User login
FDA revokes emergency use of hydroxychloroquine
The U.S. Food and Drug Administration revoked its decision from March 28 allowing use of hydroxychloroquine and chloroquine to treat people hospitalized with COVID-19 under an emergency use authorization (EUA).
“Based on its ongoing analysis of the EUA and emerging scientific data, the FDA determined that chloroquine and hydroxychloroquine are unlikely to be effective in treating COVID-19 for the authorized uses in the EUA,” the agency announced in a June 15 statement.
The FDA also warned today that the use of hydroxychloroquine or chloroquine may have a potential drug interaction with the investigational antiviral drug remdesivir that limits its effectiveness against COVID-19.
Remdesivir was granted emergency use authorization by the FDA on May 1.
“Based on a recently completed nonclinical laboratory study, the FDA is revising the fact sheet for healthcare providers that accompanies the drug to state that coadministration of remdesivir and chloroquine phosphate or hydroxychloroquine sulfate is not recommended as it may result in reduced antiviral activity of remdesivir. The agency is not aware of instances of this reduced activity occurring in the clinical setting but is continuing to evaluate all data related to remdesivir,” the FDA said in a news release.
Controversy over hydroxychloroquine
Even with such federal permission, since late March the use of these two agents has been mired in controversy.
President Donald J. Trump promoted the use of hydroxychloroquine and chloroquine to treat Americans with COVID-19, while scientific studies raised questions about their safety and effectiveness. Recent research, for example, pointed to elevated cardiovascular risks, as reported by Medscape Medical News.
The FDA acknowledged this recent evidence. “Additionally, in light of ongoing serious cardiac adverse events and other potential serious side effects, the known and potential benefits of chloroquine and hydroxychloroquine no longer outweigh the known and potential risks for the authorized use.”
The full suspension of the EUA follows a warning the agency issued on April 24. The FDA’s Safety Communication cautioned against use of the two agents outside of a hospital setting, citing an increase in outpatient prescriptions and “reports of serious heart rhythm problems.”
“While additional clinical trials continue to evaluate the potential benefit of these drugs in treating or preventing COVID-19, we determined the emergency use authorization was no longer appropriate,” based on a rigorous assessment by scientists in our Center for Drug Evaluation and Research,” Patrizia Cavazzoni, MD, acting director of CDER, noted in the FDA statement.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration revoked its decision from March 28 allowing use of hydroxychloroquine and chloroquine to treat people hospitalized with COVID-19 under an emergency use authorization (EUA).
“Based on its ongoing analysis of the EUA and emerging scientific data, the FDA determined that chloroquine and hydroxychloroquine are unlikely to be effective in treating COVID-19 for the authorized uses in the EUA,” the agency announced in a June 15 statement.
The FDA also warned today that the use of hydroxychloroquine or chloroquine may have a potential drug interaction with the investigational antiviral drug remdesivir that limits its effectiveness against COVID-19.
Remdesivir was granted emergency use authorization by the FDA on May 1.
“Based on a recently completed nonclinical laboratory study, the FDA is revising the fact sheet for healthcare providers that accompanies the drug to state that coadministration of remdesivir and chloroquine phosphate or hydroxychloroquine sulfate is not recommended as it may result in reduced antiviral activity of remdesivir. The agency is not aware of instances of this reduced activity occurring in the clinical setting but is continuing to evaluate all data related to remdesivir,” the FDA said in a news release.
Controversy over hydroxychloroquine
Even with such federal permission, since late March the use of these two agents has been mired in controversy.
President Donald J. Trump promoted the use of hydroxychloroquine and chloroquine to treat Americans with COVID-19, while scientific studies raised questions about their safety and effectiveness. Recent research, for example, pointed to elevated cardiovascular risks, as reported by Medscape Medical News.
The FDA acknowledged this recent evidence. “Additionally, in light of ongoing serious cardiac adverse events and other potential serious side effects, the known and potential benefits of chloroquine and hydroxychloroquine no longer outweigh the known and potential risks for the authorized use.”
The full suspension of the EUA follows a warning the agency issued on April 24. The FDA’s Safety Communication cautioned against use of the two agents outside of a hospital setting, citing an increase in outpatient prescriptions and “reports of serious heart rhythm problems.”
“While additional clinical trials continue to evaluate the potential benefit of these drugs in treating or preventing COVID-19, we determined the emergency use authorization was no longer appropriate,” based on a rigorous assessment by scientists in our Center for Drug Evaluation and Research,” Patrizia Cavazzoni, MD, acting director of CDER, noted in the FDA statement.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration revoked its decision from March 28 allowing use of hydroxychloroquine and chloroquine to treat people hospitalized with COVID-19 under an emergency use authorization (EUA).
“Based on its ongoing analysis of the EUA and emerging scientific data, the FDA determined that chloroquine and hydroxychloroquine are unlikely to be effective in treating COVID-19 for the authorized uses in the EUA,” the agency announced in a June 15 statement.
The FDA also warned today that the use of hydroxychloroquine or chloroquine may have a potential drug interaction with the investigational antiviral drug remdesivir that limits its effectiveness against COVID-19.
Remdesivir was granted emergency use authorization by the FDA on May 1.
“Based on a recently completed nonclinical laboratory study, the FDA is revising the fact sheet for healthcare providers that accompanies the drug to state that coadministration of remdesivir and chloroquine phosphate or hydroxychloroquine sulfate is not recommended as it may result in reduced antiviral activity of remdesivir. The agency is not aware of instances of this reduced activity occurring in the clinical setting but is continuing to evaluate all data related to remdesivir,” the FDA said in a news release.
