing 50 cc of blood every 2 weeks may be safe and effective. Furthermore, 4-aminoquinolines in patients with severe iron overload and phlebotomy have been used together. Plasmapheresis is still another option in patients with ESRD.
The use of direct antiviral agents (DAA) in the treatment of HCV has shown promising results in maintaining undetectable viral loads and concurrent remission of PCT. Several studies have shown that treatment of HCV with a DAA obviates the need for treatment PCT.3-5 Treatment of HCV with interferon (IFN) and ribavirin have shown mixed results in controlling PCT, possibly due to their ineffectiveness in maintaining a suppressed viral load. Some studies even showed worsening of PCT with IFN/ribavirin.6
Hemochromatosis
Human cells need iron for aerobic respiration. The intestinal mucosa controls iron uptake and its transfer to the blood stream. Aside from variations in intestinal absorption with fecal excretion, humans do not have another pathway to excrete excess iron. HH is the most common genetic disorder in whites.7 It is an autosomal recessive disorder that increases the intestinal absorption of iron. The most common mutation in the hemochromatosis (HFE) gene results in a substitution of tyrosine for cysteine at amino acid number 282 and is referred to as the C282Y mutation. A second mutation changes histidine at position 63 to aspartic acid and is referred to as a H63D mutation. H63D is present in a minority of the patients with phenotypically expressed HH and its clinical impact is unknown.
Homozygosity of the C282Y mutation is the most common genotype associated with clinical hemochromatosis. While carriers of the C282Y gene heterozygote mutation typically do not develop enough iron overload to cause clinical hemochromatosis, they can if other risk factors, such as PCT, excess alcohol use, liver disease, or HCV, are present.8 Additionally, an associated genetic defect, like a compound heterozygotes C282Y/H63D mutation, a private HFE mutation in trans, or other iron-related genes, can cause manifestations of iron overload. Lastly, about 20% of patients that are heterozygous for both mutations can express the HH phenotype.8
Clinical Manifestation
Patients with HH absorb only a few extra milligrams of iron daily. The clinical manifestation begins to occur when the total body iron store reaches 15-40 g (normal, 4 g). While the genetic mutation is present from birth, iron stores start to rise slowly to around 10 g > age 15 years, at which point serum iron levels are elevated. After age 20 years, the speed with which the iron is stored increases, and by 30 years, liver damage and tissue injury will occur. Cirrhosis is possible by 40 years.7 Age, sex, dietary iron intake, blood loss (menstruation), pregnancy, and other unknown factors greatly influence the disease progression. Homozygote C282Y mutation is as common in women as it is in men, but women are less likely to express the HH phenotype, presumably due, in part, to menstruation. When diagnosed early, most of the clinical manifestations of HH are preventable. Additional manifestations of HH include hyperpigmentation, cardiomyopathy, diabetes mellitus, hypogonadism, hypothyroidism, and arthropathy due to pseudogout.
Iron overload due to HH should be distinguished from other causes of iron overload including exogenous iron overload, anemia (thalassemia, sideroblastic), and chronic liver diseases like PCT, viral hepatitis, nonalcoholic steatohepatitis, and alcoholic liver disease.
Diagnosis
HH should be suspected in patients with a high serum transferrin saturation and elevated serum ferritin concentrations. Typically, transferrin saturation is > 50% and ferritin levels are > 300 ng/mL in men and > 200 ng/mL in women. In early stages of the disease, transferrin saturation can be normal. Additionally, in patients with chronic inflammation, ferritin may be high due to acute-phase reactants and the iron panel should be interpreted with caution. When the secondary causes of abnormalities in a patient’s iron studies are excluded, genetic testing for HFE gene is recommended.
The majority of patients (60-93%) with clinically evident hemochromatosis are homozygous for C282Y mutation. In a heterozygous C282Y mutation with a high transferrin saturation and HH phenotype, additional genetic testing for a heterozygous compound mutation C282Y/H63D is recommended.8 Additional studies could include evaluation for a private HFE mutation in trans or other iron-related genes. Liver biopsy is the gold standard for assessing the degree of hepatic fibrosis. Determining the degree of fibrosis by some means is needed due to the increased risk of hepatocellular carcinoma (HCC) in HH patients with advanced fibrosis and cirrhosis.9
Treatment
Iron depletion with phlebotomy is the cornerstone of treatment in HH. Phlebotomy initially is done weekly with goal of achieving a transferrin saturation < 50%, a serum ferritin level < 50 ng/mL, and a hemoglobin of 12 to 13 ng/mL. When these goals are achieved, patients typically need 4 to 8 phlebotomies per year to maintain a transferrin saturation < 50% (Figure 3).
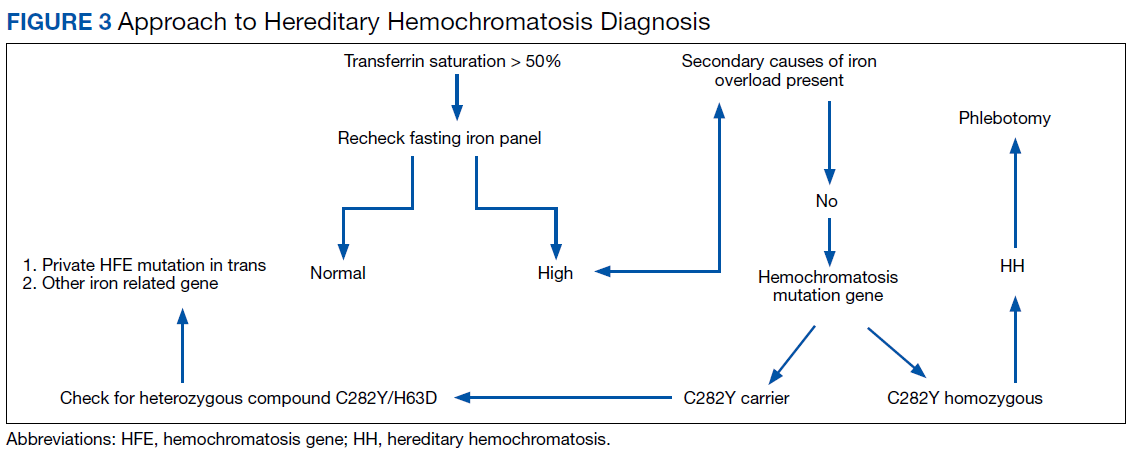
Hemochromatosis and PCT
Many studies have investigated the relevance of C282Y and/or H63D mutations in patients with PCT.9,10 It appears that ≥ 1 mutation of the HFE gene in PCT may be an important susceptibility factor in the development of clinical PCT. Various studies have shown an incidence of C282Y mutations of 44 to 47% in patients with PCT, compared with 9 to 12% in control populations.9,10 The incidence of the H63D mutation in PCT has been more variable, with some studies showing no difference between patients with PCT and a control group, while other studies showed 31% incidence of H63D mutation in patients with PCT.9,10 A higher incidence of C282Y and H63D mutations in PCT may be a sign that the HFE mutation could be an important factor in developing PCT.