Controversy over hydroxychloroquine
Even with such federal permission, since late March the use of these two agents has been mired in controversy.
President Donald J. Trump promoted the use of hydroxychloroquine and chloroquine to treat Americans with COVID-19, while scientific studies raised questions about their safety and effectiveness. Recent research, for example, pointed to elevated cardiovascular risks, as reported by Medscape Medical News.
The FDA acknowledged this recent evidence. “Additionally, in light of ongoing serious cardiac adverse events and other potential serious side effects, the known and potential benefits of chloroquine and hydroxychloroquine no longer outweigh the known and potential risks for the authorized use.”
The full suspension of the EUA follows a warning the agency issued on April 24. The FDA’s Safety Communication cautioned against use of the two agents outside of a hospital setting, citing an increase in outpatient prescriptions and “reports of serious heart rhythm problems.”
“While additional clinical trials continue to evaluate the potential benefit of these drugs in treating or preventing COVID-19, we determined the emergency use authorization was no longer appropriate,” based on a rigorous assessment by scientists in our Center for Drug Evaluation and Research,” Patrizia Cavazzoni, MD, acting director of CDER, noted in the FDA statement.
This article first appeared on Medscape.com.
No OS benefit with gefitinib vs. chemo for EGFR+ NSCLC
The median OS was 75.5 months in patients randomized to adjuvant gefitinib and 62.8 months in patients randomized to vinorelbine plus cisplatin.
Yi-Long Wu, MD, of Guangdong Lung Cancer Institute in Guangzhou, China, reported these results as part of the American Society of Clinical Oncology virtual scientific program.
Prior results from this trial had shown a disease-free survival (DFS) benefit with gefitinib, but this did not translate to an OS benefit at the final analysis, Dr. Wu said.
He noted, however, that the median OS of 75.5 months in the gefitinib arm “was one of the best in resected EGFR-mutant non–small cell lung cancer, compared with historical data.”
The findings also suggest a possible benefit with at least 18 months of gefitinib and show that adjuvant EGFR tyrosine kinase inhibitors (TKIs) should be considered the optimal therapy to improve DFS and achieve potentially better OS in this setting, Dr. Wu said.
Study details and DFS
The ADJUVANT trial (NCT01405079) randomized 222 patients, aged 18-75 years, with EGFR-mutant, stage II-IIIA (N1-N2) NSCLC who had undergone complete resection. Patients were enrolled at 27 sites between September 2011 and April 2014.
The patients were randomized 1:1 to receive 250 mg of gefitinib once daily for 24 months, or 25 mg/m2 of vinorelbine on days 1 and 8 plus 75 mg/m2 of cisplatin on day 1 every 3 weeks for 4 cycles.
The intent-to-treat (ITT) population included 111 patients in each arm. The per-protocol population included 106 patients in the gefitinib arm and 87 patients in the chemotherapy arm.
Primary results from this trial showed a significant improvement in DFS with gefitinib (Lancet Oncol. 2018 Jan;19[1]:139-48). That improvement was maintained in the final analysis.
The median DFS was 30.8 months in the gefitinib arm and 19.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The hazard ratio (HR) was 0.56 (P = .001) in the ITT population and 0.51 (P < .001) in the per-protocol population.
In the ITT population, the 5-year DFS rates were 22.6% in the gefitinib arm and 23.2% in the chemotherapy arm. In the per-protocol population, the 5-year DFS rates were 22.6% and 22.8%, respectively.
OS results
The median OS was 75.5 months in the gefitinib arm and 62.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The HR was 0.92 in both the ITT (P = .674) and per-protocol populations (P = .686).
In the ITT population, the 5-year OS rates were 53.2% in the gefitinib arm and 51.2% in the chemotherapy arm. In the per-protocol population, the 5-year OS rates were 53.2% and 50.7%, respectively.
Subgroup analyses by age, gender, lymph node status, and EGFR mutation showed trends toward improved OS with gefitinib, but the differences were not statistically significant.
The researchers conducted a post hoc analysis to assess the effect of subsequent treatment on patient outcomes. The analysis showed that patients who received gefitinib with subsequent EGFR-TKIs had the best responses and OS.
The median OS was not reached among patients who received gefitinib and subsequent EGFR-TKIs, whereas the median OS ranged from 15.6 months to 62.8 months in other groups. The shortest OS was observed in patients who received adjuvant chemotherapy without subsequent therapy.
The duration of gefitinib treatment also appeared to affect OS. The median OS was 35.7 months in patients who received gefitinib for less than 18 months, and the median OS was not reached in patients who received gefitinib for 18 months or longer (HR, 0.38; P < .001).
Implications and potential next steps
Despite the lack of OS improvement with gefitinib, “all of the patients on this study did much, much better than historical non–small cell lung cancer not specified by the EGFR mutation, with 70 months median survival compared to 35 months median survival for N2-positive disease,” said invited discussant Christopher G. Azzoli, MD, director of thoracic oncology at Lifespan Cancer Institute at Brown University in Providence, R.I.
“But you can’t avoid noticing how the curves come back together in terms of disease-free survival when your effective treatment is limited to 24 months,” he added.
An apparent risk of late brain recurrence in the gefitinib arm is also a concern, Dr. Azzoli said. “So ... longer duration of treatment with a drug that has better control of CNS [central nervous system] disease, such as osimertinib, may improve both DFS and OS,” he added.
Only about 50% of patients in the chemotherapy arm received a TKI at recurrence. The post hoc analysis showing that TKI recipients had the best outcomes raises the question of whether “the survival benefit could be conferred by delivering a superior drug merely at recurrence, or is there benefit to earlier delivery of an effective drug,” Dr. Azzoli said.