Hemochromatosis and Hepatitis C
Transferrin saturation is frequently elevated in patients with HCV. It is yet unclear whether the pathology of liver disease in patients with HCV is influenced by iron overload or limited to the direct cell damage from replication of the virus and subsequent inflammation. It is believed that the pathology of iron overload in the patients with HCV is different from HH. Like other secondary causes of iron overload, the excess iron is stored in the Kupffer cells of patients with HCV. In HH, excess iron is stored in hepatocytes.
The prevalence of the HFE mutation is the same in the patients with chronic HCV and healthy individuals.10,11 However, HFE mutations are more prevalent in 30 to 60% of the patients with chronic HCV who have elevated transferrin saturations. Alone, C282Y heterozygosity, H63D heterozygosity, or C282Y/H63D compound heterozygosity could not lead to clinically significant iron overload in otherwise healthy individuals; however, these could be a significant cause of iron overload in patients with chronic HCV. Theoretically, the combination of iron overload and HFE gene mutations could increase the rate of advanced fibrosis/cirrhosis in chronic HCV. An increase serum ferritin level of 200 ng/dL in women and 250 ng/dL in men has been observed in 32% of patients with chronic HCV. In this subset of patients, phlebotomy reduced the progression of their liver disease and reduction in their liver enzymes.
Iron Overload and Cardiovascular Risk
In 1987, a Framingham cohort of > 2,800 patients showed a higher incident of CAD in postmenopausal women when compared with premenopausal women.12 In the 1980s, Sullivan hypothesized that the reason for higher incidence of CAD in men when compared with premenopausal women was due to their higher body iron storage.13-16 A study of 1,930 Finnish men reported that the men with ferritin level ≥ 200 ng/dL had a risk 2.2 times higher of acute myocardial infarction when compared to men with lower serum ferritin level.17
A prospective study published in 1997 by Klechl showed the role of iron stores in early atherogenesis via promotion of lipid oxidation.18 Other epidemiological studies have shown a decreased risk of myocardial infarction in blood donors, and while arguments have been made that the blood donors tend to be healthier individuals, 2 studies were published in 1997 matching healthy blood donors to healthy nonblood donors, and both showed a lower risk of CVD in the donors when compared with nondonors.19,20 Furthermore, in an animal model of atherosclerosis, an iron depleted diet showed a reduction of atherosclerosis progression.21 Multiple studies have shown that the heterozygosity for HFE is significantly linked to the risk of cardiovascular events, including the fact that heterozygosity for C282Y has been shown to be a risk factor for myocardial infarction in men and cerebrovascular death in women.22-25
Conclusion
Multiple studies have shown an association between the elevated iron levels associated with the HFE genotype and the disease states of our patient. These include an increased risk of CAD, the increased risk of cirrhosis in HCV and the development of PCT. Indeed, in this case, our patient likely acquired PCT from the combined risks of HCV and his heterozygous HFE genetic mutation.
With regard to Mr. M’s treatment, the use of an antiviral agent in the treatment of his HCV is fundamental, along with avoidance of alcohol and smoking. If he were to accept HCV treatment, we would anticipate resolution of the PCT, but the ongoing progression of his liver and cardiovascular conditions, due perhaps in part, to relative iron overload from his heterozygous HFE mutation. In this situation, we expect that an ongoing course of therapeutic phlebotomy could help to delay the progression of his chronic liver and cardiovascular diseases.
1. Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab. 2019;128(3):271-281.
2. Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012;32(6):880-893.
3. Wiznia LE, Laird ME, Franks AG Jr. Hepatitis C virus and its cutaneous manifestations: treatment in the direct-acting antiviral era. J Eur Acad Dermatol Venereol. 2017;31(8):1260-1270.
4. Nihei T, Kiniwa Y, Mikoshiba Y, Joshita S, Okuyama R. Improvement of porphyria cutanea tarda following treatment of hepatitis C virus by direct-acting antivirals: a case report. J Dermatol. 2019;46(5):e149-e151.
5. Combalia A, To-Figueras J, Laguno M, Martínez-Rebollar M, Aguilera P. Direct-acting antivirals for hepatitis C virus induce a rapid clinical and biochemical remission of porphyria cutanea tarda. Br J Dermatol. 2017;177(5):e183-e184. 6. Singal AK, Venkata KVR, Jampana S, Islam FU, Anderson KE. Hepatitis C treatment in patients with porphyria cutanea tarda. Am J Med Sci. 2017;353 (6):523-528.
7. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65(5):853-860.
8. Aguilar-Martinez P, Grandchamp B, Cunat S, et al. Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants. Haematologica. 2011;96(4):507-514.
9. Erhardt A, Maschner-Olberg A, Mellenthin C, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003;38(3):335-342.
10. Mehrany K, Drage LA, Brandhagen DJ, Pittelkow MR. Association of porphyria cutanea tarda with hereditary hemochromatosis. J Am Acad Dermatol. 2004;51(2):205-211.
11. Pietrangelo A. Hemochromatosis gene modifies course of hepatitis C viral infection. Gastroenterology. 2003;124(5):1509-1523.
12. Gordon T, Kannel WB, Hjortland MC, McNamara PM. Menopause and coronary heart disease. The Framingham Study. Ann Intern Med. 1978;89(2):157-161.
13. Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1(8233):1293-1294.
14. Sullivan JL. The sex difference in ischemic heart disease. Perspect Biol Med. 1983;26(4):657-671.
15. Sullivan JL. The iron paradigm of ischemic heart disease. Am Heart J. 1989;117(5):1177-1188.
16. Sullivan JL. Stored iron and ischemic heart disease: empirical support for a new paradigm. Circulation. 1992;86(3):1036-1037.
17. Salonen JT, Nyyssönen K, Korpela H, Tuomilehto J, Seppänen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86(3):803-811.
18. Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation. 1997;96(10):3300-3307.
19. Tuomainen TP, Salonen R, Nyyssönen K, Salonen JT. Cohort study of relation between donating blood and risk of myocardial infarction in 2682 men in eastern Finland. BMJ. 1997;314(7083):793-794.
20. Meyers DG, Strickland D, Maloley PA, Seburg JK, Wilson JE, McManus BF. Possible association of a reduction in cardiovascular events with blood donation. Heart. 1997;78(2):188-193.
21. Lee TS, Shiao MS, Pan CC, Chau LY. Iron-deficient diet reduces atherosclerotic lesions in apoE-deficient mice. Circulation. 1999;99(9):1222-1229.
22. Surber R, Sigusch HH, Kuehnert H, Figulla HR. Haemochromatosis (HFE) gene C282Y mutation and the risk of coronary artery disease and myocardial infarction: a study in 1279 patients undergoing coronary angiography. J Med Genet. 2003;40(5):e58.
23. Tuomainen TP, Kontula K, Nyyssönen K, Lakka TA, Heliö T, Salonen JT. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: a prospective cohort study in men in eastern Finland. Circulation. 1999;100(12):1274-1279.
24. Roest M, van der Schouw YT, de Valk B, et al. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation. 1999;100(12):1268-1273.
25. Pourmoghaddas A, Sanei H, Garakyaraghi M, Esteki-Ghashghaei F, Gharaati M. The relation between body iron store and ferritin, and coronary artery disease. ARYA Atheroscler. 2014;10(1):32-36.
Sporadic porphyria cutanea tarda (PCT) is the most common cause of porphyria worldwide.1,2 Unlike other forms of porphyria, PCT usually is an acquired disease precipitated by extrinsic risk factors that commonly include excessive alcohol consumption, smoking, and chronic hepatitis C virus (HCV) infection. Additional risk factors include myeloproliferative disorders, exposure to polyhalogenated compounds, estrogen therapy, diseases of iron overload like hereditary hemochromatosis (HH), and potentially, HIV infection.1-3
In this case report, we present a patient with an iron overload (due in part to an HFE gene mutation) and concomitant PCT,
Case Presentation
Mr. M is a 59-year-old white male of Irish background with a medical history that includes coronary artery disease. He is status post ST-elevation myocardial infarction and percutaneous coronary intervention with placement of 2 drug-eluting stents. Additional medical issues include PCT and HCV infection with cirrhosis. He is an active smoker.
The patient has a long history of developing blisters with minor trauma, such as rubbing against his mattress/bed sheets or bumping into doors. These blisters primarily occur on his upper extremities, but also can occur on his face after shaving. Mr. M was diagnosed with HCV infection in 1979 while on active military duty. At that time, he had an acute HCV infection and jaundice that required a prolonged hospitalization. He reported no IV drug use and that many others on his military base had similar manifestations. He drinks 1 to 2 beers daily, but reports no binge drinking.
His laboratory studies were notable for ferritin, 2,069 ng/mL; serum iron, 317 mcg/dL; total iron binding capacity, 320 mcg/dL; transferrin, 239 mg/dl; liver function test alanine aminotransferase, 151 U/L; aspartate aminotransferase, 159 U/L; total bilirubin, 1.73 mg/dL; albumin, 3.6 g/dL; alkaline phosphatase, 119 U/L; INR, 1.1; and transferrin saturation, 99%. Mr. M’s HCV viral load was 28,700 IU/L with genotype 1b. Hemochromatosis genetic studies were notable for a heterozygous C282Y gene mutation and negative for H63D and S65C mutations. He repeatedly declined completing a 24-hour urine study of porphyrins. Ultrasonography was consistent with cirrhosis and splenomegaly. The patient was treatment naïve for HCV. He declined multiple offers for treatment of his HCV, citing financial considerations.
Porphyria Cutanea Tarda
The pathogenesis of PCT is related to the intrahepatic deficiency of uroporphyrinogen decarboxylase (UROD), an enzyme in the heme biosynthetic pathway (Figure 1). Decreased activity of UROD leads to accumulation of uroporphyrinogen and its derivatives, which most likely are oxidized in presence of cytochrome P450 1A2. Up to 80% of PCT cases are sporadic, in which the deficiency of UROD is acquired by exogenous risk factors as mentioned above. However, the remaining 20% of PCT cases are due to an autosomal dominant mutation of UROD that causes the partial deficiency (up to 50%) of UROD. In these cases, additional risk factors are needed to decrease UROD activity to < 75% for symptoms to occur.
Clinical Manifestation
Patients with PCT typically develop blisters, skin fragility, and peeling with sun exposure or minor trauma. They also may experience delayed wound healing in sun-exposed skin.3 The photosensitivity of PCT is believed to be related to the saturation of highly carboxylated uroporphyrins in the liver, which are then released into the circulation. Sun exposure then activates these products facilitating an immune reaction and subsequent skin damage.2 In chronic cases, fibrotic reactions and scaring occur which can be mistaken for scleroderma. Other skin manifestations include hyperpigmentation, hypertrichosis, alopecia due to scaring and purplish heliotrope suffusion of periorbital areas.
Patients can develop cirrhosis due to accumulation of porphyria in the hepatocytes and subsequent parenchymal damage. Hepatocellular carcinoma surveillance is recommended for patients with PCT, although its incidence is rare in those patients.
Diagnosis and Treatment
PCT is mainly a clinical diagnosis. Physicians should consider PCT in patients with photosensitivity and blisters after minor trauma (Figure 2). The urine of a patient with PCT is often pink or red when exposed to air or light due to its high concentration of porphyrin products. Mild elevation of liver enzymes and fatty liver on ultrasound are also noted. Evidence of iron overload is seen in most cases. Screening for risk factors like HCV, HIV, hepatitis B virus, and HH is recommended. Confirmation of PCT typically requires measurement of the porphyria level in a 24-hour urine collection.
Avoiding sun exposure is fundamental in decreasing the development of skin lesions and scaring. Additionally, patients should be advised about the adverse effects of alcohol, smoking, and estrogen therapy on PCT. Treatment of PCT is frequently focused on iron overload and subsequent increased porphyrin oxidation.1,2 Iron can increase reactive oxygen species (ROS), which, in turn, increases the rate of oxidation of uroporphyrinogens. Excess iron also decreases the activity of UROD and increases δ-aminolevulinic acid (ALA) production (the precursor of uroporphyrinogen). Phlebotomy to treat iron overload should be done to a target ferritin level of 20 ng/mL. Clinical manifestations, including skin lesions, typically will normalize before the laboratory findings. Therapeutic remission is expected after 6 to 7 phlebotomy attempts, while clinical improvement can occur after 2 to 3 phlebotomies.