Given the high cost of continuous therapy, biomarker refinement could help improve treatment decision-making, he said, noting that “early testing of blood DNA to detect cancer in the body as minimal residual disease is showing promise,” and that many phase 3 studies of EGFR-TKIs are ongoing.
The current trial was sponsored by the Guangdong Association of Clinical Trials. Dr. Wu disclosed relationships with AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb/China, Lilly, MSD Oncology, Pfizer, and Roche. Dr. Azzoli reported having no disclosures.
SOURCE: Wu Y et al. ASCO 2020, Abstract 9005.
The median OS was 75.5 months in patients randomized to adjuvant gefitinib and 62.8 months in patients randomized to vinorelbine plus cisplatin.
Yi-Long Wu, MD, of Guangdong Lung Cancer Institute in Guangzhou, China, reported these results as part of the American Society of Clinical Oncology virtual scientific program.
Prior results from this trial had shown a disease-free survival (DFS) benefit with gefitinib, but this did not translate to an OS benefit at the final analysis, Dr. Wu said.
He noted, however, that the median OS of 75.5 months in the gefitinib arm “was one of the best in resected EGFR-mutant non–small cell lung cancer, compared with historical data.”
The findings also suggest a possible benefit with at least 18 months of gefitinib and show that adjuvant EGFR tyrosine kinase inhibitors (TKIs) should be considered the optimal therapy to improve DFS and achieve potentially better OS in this setting, Dr. Wu said.
Study details and DFS
The ADJUVANT trial (NCT01405079) randomized 222 patients, aged 18-75 years, with EGFR-mutant, stage II-IIIA (N1-N2) NSCLC who had undergone complete resection. Patients were enrolled at 27 sites between September 2011 and April 2014.
The patients were randomized 1:1 to receive 250 mg of gefitinib once daily for 24 months, or 25 mg/m2 of vinorelbine on days 1 and 8 plus 75 mg/m2 of cisplatin on day 1 every 3 weeks for 4 cycles.
The intent-to-treat (ITT) population included 111 patients in each arm. The per-protocol population included 106 patients in the gefitinib arm and 87 patients in the chemotherapy arm.
Primary results from this trial showed a significant improvement in DFS with gefitinib (Lancet Oncol. 2018 Jan;19[1]:139-48). That improvement was maintained in the final analysis.
The median DFS was 30.8 months in the gefitinib arm and 19.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The hazard ratio (HR) was 0.56 (P = .001) in the ITT population and 0.51 (P < .001) in the per-protocol population.
In the ITT population, the 5-year DFS rates were 22.6% in the gefitinib arm and 23.2% in the chemotherapy arm. In the per-protocol population, the 5-year DFS rates were 22.6% and 22.8%, respectively.
OS results
The median OS was 75.5 months in the gefitinib arm and 62.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The HR was 0.92 in both the ITT (P = .674) and per-protocol populations (P = .686).
In the ITT population, the 5-year OS rates were 53.2% in the gefitinib arm and 51.2% in the chemotherapy arm. In the per-protocol population, the 5-year OS rates were 53.2% and 50.7%, respectively.
Subgroup analyses by age, gender, lymph node status, and EGFR mutation showed trends toward improved OS with gefitinib, but the differences were not statistically significant.
The researchers conducted a post hoc analysis to assess the effect of subsequent treatment on patient outcomes. The analysis showed that patients who received gefitinib with subsequent EGFR-TKIs had the best responses and OS.
The median OS was not reached among patients who received gefitinib and subsequent EGFR-TKIs, whereas the median OS ranged from 15.6 months to 62.8 months in other groups. The shortest OS was observed in patients who received adjuvant chemotherapy without subsequent therapy.
The duration of gefitinib treatment also appeared to affect OS. The median OS was 35.7 months in patients who received gefitinib for less than 18 months, and the median OS was not reached in patients who received gefitinib for 18 months or longer (HR, 0.38; P < .001).
Implications and potential next steps
Despite the lack of OS improvement with gefitinib, “all of the patients on this study did much, much better than historical non–small cell lung cancer not specified by the EGFR mutation, with 70 months median survival compared to 35 months median survival for N2-positive disease,” said invited discussant Christopher G. Azzoli, MD, director of thoracic oncology at Lifespan Cancer Institute at Brown University in Providence, R.I.
“But you can’t avoid noticing how the curves come back together in terms of disease-free survival when your effective treatment is limited to 24 months,” he added.
An apparent risk of late brain recurrence in the gefitinib arm is also a concern, Dr. Azzoli said. “So ... longer duration of treatment with a drug that has better control of CNS [central nervous system] disease, such as osimertinib, may improve both DFS and OS,” he added.
Only about 50% of patients in the chemotherapy arm received a TKI at recurrence. The post hoc analysis showing that TKI recipients had the best outcomes raises the question of whether “the survival benefit could be conferred by delivering a superior drug merely at recurrence, or is there benefit to earlier delivery of an effective drug,” Dr. Azzoli said.
Given the high cost of continuous therapy, biomarker refinement could help improve treatment decision-making, he said, noting that “early testing of blood DNA to detect cancer in the body as minimal residual disease is showing promise,” and that many phase 3 studies of EGFR-TKIs are ongoing.
The current trial was sponsored by the Guangdong Association of Clinical Trials. Dr. Wu disclosed relationships with AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb/China, Lilly, MSD Oncology, Pfizer, and Roche. Dr. Azzoli reported having no disclosures.
SOURCE: Wu Y et al. ASCO 2020, Abstract 9005.