In addition to phlebotomy, 4-aminoquinoline medications (chloroquine and hydroxychloroquine) can be used effectively to treat PCT. Hydroxychloroquine is generally preferred due to its better safety profile. Although the exact mechanism of action of 4-aminoquinolines is not clear, it has been suggested that they bind to porphyrins and form water-soluble products, which are then excreted in the urine. Again, clinical remission occurs much sooner than chemical remission, (3 months vs 12 months). A 4-aminoquinoline should not be used in patients with severe liver disease, renal insufficiency, pregnancy, or G6PD deficiency. When used, they should be used in lower than typical doses due to the rapid removal of accumulated porphyrin from the hepatocytes potentially causing necrosis and acute hepatitis.
Iron chelation also is effective, but it is slower in achieving remission and more expensive than phlebotomy. Treatment of PCT should be individualized. For example, 4-aminoquinolines are contraindicated for patients with end-stage renal disease (ESRD), while phlebotomy could present a problem for patients with preexisting anemia. In this instance, removing 50 cc of blood every 2 weeks may be safe and effective. Furthermore, 4-aminoquinolines in patients with severe iron overload and phlebotomy have been used together. Plasmapheresis is still another option in patients with ESRD.
The use of direct antiviral agents (DAA) in the treatment of HCV has shown promising results in maintaining undetectable viral loads and concurrent remission of PCT. Several studies have shown that treatment of HCV with a DAA obviates the need for treatment PCT.3-5 Treatment of HCV with interferon (IFN) and ribavirin have shown mixed results in controlling PCT, possibly due to their ineffectiveness in maintaining a suppressed viral load. Some studies even showed worsening of PCT with IFN/ribavirin.6
Hemochromatosis
Human cells need iron for aerobic respiration. The intestinal mucosa controls iron uptake and its transfer to the blood stream. Aside from variations in intestinal absorption with fecal excretion, humans do not have another pathway to excrete excess iron. HH is the most common genetic disorder in whites.7 It is an autosomal recessive disorder that increases the intestinal absorption of iron. The most common mutation in the hemochromatosis (HFE) gene results in a substitution of tyrosine for cysteine at amino acid number 282 and is referred to as the C282Y mutation. A second mutation changes histidine at position 63 to aspartic acid and is referred to as a H63D mutation. H63D is present in a minority of the patients with phenotypically expressed HH and its clinical impact is unknown.
Homozygosity of the C282Y mutation is the most common genotype associated with clinical hemochromatosis. While carriers of the C282Y gene heterozygote mutation typically do not develop enough iron overload to cause clinical hemochromatosis, they can if other risk factors, such as PCT, excess alcohol use, liver disease, or HCV, are present.8 Additionally, an associated genetic defect, like a compound heterozygotes C282Y/H63D mutation, a private HFE mutation in trans, or other iron-related genes, can cause manifestations of iron overload. Lastly, about 20% of patients that are heterozygous for both mutations can express the HH phenotype.8
Clinical Manifestation
Patients with HH absorb only a few extra milligrams of iron daily. The clinical manifestation begins to occur when the total body iron store reaches 15-40 g (normal, 4 g). While the genetic mutation is present from birth, iron stores start to rise slowly to around 10 g > age 15 years, at which point serum iron levels are elevated. After age 20 years, the speed with which the iron is stored increases, and by 30 years, liver damage and tissue injury will occur. Cirrhosis is possible by 40 years.7 Age, sex, dietary iron intake, blood loss (menstruation), pregnancy, and other unknown factors greatly influence the disease progression. Homozygote C282Y mutation is as common in women as it is in men, but women are less likely to express the HH phenotype, presumably due, in part, to menstruation. When diagnosed early, most of the clinical manifestations of HH are preventable. Additional manifestations of HH include hyperpigmentation, cardiomyopathy, diabetes mellitus, hypogonadism, hypothyroidism, and arthropathy due to pseudogout.
Iron overload due to HH should be distinguished from other causes of iron overload including exogenous iron overload, anemia (thalassemia, sideroblastic), and chronic liver diseases like PCT, viral hepatitis, nonalcoholic steatohepatitis, and alcoholic liver disease.
Diagnosis
HH should be suspected in patients with a high serum transferrin saturation and elevated serum ferritin concentrations. Typically, transferrin saturation is > 50% and ferritin levels are > 300 ng/mL in men and > 200 ng/mL in women. In early stages of the disease, transferrin saturation can be normal. Additionally, in patients with chronic inflammation, ferritin may be high due to acute-phase reactants and the iron panel should be interpreted with caution. When the secondary causes of abnormalities in a patient’s iron studies are excluded, genetic testing for HFE gene is recommended.
The majority of patients (60-93%) with clinically evident hemochromatosis are homozygous for C282Y mutation. In a heterozygous C282Y mutation with a high transferrin saturation and HH phenotype, additional genetic testing for a heterozygous compound mutation C282Y/H63D is recommended.8 Additional studies could include evaluation for a private HFE mutation in trans or other iron-related genes. Liver biopsy is the gold standard for assessing the degree of hepatic fibrosis. Determining the degree of fibrosis by some means is needed due to the increased risk of hepatocellular carcinoma (HCC) in HH patients with advanced fibrosis and cirrhosis.9
Treatment
Iron depletion with phlebotomy is the cornerstone of treatment in HH. Phlebotomy initially is done weekly with goal of achieving a transferrin saturation < 50%, a serum ferritin level < 50 ng/mL, and a hemoglobin of 12 to 13 ng/mL. When these goals are achieved, patients typically need 4 to 8 phlebotomies per year to maintain a transferrin saturation < 50% (Figure 3).
Hemochromatosis and PCT
Many studies have investigated the relevance of C282Y and/or H63D mutations in patients with PCT.9,10 It appears that ≥ 1 mutation of the HFE gene in PCT may be an important susceptibility factor in the development of clinical PCT. Various studies have shown an incidence of C282Y mutations of 44 to 47% in patients with PCT, compared with 9 to 12% in control populations.9,10 The incidence of the H63D mutation in PCT has been more variable, with some studies showing no difference between patients with PCT and a control group, while other studies showed 31% incidence of H63D mutation in patients with PCT.9,10 A higher incidence of C282Y and H63D mutations in PCT may be a sign that the HFE mutation could be an important factor in developing PCT.