The median OS was 75.5 months in patients randomized to adjuvant gefitinib and 62.8 months in patients randomized to vinorelbine plus cisplatin.
Yi-Long Wu, MD, of Guangdong Lung Cancer Institute in Guangzhou, China, reported these results as part of the American Society of Clinical Oncology virtual scientific program.
Prior results from this trial had shown a disease-free survival (DFS) benefit with gefitinib, but this did not translate to an OS benefit at the final analysis, Dr. Wu said.
He noted, however, that the median OS of 75.5 months in the gefitinib arm “was one of the best in resected EGFR-mutant non–small cell lung cancer, compared with historical data.”
The findings also suggest a possible benefit with at least 18 months of gefitinib and show that adjuvant EGFR tyrosine kinase inhibitors (TKIs) should be considered the optimal therapy to improve DFS and achieve potentially better OS in this setting, Dr. Wu said.
Study details and DFS
The ADJUVANT trial (NCT01405079) randomized 222 patients, aged 18-75 years, with EGFR-mutant, stage II-IIIA (N1-N2) NSCLC who had undergone complete resection. Patients were enrolled at 27 sites between September 2011 and April 2014.
The patients were randomized 1:1 to receive 250 mg of gefitinib once daily for 24 months, or 25 mg/m2 of vinorelbine on days 1 and 8 plus 75 mg/m2 of cisplatin on day 1 every 3 weeks for 4 cycles.
The intent-to-treat (ITT) population included 111 patients in each arm. The per-protocol population included 106 patients in the gefitinib arm and 87 patients in the chemotherapy arm.
Primary results from this trial showed a significant improvement in DFS with gefitinib (Lancet Oncol. 2018 Jan;19[1]:139-48). That improvement was maintained in the final analysis.
The median DFS was 30.8 months in the gefitinib arm and 19.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The hazard ratio (HR) was 0.56 (P = .001) in the ITT population and 0.51 (P < .001) in the per-protocol population.
In the ITT population, the 5-year DFS rates were 22.6% in the gefitinib arm and 23.2% in the chemotherapy arm. In the per-protocol population, the 5-year DFS rates were 22.6% and 22.8%, respectively.
OS results
The median OS was 75.5 months in the gefitinib arm and 62.8 months in the chemotherapy arm for both the ITT and per-protocol populations. The HR was 0.92 in both the ITT (P = .674) and per-protocol populations (P = .686).
In the ITT population, the 5-year OS rates were 53.2% in the gefitinib arm and 51.2% in the chemotherapy arm. In the per-protocol population, the 5-year OS rates were 53.2% and 50.7%, respectively.
Subgroup analyses by age, gender, lymph node status, and EGFR mutation showed trends toward improved OS with gefitinib, but the differences were not statistically significant.
The researchers conducted a post hoc analysis to assess the effect of subsequent treatment on patient outcomes. The analysis showed that patients who received gefitinib with subsequent EGFR-TKIs had the best responses and OS.
The median OS was not reached among patients who received gefitinib and subsequent EGFR-TKIs, whereas the median OS ranged from 15.6 months to 62.8 months in other groups. The shortest OS was observed in patients who received adjuvant chemotherapy without subsequent therapy.
The duration of gefitinib treatment also appeared to affect OS. The median OS was 35.7 months in patients who received gefitinib for less than 18 months, and the median OS was not reached in patients who received gefitinib for 18 months or longer (HR, 0.38; P < .001).
Implications and potential next steps
Despite the lack of OS improvement with gefitinib, “all of the patients on this study did much, much better than historical non–small cell lung cancer not specified by the EGFR mutation, with 70 months median survival compared to 35 months median survival for N2-positive disease,” said invited discussant Christopher G. Azzoli, MD, director of thoracic oncology at Lifespan Cancer Institute at Brown University in Providence, R.I.
“But you can’t avoid noticing how the curves come back together in terms of disease-free survival when your effective treatment is limited to 24 months,” he added.
An apparent risk of late brain recurrence in the gefitinib arm is also a concern, Dr. Azzoli said. “So ... longer duration of treatment with a drug that has better control of CNS [central nervous system] disease, such as osimertinib, may improve both DFS and OS,” he added.
Only about 50% of patients in the chemotherapy arm received a TKI at recurrence. The post hoc analysis showing that TKI recipients had the best outcomes raises the question of whether “the survival benefit could be conferred by delivering a superior drug merely at recurrence, or is there benefit to earlier delivery of an effective drug,” Dr. Azzoli said.
Given the high cost of continuous therapy, biomarker refinement could help improve treatment decision-making, he said, noting that “early testing of blood DNA to detect cancer in the body as minimal residual disease is showing promise,” and that many phase 3 studies of EGFR-TKIs are ongoing.
The current trial was sponsored by the Guangdong Association of Clinical Trials. Dr. Wu disclosed relationships with AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb/China, Lilly, MSD Oncology, Pfizer, and Roche. Dr. Azzoli reported having no disclosures.
SOURCE: Wu Y et al. ASCO 2020, Abstract 9005.
FROM ASCO 2020
Preliminary evidence indicates famotidine might improve COVID-19 symptoms
High-dose oral famotidine might improve cardinal symptoms of COVID-19 infection, according to the findings of a small outpatient case series and a subsequent retrospective study.
After developing COVID-19 symptoms, the 10 patients in the case series began self-medicating with 60-240 mg famotidine daily over a median of 11 days. “All patients reported marked improvements of disease-related symptoms after starting famotidine,” first author Tobias Janowitz, MD, PhD, of Cold Spring Harbor Laboratory, N.Y., and associates wrote in Gut.