Hemochromatosis and Hepatitis C
Transferrin saturation is frequently elevated in patients with HCV. It is yet unclear whether the pathology of liver disease in patients with HCV is influenced by iron overload or limited to the direct cell damage from replication of the virus and subsequent inflammation. It is believed that the pathology of iron overload in the patients with HCV is different from HH. Like other secondary causes of iron overload, the excess iron is stored in the Kupffer cells of patients with HCV. In HH, excess iron is stored in hepatocytes.
The prevalence of the HFE mutation is the same in the patients with chronic HCV and healthy individuals.10,11 However, HFE mutations are more prevalent in 30 to 60% of the patients with chronic HCV who have elevated transferrin saturations. Alone, C282Y heterozygosity, H63D heterozygosity, or C282Y/H63D compound heterozygosity could not lead to clinically significant iron overload in otherwise healthy individuals; however, these could be a significant cause of iron overload in patients with chronic HCV. Theoretically, the combination of iron overload and HFE gene mutations could increase the rate of advanced fibrosis/cirrhosis in chronic HCV. An increase serum ferritin level of 200 ng/dL in women and 250 ng/dL in men has been observed in 32% of patients with chronic HCV. In this subset of patients, phlebotomy reduced the progression of their liver disease and reduction in their liver enzymes.
Iron Overload and Cardiovascular Risk
In 1987, a Framingham cohort of > 2,800 patients showed a higher incident of CAD in postmenopausal women when compared with premenopausal women.12 In the 1980s, Sullivan hypothesized that the reason for higher incidence of CAD in men when compared with premenopausal women was due to their higher body iron storage.13-16 A study of 1,930 Finnish men reported that the men with ferritin level ≥ 200 ng/dL had a risk 2.2 times higher of acute myocardial infarction when compared to men with lower serum ferritin level.17
A prospective study published in 1997 by Klechl showed the role of iron stores in early atherogenesis via promotion of lipid oxidation.18 Other epidemiological studies have shown a decreased risk of myocardial infarction in blood donors, and while arguments have been made that the blood donors tend to be healthier individuals, 2 studies were published in 1997 matching healthy blood donors to healthy nonblood donors, and both showed a lower risk of CVD in the donors when compared with nondonors.19,20 Furthermore, in an animal model of atherosclerosis, an iron depleted diet showed a reduction of atherosclerosis progression.21 Multiple studies have shown that the heterozygosity for HFE is significantly linked to the risk of cardiovascular events, including the fact that heterozygosity for C282Y has been shown to be a risk factor for myocardial infarction in men and cerebrovascular death in women.22-25
Conclusion
Multiple studies have shown an association between the elevated iron levels associated with the HFE genotype and the disease states of our patient. These include an increased risk of CAD, the increased risk of cirrhosis in HCV and the development of PCT. Indeed, in this case, our patient likely acquired PCT from the combined risks of HCV and his heterozygous HFE genetic mutation.
With regard to Mr. M’s treatment, the use of an antiviral agent in the treatment of his HCV is fundamental, along with avoidance of alcohol and smoking. If he were to accept HCV treatment, we would anticipate resolution of the PCT, but the ongoing progression of his liver and cardiovascular conditions, due perhaps in part, to relative iron overload from his heterozygous HFE mutation. In this situation, we expect that an ongoing course of therapeutic phlebotomy could help to delay the progression of his chronic liver and cardiovascular diseases.
Sporadic porphyria cutanea tarda (PCT) is the most common cause of porphyria worldwide.1,2 Unlike other forms of porphyria, PCT usually is an acquired disease precipitated by extrinsic risk factors that commonly include excessive alcohol consumption, smoking, and chronic hepatitis C virus (HCV) infection. Additional risk factors include myeloproliferative disorders, exposure to polyhalogenated compounds, estrogen therapy, diseases of iron overload like hereditary hemochromatosis (HH), and potentially, HIV infection.1-3
In this case report, we present a patient with an iron overload (due in part to an HFE gene mutation) and concomitant PCT,
Case Presentation
Mr. M is a 59-year-old white male of Irish background with a medical history that includes coronary artery disease. He is status post ST-elevation myocardial infarction and percutaneous coronary intervention with placement of 2 drug-eluting stents. Additional medical issues include PCT and HCV infection with cirrhosis. He is an active smoker.
The patient has a long history of developing blisters with minor trauma, such as rubbing against his mattress/bed sheets or bumping into doors. These blisters primarily occur on his upper extremities, but also can occur on his face after shaving. Mr. M was diagnosed with HCV infection in 1979 while on active military duty. At that time, he had an acute HCV infection and jaundice that required a prolonged hospitalization. He reported no IV drug use and that many others on his military base had similar manifestations. He drinks 1 to 2 beers daily, but reports no binge drinking.
His laboratory studies were notable for ferritin, 2,069 ng/mL; serum iron, 317 mcg/dL; total iron binding capacity, 320 mcg/dL; transferrin, 239 mg/dl; liver function test alanine aminotransferase, 151 U/L; aspartate aminotransferase, 159 U/L; total bilirubin, 1.73 mg/dL; albumin, 3.6 g/dL; alkaline phosphatase, 119 U/L; INR, 1.1; and transferrin saturation, 99%. Mr. M’s HCV viral load was 28,700 IU/L with genotype 1b. Hemochromatosis genetic studies were notable for a heterozygous C282Y gene mutation and negative for H63D and S65C mutations. He repeatedly declined completing a 24-hour urine study of porphyrins. Ultrasonography was consistent with cirrhosis and splenomegaly. The patient was treatment naïve for HCV. He declined multiple offers for treatment of his HCV, citing financial considerations.
Porphyria Cutanea Tarda
The pathogenesis of PCT is related to the intrahepatic deficiency of uroporphyrinogen decarboxylase (UROD), an enzyme in the heme biosynthetic pathway (Figure 1). Decreased activity of UROD leads to accumulation of uroporphyrinogen and its derivatives, which most likely are oxidized in presence of cytochrome P450 1A2. Up to 80% of PCT cases are sporadic, in which the deficiency of UROD is acquired by exogenous risk factors as mentioned above. However, the remaining 20% of PCT cases are due to an autosomal dominant mutation of UROD that causes the partial deficiency (up to 50%) of UROD. In these cases, additional risk factors are needed to decrease UROD activity to < 75% for symptoms to occur.
Clinical Manifestation
Patients with PCT typically develop blisters, skin fragility, and peeling with sun exposure or minor trauma. They also may experience delayed wound healing in sun-exposed skin.3 The photosensitivity of PCT is believed to be related to the saturation of highly carboxylated uroporphyrins in the liver, which are then released into the circulation. Sun exposure then activates these products facilitating an immune reaction and subsequent skin damage.2 In chronic cases, fibrotic reactions and scaring occur which can be mistaken for scleroderma. Other skin manifestations include hyperpigmentation, hypertrichosis, alopecia due to scaring and purplish heliotrope suffusion of periorbital areas.
Patients can develop cirrhosis due to accumulation of porphyria in the hepatocytes and subsequent parenchymal damage. Hepatocellular carcinoma surveillance is recommended for patients with PCT, although its incidence is rare in those patients.
Diagnosis and Treatment
PCT is mainly a clinical diagnosis. Physicians should consider PCT in patients with photosensitivity and blisters after minor trauma (Figure 2). The urine of a patient with PCT is often pink or red when exposed to air or light due to its high concentration of porphyrin products. Mild elevation of liver enzymes and fatty liver on ultrasound are also noted. Evidence of iron overload is seen in most cases. Screening for risk factors like HCV, HIV, hepatitis B virus, and HH is recommended. Confirmation of PCT typically requires measurement of the porphyria level in a 24-hour urine collection.
Avoiding sun exposure is fundamental in decreasing the development of skin lesions and scaring. Additionally, patients should be advised about the adverse effects of alcohol, smoking, and estrogen therapy on PCT. Treatment of PCT is frequently focused on iron overload and subsequent increased porphyrin oxidation.1,2 Iron can increase reactive oxygen species (ROS), which, in turn, increases the rate of oxidation of uroporphyrinogens. Excess iron also decreases the activity of UROD and increases δ-aminolevulinic acid (ALA) production (the precursor of uroporphyrinogen). Phlebotomy to treat iron overload should be done to a target ferritin level of 20 ng/mL. Clinical manifestations, including skin lesions, typically will normalize before the laboratory findings. Therapeutic remission is expected after 6 to 7 phlebotomy attempts, while clinical improvement can occur after 2 to 3 phlebotomies.
In addition to phlebotomy, 4-aminoquinoline medications (chloroquine and hydroxychloroquine) can be used effectively to treat PCT. Hydroxychloroquine is generally preferred due to its better safety profile. Although the exact mechanism of action of 4-aminoquinolines is not clear, it has been suggested that they bind to porphyrins and form water-soluble products, which are then excreted in the urine. Again, clinical remission occurs much sooner than chemical remission, (3 months vs 12 months). A 4-aminoquinoline should not be used in patients with severe liver disease, renal insufficiency, pregnancy, or G6PD deficiency. When used, they should be used in lower than typical doses due to the rapid removal of accumulated porphyrin from the hepatocytes potentially causing necrosis and acute hepatitis.
Iron chelation also is effective, but it is slower in achieving remission and more expensive than phlebotomy. Treatment of PCT should be individualized. For example, 4-aminoquinolines are contraindicated for patients with end-stage renal disease (ESRD), while phlebotomy could present a problem for patients with preexisting anemia. In this instance, removing 50 cc of blood every 2 weeks may be safe and effective. Furthermore, 4-aminoquinolines in patients with severe iron overload and phlebotomy have been used together. Plasmapheresis is still another option in patients with ESRD.
The use of direct antiviral agents (DAA) in the treatment of HCV has shown promising results in maintaining undetectable viral loads and concurrent remission of PCT. Several studies have shown that treatment of HCV with a DAA obviates the need for treatment PCT.3-5 Treatment of HCV with interferon (IFN) and ribavirin have shown mixed results in controlling PCT, possibly due to their ineffectiveness in maintaining a suppressed viral load. Some studies even showed worsening of PCT with IFN/ribavirin.6
Hemochromatosis
Human cells need iron for aerobic respiration. The intestinal mucosa controls iron uptake and its transfer to the blood stream. Aside from variations in intestinal absorption with fecal excretion, humans do not have another pathway to excrete excess iron. HH is the most common genetic disorder in whites.7 It is an autosomal recessive disorder that increases the intestinal absorption of iron. The most common mutation in the hemochromatosis (HFE) gene results in a substitution of tyrosine for cysteine at amino acid number 282 and is referred to as the C282Y mutation. A second mutation changes histidine at position 63 to aspartic acid and is referred to as a H63D mutation. H63D is present in a minority of the patients with phenotypically expressed HH and its clinical impact is unknown.
Homozygosity of the C282Y mutation is the most common genotype associated with clinical hemochromatosis. While carriers of the C282Y gene heterozygote mutation typically do not develop enough iron overload to cause clinical hemochromatosis, they can if other risk factors, such as PCT, excess alcohol use, liver disease, or HCV, are present.8 Additionally, an associated genetic defect, like a compound heterozygotes C282Y/H63D mutation, a private HFE mutation in trans, or other iron-related genes, can cause manifestations of iron overload. Lastly, about 20% of patients that are heterozygous for both mutations can express the HH phenotype.8
Clinical Manifestation
Patients with HH absorb only a few extra milligrams of iron daily. The clinical manifestation begins to occur when the total body iron store reaches 15-40 g (normal, 4 g). While the genetic mutation is present from birth, iron stores start to rise slowly to around 10 g > age 15 years, at which point serum iron levels are elevated. After age 20 years, the speed with which the iron is stored increases, and by 30 years, liver damage and tissue injury will occur. Cirrhosis is possible by 40 years.7 Age, sex, dietary iron intake, blood loss (menstruation), pregnancy, and other unknown factors greatly influence the disease progression. Homozygote C282Y mutation is as common in women as it is in men, but women are less likely to express the HH phenotype, presumably due, in part, to menstruation. When diagnosed early, most of the clinical manifestations of HH are preventable. Additional manifestations of HH include hyperpigmentation, cardiomyopathy, diabetes mellitus, hypogonadism, hypothyroidism, and arthropathy due to pseudogout.
Iron overload due to HH should be distinguished from other causes of iron overload including exogenous iron overload, anemia (thalassemia, sideroblastic), and chronic liver diseases like PCT, viral hepatitis, nonalcoholic steatohepatitis, and alcoholic liver disease.
Diagnosis
HH should be suspected in patients with a high serum transferrin saturation and elevated serum ferritin concentrations. Typically, transferrin saturation is > 50% and ferritin levels are > 300 ng/mL in men and > 200 ng/mL in women. In early stages of the disease, transferrin saturation can be normal. Additionally, in patients with chronic inflammation, ferritin may be high due to acute-phase reactants and the iron panel should be interpreted with caution. When the secondary causes of abnormalities in a patient’s iron studies are excluded, genetic testing for HFE gene is recommended.