Improvements began within 24-48 hours of starting on the histamine-2 receptor antagonist. By 14 days after treatment initiation, all patients reported near-normalization of both respiratory and systemic symptoms, the researchers reported.
The patients were 23-71 years old. Seven tested positive for COVID-19, two had antibodies to COVID-19, and one had a clinical diagnosis of COVID-19 without laboratory confirmation. Over a median of 11 days (range, 5-21 days), six patients self-administered 80 mg famotidine three times daily and four self-administered lower amounts – from 60 to 150 mg of famotidine daily, divided into two or three doses. Patients started on famotidine between 2 and 26 days after symptom onset.
Through phone interviews and questionnaires, the researchers ascertained changes in cough, dyspnea, fatigue, headache, anosmia, and general unwellness by using a modified four-point Eastern Cooperative Oncology Group (ECOG) performance status scale. Improvements were seen across all symptom categories, and respiratory symptoms improved faster than systemic symptoms. Apart from two cases of persistent anosmia, symptoms resolved completely within 14 days of starting famotidine.
Seven patients reported no side effects of famotidine; one reported grade 1 dizziness and infrequent perceptions of tachycardia; one reported grade 1 dizziness, dry skin, and insomnia; and one reported grade 1 gastrointestinal symptoms and temporary forgetfulness. “Other than forgetfulness, all of these side effects are listed in the prescription information for famotidine, and all side effects resolved on discontinuation of famotidine,” the investigators wrote.
While the findings are intriguing, Dr. Janowitz and associates cautioned against overinterpretation of them. Another expert agreed: “This is a preliminary study based on a hypothesized antiviral effect. It’s important to know that it doesn’t really prove it works,” said Amesh Adalja, MD, senior scholar at the Johns Hopkins University Center for Health Security, Baltimore, and a spokesperson for the Infectious Diseases Society of America, during an interview with MDedge.
These patients might have improved anyway, without self-administering famotidine, said Dr. Adalja, who was not involved in the study.
Furthermore, the mechanism by which famotidine might act on COVID-19 remains unclear. The drug “could have a viral target, for example, one of the viral proteases, or a host target, resulting, for example, in modulation of the immunological response to the virus,” Dr. Janowitz and associates wrote.
Dr. Adalja noted that many compounds show effects against COVID-19 that are not well understood. He called for randomized trials to evaluate the biological plausibility of famotidine use, and its potential efficacy.
“This is a cheap, over-the-counter drug, but no drug is without side effects,” he added. “We need to know whether it works.”
Based on the case series findings, researchers conducted another retrospective study of patients hospitalized with COVID-19 infection. Those who were incidentally taking famotidine before or at hospitalization had a significantly reduced risk of intubation or death, with a hazard ratio of 0.43 (Gastroenterology. 2020 May 22. doi: 10.1053/j.gastro.2020.05.053)
The National Institutes of Health provided partial support. The investigators reported having no conflicts of interest.
SOURCE: Janowitz T et al. Gut. 2020 Jun 4. doi: 10.1136/gutjnl-2020-321852.
High-dose oral famotidine might improve cardinal symptoms of COVID-19 infection, according to the findings of a small outpatient case series and a subsequent retrospective study.
After developing COVID-19 symptoms, the 10 patients in the case series began self-medicating with 60-240 mg famotidine daily over a median of 11 days. “All patients reported marked improvements of disease-related symptoms after starting famotidine,” first author Tobias Janowitz, MD, PhD, of Cold Spring Harbor Laboratory, N.Y., and associates wrote in Gut.
Improvements began within 24-48 hours of starting on the histamine-2 receptor antagonist. By 14 days after treatment initiation, all patients reported near-normalization of both respiratory and systemic symptoms, the researchers reported.
The patients were 23-71 years old. Seven tested positive for COVID-19, two had antibodies to COVID-19, and one had a clinical diagnosis of COVID-19 without laboratory confirmation. Over a median of 11 days (range, 5-21 days), six patients self-administered 80 mg famotidine three times daily and four self-administered lower amounts – from 60 to 150 mg of famotidine daily, divided into two or three doses. Patients started on famotidine between 2 and 26 days after symptom onset.
Through phone interviews and questionnaires, the researchers ascertained changes in cough, dyspnea, fatigue, headache, anosmia, and general unwellness by using a modified four-point Eastern Cooperative Oncology Group (ECOG) performance status scale. Improvements were seen across all symptom categories, and respiratory symptoms improved faster than systemic symptoms. Apart from two cases of persistent anosmia, symptoms resolved completely within 14 days of starting famotidine.
Seven patients reported no side effects of famotidine; one reported grade 1 dizziness and infrequent perceptions of tachycardia; one reported grade 1 dizziness, dry skin, and insomnia; and one reported grade 1 gastrointestinal symptoms and temporary forgetfulness. “Other than forgetfulness, all of these side effects are listed in the prescription information for famotidine, and all side effects resolved on discontinuation of famotidine,” the investigators wrote.
While the findings are intriguing, Dr. Janowitz and associates cautioned against overinterpretation of them. Another expert agreed: “This is a preliminary study based on a hypothesized antiviral effect. It’s important to know that it doesn’t really prove it works,” said Amesh Adalja, MD, senior scholar at the Johns Hopkins University Center for Health Security, Baltimore, and a spokesperson for the Infectious Diseases Society of America, during an interview with MDedge.
These patients might have improved anyway, without self-administering famotidine, said Dr. Adalja, who was not involved in the study.
Furthermore, the mechanism by which famotidine might act on COVID-19 remains unclear. The drug “could have a viral target, for example, one of the viral proteases, or a host target, resulting, for example, in modulation of the immunological response to the virus,” Dr. Janowitz and associates wrote.