The majority of patients (60-93%) with clinically evident hemochromatosis are homozygous for C282Y mutation. In a heterozygous C282Y mutation with a high transferrin saturation and HH phenotype, additional genetic testing for a heterozygous compound mutation C282Y/H63D is recommended.8 Additional studies could include evaluation for a private HFE mutation in trans or other iron-related genes. Liver biopsy is the gold standard for assessing the degree of hepatic fibrosis. Determining the degree of fibrosis by some means is needed due to the increased risk of hepatocellular carcinoma (HCC) in HH patients with advanced fibrosis and cirrhosis.9
Treatment
Iron depletion with phlebotomy is the cornerstone of treatment in HH. Phlebotomy initially is done weekly with goal of achieving a transferrin saturation < 50%, a serum ferritin level < 50 ng/mL, and a hemoglobin of 12 to 13 ng/mL. When these goals are achieved, patients typically need 4 to 8 phlebotomies per year to maintain a transferrin saturation < 50% (Figure 3).
Hemochromatosis and PCT
Many studies have investigated the relevance of C282Y and/or H63D mutations in patients with PCT.9,10 It appears that ≥ 1 mutation of the HFE gene in PCT may be an important susceptibility factor in the development of clinical PCT. Various studies have shown an incidence of C282Y mutations of 44 to 47% in patients with PCT, compared with 9 to 12% in control populations.9,10 The incidence of the H63D mutation in PCT has been more variable, with some studies showing no difference between patients with PCT and a control group, while other studies showed 31% incidence of H63D mutation in patients with PCT.9,10 A higher incidence of C282Y and H63D mutations in PCT may be a sign that the HFE mutation could be an important factor in developing PCT.
Hemochromatosis and Hepatitis C
Transferrin saturation is frequently elevated in patients with HCV. It is yet unclear whether the pathology of liver disease in patients with HCV is influenced by iron overload or limited to the direct cell damage from replication of the virus and subsequent inflammation. It is believed that the pathology of iron overload in the patients with HCV is different from HH. Like other secondary causes of iron overload, the excess iron is stored in the Kupffer cells of patients with HCV. In HH, excess iron is stored in hepatocytes.
The prevalence of the HFE mutation is the same in the patients with chronic HCV and healthy individuals.10,11 However, HFE mutations are more prevalent in 30 to 60% of the patients with chronic HCV who have elevated transferrin saturations. Alone, C282Y heterozygosity, H63D heterozygosity, or C282Y/H63D compound heterozygosity could not lead to clinically significant iron overload in otherwise healthy individuals; however, these could be a significant cause of iron overload in patients with chronic HCV. Theoretically, the combination of iron overload and HFE gene mutations could increase the rate of advanced fibrosis/cirrhosis in chronic HCV. An increase serum ferritin level of 200 ng/dL in women and 250 ng/dL in men has been observed in 32% of patients with chronic HCV. In this subset of patients, phlebotomy reduced the progression of their liver disease and reduction in their liver enzymes.
Iron Overload and Cardiovascular Risk
In 1987, a Framingham cohort of > 2,800 patients showed a higher incident of CAD in postmenopausal women when compared with premenopausal women.12 In the 1980s, Sullivan hypothesized that the reason for higher incidence of CAD in men when compared with premenopausal women was due to their higher body iron storage.13-16 A study of 1,930 Finnish men reported that the men with ferritin level ≥ 200 ng/dL had a risk 2.2 times higher of acute myocardial infarction when compared to men with lower serum ferritin level.17
A prospective study published in 1997 by Klechl showed the role of iron stores in early atherogenesis via promotion of lipid oxidation.18 Other epidemiological studies have shown a decreased risk of myocardial infarction in blood donors, and while arguments have been made that the blood donors tend to be healthier individuals, 2 studies were published in 1997 matching healthy blood donors to healthy nonblood donors, and both showed a lower risk of CVD in the donors when compared with nondonors.19,20 Furthermore, in an animal model of atherosclerosis, an iron depleted diet showed a reduction of atherosclerosis progression.21 Multiple studies have shown that the heterozygosity for HFE is significantly linked to the risk of cardiovascular events, including the fact that heterozygosity for C282Y has been shown to be a risk factor for myocardial infarction in men and cerebrovascular death in women.22-25
Conclusion
Multiple studies have shown an association between the elevated iron levels associated with the HFE genotype and the disease states of our patient. These include an increased risk of CAD, the increased risk of cirrhosis in HCV and the development of PCT. Indeed, in this case, our patient likely acquired PCT from the combined risks of HCV and his heterozygous HFE genetic mutation.
With regard to Mr. M’s treatment, the use of an antiviral agent in the treatment of his HCV is fundamental, along with avoidance of alcohol and smoking. If he were to accept HCV treatment, we would anticipate resolution of the PCT, but the ongoing progression of his liver and cardiovascular conditions, due perhaps in part, to relative iron overload from his heterozygous HFE mutation. In this situation, we expect that an ongoing course of therapeutic phlebotomy could help to delay the progression of his chronic liver and cardiovascular diseases.
1. Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab. 2019;128(3):271-281.
2. Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012;32(6):880-893.
3. Wiznia LE, Laird ME, Franks AG Jr. Hepatitis C virus and its cutaneous manifestations: treatment in the direct-acting antiviral era. J Eur Acad Dermatol Venereol. 2017;31(8):1260-1270.
4. Nihei T, Kiniwa Y, Mikoshiba Y, Joshita S, Okuyama R. Improvement of porphyria cutanea tarda following treatment of hepatitis C virus by direct-acting antivirals: a case report. J Dermatol. 2019;46(5):e149-e151.
5. Combalia A, To-Figueras J, Laguno M, Martínez-Rebollar M, Aguilera P. Direct-acting antivirals for hepatitis C virus induce a rapid clinical and biochemical remission of porphyria cutanea tarda. Br J Dermatol. 2017;177(5):e183-e184. 6. Singal AK, Venkata KVR, Jampana S, Islam FU, Anderson KE. Hepatitis C treatment in patients with porphyria cutanea tarda. Am J Med Sci. 2017;353 (6):523-528.
7. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65(5):853-860.
8. Aguilar-Martinez P, Grandchamp B, Cunat S, et al. Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants. Haematologica. 2011;96(4):507-514.
9. Erhardt A, Maschner-Olberg A, Mellenthin C, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003;38(3):335-342.
10. Mehrany K, Drage LA, Brandhagen DJ, Pittelkow MR. Association of porphyria cutanea tarda with hereditary hemochromatosis. J Am Acad Dermatol. 2004;51(2):205-211.
11. Pietrangelo A. Hemochromatosis gene modifies course of hepatitis C viral infection. Gastroenterology. 2003;124(5):1509-1523.
12. Gordon T, Kannel WB, Hjortland MC, McNamara PM. Menopause and coronary heart disease. The Framingham Study. Ann Intern Med. 1978;89(2):157-161.
13. Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1(8233):1293-1294.
14. Sullivan JL. The sex difference in ischemic heart disease. Perspect Biol Med. 1983;26(4):657-671.
15. Sullivan JL. The iron paradigm of ischemic heart disease. Am Heart J. 1989;117(5):1177-1188.
16. Sullivan JL. Stored iron and ischemic heart disease: empirical support for a new paradigm. Circulation. 1992;86(3):1036-1037.
17. Salonen JT, Nyyssönen K, Korpela H, Tuomilehto J, Seppänen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86(3):803-811.
18. Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation. 1997;96(10):3300-3307.
19. Tuomainen TP, Salonen R, Nyyssönen K, Salonen JT. Cohort study of relation between donating blood and risk of myocardial infarction in 2682 men in eastern Finland. BMJ. 1997;314(7083):793-794.
20. Meyers DG, Strickland D, Maloley PA, Seburg JK, Wilson JE, McManus BF. Possible association of a reduction in cardiovascular events with blood donation. Heart. 1997;78(2):188-193.
21. Lee TS, Shiao MS, Pan CC, Chau LY. Iron-deficient diet reduces atherosclerotic lesions in apoE-deficient mice. Circulation. 1999;99(9):1222-1229.
22. Surber R, Sigusch HH, Kuehnert H, Figulla HR. Haemochromatosis (HFE) gene C282Y mutation and the risk of coronary artery disease and myocardial infarction: a study in 1279 patients undergoing coronary angiography. J Med Genet. 2003;40(5):e58.
23. Tuomainen TP, Kontula K, Nyyssönen K, Lakka TA, Heliö T, Salonen JT. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: a prospective cohort study in men in eastern Finland. Circulation. 1999;100(12):1274-1279.
24. Roest M, van der Schouw YT, de Valk B, et al. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation. 1999;100(12):1268-1273.
25. Pourmoghaddas A, Sanei H, Garakyaraghi M, Esteki-Ghashghaei F, Gharaati M. The relation between body iron store and ferritin, and coronary artery disease. ARYA Atheroscler. 2014;10(1):32-36.
1. Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab. 2019;128(3):271-281.
2. Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012;32(6):880-893.
3. Wiznia LE, Laird ME, Franks AG Jr. Hepatitis C virus and its cutaneous manifestations: treatment in the direct-acting antiviral era. J Eur Acad Dermatol Venereol. 2017;31(8):1260-1270.
4. Nihei T, Kiniwa Y, Mikoshiba Y, Joshita S, Okuyama R. Improvement of porphyria cutanea tarda following treatment of hepatitis C virus by direct-acting antivirals: a case report. J Dermatol. 2019;46(5):e149-e151.
5. Combalia A, To-Figueras J, Laguno M, Martínez-Rebollar M, Aguilera P. Direct-acting antivirals for hepatitis C virus induce a rapid clinical and biochemical remission of porphyria cutanea tarda. Br J Dermatol. 2017;177(5):e183-e184. 6. Singal AK, Venkata KVR, Jampana S, Islam FU, Anderson KE. Hepatitis C treatment in patients with porphyria cutanea tarda. Am J Med Sci. 2017;353 (6):523-528.
7. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65(5):853-860.
8. Aguilar-Martinez P, Grandchamp B, Cunat S, et al. Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants. Haematologica. 2011;96(4):507-514.
9. Erhardt A, Maschner-Olberg A, Mellenthin C, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003;38(3):335-342.
10. Mehrany K, Drage LA, Brandhagen DJ, Pittelkow MR. Association of porphyria cutanea tarda with hereditary hemochromatosis. J Am Acad Dermatol. 2004;51(2):205-211.
11. Pietrangelo A. Hemochromatosis gene modifies course of hepatitis C viral infection. Gastroenterology. 2003;124(5):1509-1523.
12. Gordon T, Kannel WB, Hjortland MC, McNamara PM. Menopause and coronary heart disease. The Framingham Study. Ann Intern Med. 1978;89(2):157-161.
13. Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1(8233):1293-1294.
14. Sullivan JL. The sex difference in ischemic heart disease. Perspect Biol Med. 1983;26(4):657-671.
15. Sullivan JL. The iron paradigm of ischemic heart disease. Am Heart J. 1989;117(5):1177-1188.
16. Sullivan JL. Stored iron and ischemic heart disease: empirical support for a new paradigm. Circulation. 1992;86(3):1036-1037.
17. Salonen JT, Nyyssönen K, Korpela H, Tuomilehto J, Seppänen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86(3):803-811.
18. Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation. 1997;96(10):3300-3307.
19. Tuomainen TP, Salonen R, Nyyssönen K, Salonen JT. Cohort study of relation between donating blood and risk of myocardial infarction in 2682 men in eastern Finland. BMJ. 1997;314(7083):793-794.
20. Meyers DG, Strickland D, Maloley PA, Seburg JK, Wilson JE, McManus BF. Possible association of a reduction in cardiovascular events with blood donation. Heart. 1997;78(2):188-193.
21. Lee TS, Shiao MS, Pan CC, Chau LY. Iron-deficient diet reduces atherosclerotic lesions in apoE-deficient mice. Circulation. 1999;99(9):1222-1229.
22. Surber R, Sigusch HH, Kuehnert H, Figulla HR. Haemochromatosis (HFE) gene C282Y mutation and the risk of coronary artery disease and myocardial infarction: a study in 1279 patients undergoing coronary angiography. J Med Genet. 2003;40(5):e58.
23. Tuomainen TP, Kontula K, Nyyssönen K, Lakka TA, Heliö T, Salonen JT. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: a prospective cohort study in men in eastern Finland. Circulation. 1999;100(12):1274-1279.
24. Roest M, van der Schouw YT, de Valk B, et al. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation. 1999;100(12):1268-1273.
25. Pourmoghaddas A, Sanei H, Garakyaraghi M, Esteki-Ghashghaei F, Gharaati M. The relation between body iron store and ferritin, and coronary artery disease. ARYA Atheroscler. 2014;10(1):32-36.