Dr. Adalja noted that many compounds show effects against COVID-19 that are not well understood. He called for randomized trials to evaluate the biological plausibility of famotidine use, and its potential efficacy.
“This is a cheap, over-the-counter drug, but no drug is without side effects,” he added. “We need to know whether it works.”
Based on the case series findings, researchers conducted another retrospective study of patients hospitalized with COVID-19 infection. Those who were incidentally taking famotidine before or at hospitalization had a significantly reduced risk of intubation or death, with a hazard ratio of 0.43 (Gastroenterology. 2020 May 22. doi: 10.1053/j.gastro.2020.05.053)
The National Institutes of Health provided partial support. The investigators reported having no conflicts of interest.
SOURCE: Janowitz T et al. Gut. 2020 Jun 4. doi: 10.1136/gutjnl-2020-321852.
High-dose oral famotidine might improve cardinal symptoms of COVID-19 infection, according to the findings of a small outpatient case series and a subsequent retrospective study.
After developing COVID-19 symptoms, the 10 patients in the case series began self-medicating with 60-240 mg famotidine daily over a median of 11 days. “All patients reported marked improvements of disease-related symptoms after starting famotidine,” first author Tobias Janowitz, MD, PhD, of Cold Spring Harbor Laboratory, N.Y., and associates wrote in Gut.
Improvements began within 24-48 hours of starting on the histamine-2 receptor antagonist. By 14 days after treatment initiation, all patients reported near-normalization of both respiratory and systemic symptoms, the researchers reported.
The patients were 23-71 years old. Seven tested positive for COVID-19, two had antibodies to COVID-19, and one had a clinical diagnosis of COVID-19 without laboratory confirmation. Over a median of 11 days (range, 5-21 days), six patients self-administered 80 mg famotidine three times daily and four self-administered lower amounts – from 60 to 150 mg of famotidine daily, divided into two or three doses. Patients started on famotidine between 2 and 26 days after symptom onset.
Through phone interviews and questionnaires, the researchers ascertained changes in cough, dyspnea, fatigue, headache, anosmia, and general unwellness by using a modified four-point Eastern Cooperative Oncology Group (ECOG) performance status scale. Improvements were seen across all symptom categories, and respiratory symptoms improved faster than systemic symptoms. Apart from two cases of persistent anosmia, symptoms resolved completely within 14 days of starting famotidine.
Seven patients reported no side effects of famotidine; one reported grade 1 dizziness and infrequent perceptions of tachycardia; one reported grade 1 dizziness, dry skin, and insomnia; and one reported grade 1 gastrointestinal symptoms and temporary forgetfulness. “Other than forgetfulness, all of these side effects are listed in the prescription information for famotidine, and all side effects resolved on discontinuation of famotidine,” the investigators wrote.
While the findings are intriguing, Dr. Janowitz and associates cautioned against overinterpretation of them. Another expert agreed: “This is a preliminary study based on a hypothesized antiviral effect. It’s important to know that it doesn’t really prove it works,” said Amesh Adalja, MD, senior scholar at the Johns Hopkins University Center for Health Security, Baltimore, and a spokesperson for the Infectious Diseases Society of America, during an interview with MDedge.
These patients might have improved anyway, without self-administering famotidine, said Dr. Adalja, who was not involved in the study.
Furthermore, the mechanism by which famotidine might act on COVID-19 remains unclear. The drug “could have a viral target, for example, one of the viral proteases, or a host target, resulting, for example, in modulation of the immunological response to the virus,” Dr. Janowitz and associates wrote.
Dr. Adalja noted that many compounds show effects against COVID-19 that are not well understood. He called for randomized trials to evaluate the biological plausibility of famotidine use, and its potential efficacy.
“This is a cheap, over-the-counter drug, but no drug is without side effects,” he added. “We need to know whether it works.”
Based on the case series findings, researchers conducted another retrospective study of patients hospitalized with COVID-19 infection. Those who were incidentally taking famotidine before or at hospitalization had a significantly reduced risk of intubation or death, with a hazard ratio of 0.43 (Gastroenterology. 2020 May 22. doi: 10.1053/j.gastro.2020.05.053)
The National Institutes of Health provided partial support. The investigators reported having no conflicts of interest.
SOURCE: Janowitz T et al. Gut. 2020 Jun 4. doi: 10.1136/gutjnl-2020-321852.
FROM GUT
Analysis of Pharmacist Interventions Used to Resolve Safety Target of Polypharmacy (STOP) Drug Interactions
Statins are one of the most common medications dispensed in the US and are associated with clinically significant drug interactions.1,2 The most common adverse drug reaction (ADR) of statin drug interactions is muscle-related toxicities.2 Despite technology advances to alert clinicians to drug interactions, updated statin manufacturer labeling, and guideline recommendations, inappropriate prescribing and dispensing of statin drug interactions continues to occur in health care systems.2-10
The medical literature has demonstrated many opportunities for pharmacists to prevent and mitigate drug interactions. At the points of prescribing and dispensing, pharmacists can reduce the number of potential drug interactions for the patient.11-13 Pharmacists also have identified and resolved drug interactions through quality assurance review after dispensing to a patient.7,8
Regardless of the time point of an intervention, the most common method pharmacists used to resolve drug interactions was through recommendations to a prescriber. The recommendations were generated through academic detailing, clinical decision support algorithms, drug conversions, or the pharmacist’s expertise. Regardless of the method the pharmacist used, the prescriber had the final authority to accept or decline the recommendation.7,8,11-13 Although these interventions were effective, pharmacists could further streamline the process by autonomously resolving drug interactions. However, these types of interventions are not well described in the medical literature.
Background
The US Department of Veterans Affairs (VA) Veterans Integrated Service Network (VISN), established the Safety Target of Polypharmacy (STOP) report in 2015. At each facility in the network, the report identified patients who were dispensed medications known to have drug interactions. The interactions were chosen by the VISN, and the severity of the interactions was based on coding parameters within the VA computerized order entry system, which uses a severity score based on First Databank data. At the Harry S. Truman Memorial Veterans’ Hospital (Truman VA) in Columbia, Missouri, > 500 drug interactions were initially active on the STOP report. The most common drug interactions were statins with gemfibrozil and statins with niacin.14-18 The Truman VA Pharmacy Service was charged with resolving the interactions for the facility.
The Truman VA employs 3 Patient Aligned Care Team (PACT) Clinical Pharmacy Specialists (CPS) practicing within primary care clinics. PACT is the patientcentered medical home model used by the VA. PACT CPS are ambulatory care pharmacists who assist providers in managing diseases using a scope of practice. Having a scope of practice would have allowed the PACT CPS to manage drug interactions with independent prescribing authority. However, due to the high volume of STOP report interactions and limited PACT CPS resources, the Pharmacy Service needed to develop an efficient, patient-centered method to resolve them. The intervention also needed to allow pharmacists, both with and without a scope of practice, to address the interactions.
Methods
The Truman VA Pharmacy Service developed protocols, approved by the Pharmacy and Therapeutics (P&T) Committee, to manage the specific gemfibrozil-statin and niacinstatin interactions chosen for the VISN 15 STOP report (Figures 1 and 2). The protocols were designed to identify patients who did not have a clear indication for gemfibrozil or niacin, were likely to maintain triglycerides (TGs) < 500 mg/dL without these medications, and would not likely require close monitoring after discontinuation.19 The protocols allowed pharmacists to autonomously discontinue gemfibrozil or niacin if patients did not have a history of pancreatitis, TGs ≥ 400 mg/dL or a nonlipid indication for niacin (eg, pellagra) after establishing care at Truman VA. Additionally, both interacting medications had to be dispensed by the VA. When pharmacists discontinued a medication, it was documented in a note in the patient electronic health record. The prescriber was notified through the note and the patient received a notification letter. Follow-up laboratory monitoring was not required as part of the protocol.
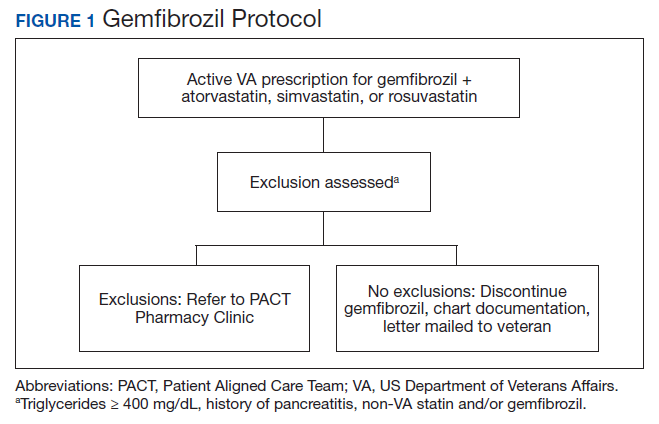
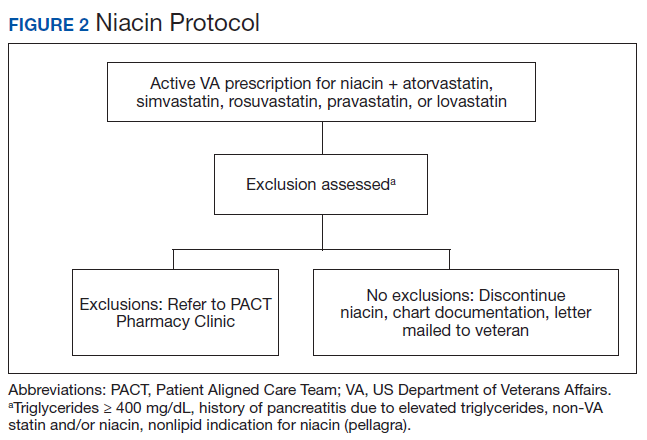
If patients met any of the exclusion criteria for discontinuation, the primary care provider (PCP) was notified to place a consult to the PACT Pharmacy Clinic for individualized interventions and close monitoring. Patients prescribed niacin for nonlipid indications were allowed to continue with their current drug regimen. At each encounter, the PACT CPS assessed for ADRs, made individualized medication changes, and arranged follow-up appointments. Once the interaction was resolved and treatment goals met, the PCP resumed monitoring of the patient’s lipid therapy.
Following all pharmacist interventions, a retrospective quality improvement analysis was conducted. The primary outcome was to evaluate the impact of discontinuing gemfibrozil and niacin by protocol on patients’ laboratory results. The coprimary endpoints were to describe the change in TG levels and the percentage of patients with TGs ≥ 500 mg/dL at least 5 weeks following the pharmacist-directed discontinuation by protocol. Secondary outcomes included the time required to resolve the interactions and a description of the PACT CPS pharmacologic interventions. Additionally, a quality assurance peer review was used to ensure the pharmacists appropriately utilized the protocols.
Data were collected from August 2016 to September 2017 for patients prescribed gemfibrozil and from May 2017 to January 2018 for patients prescribed niacin. The time spent resolving interactions was quantified based on encounter data. Descriptive statistics were used to analyze demographic information and the endpoints associated with each outcome. The project was reviewed by the University of Missouri Institutional Review Board, Truman VA privacy and information security officers, and was determined to meet guidelines for quality improvement.
Results
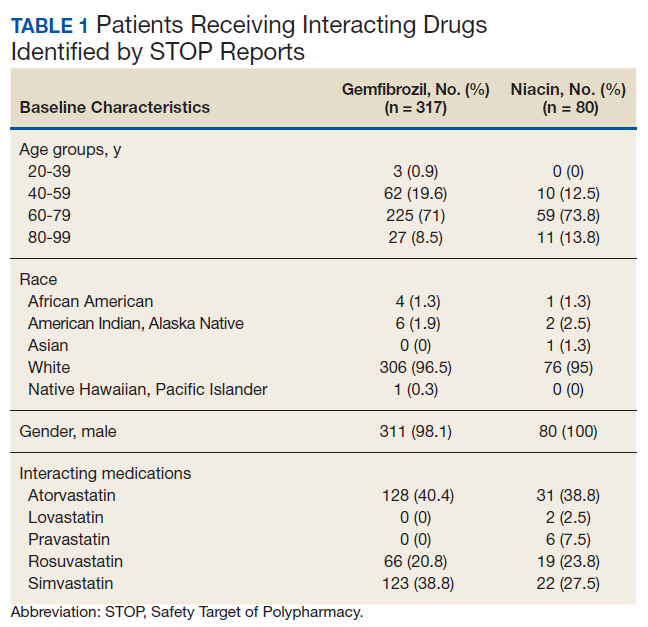
The original STOP report included 397 drug interactions involving statins with gemfibrozil or niacin (Table 1). The majority of patients were white and male aged 60 to 79 years. Gemfibrozil was the most common drug involved in all interactions (79.8%). The most common statins were atorvastatin (40%) and simvastatin (36.5%).
Gemfibrozil-Statin Interactions
Pharmacists discontinued gemfibrozil by protocol for 94 patients (29.6%), and 107 patients (33.8%) were referred to the PACT Pharmacy Clinic (Figure 3). For the remaining 116 patients (36.6%), the drug interaction was addressed outside of the protocol for the following reasons: the drug interaction was resolved prior to pharmacist review; an interacting prescription was expired and not to be continued; the patient self-discontinued ≥ 1 interacting medications; the patient was deceased; the patient moved; the patient was receiving ≥ 1 interacting medications outside of the VA; or the prescriber resolved the interaction following notification by the pharmacist.
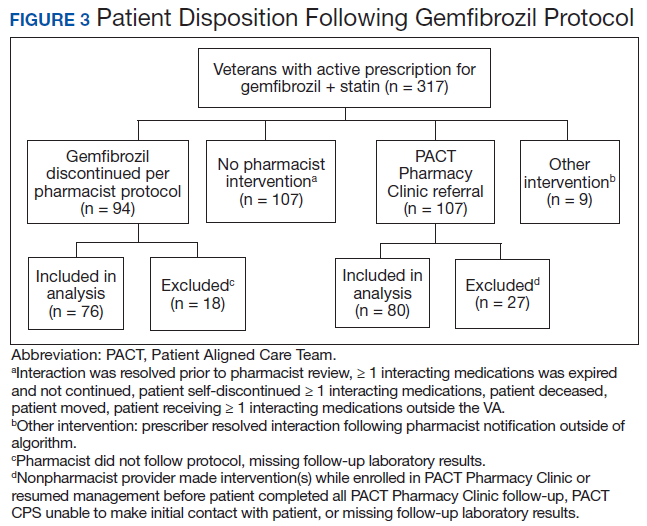
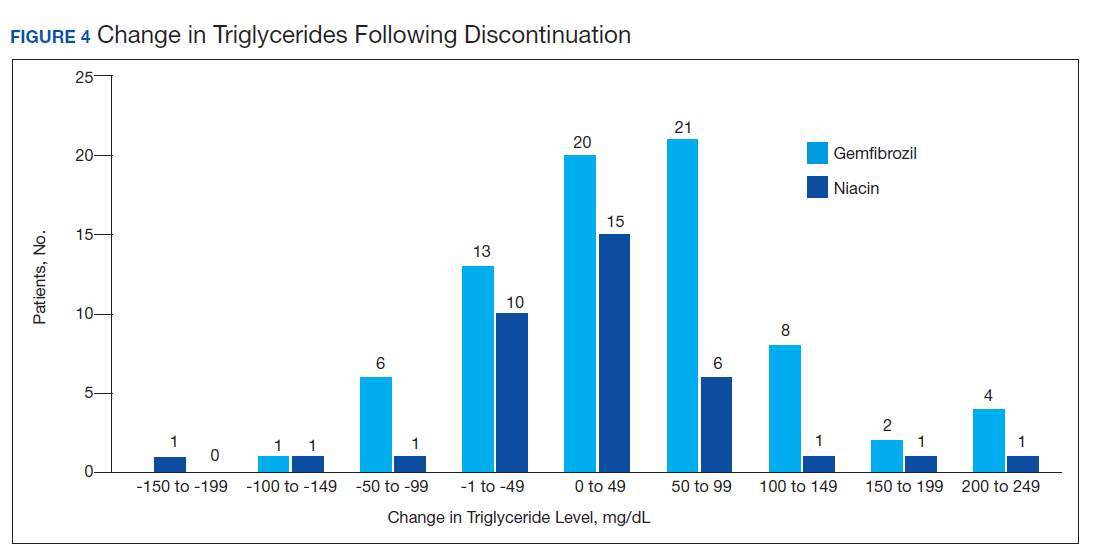
Ultimately, the interaction was resolved for all patients with a gemfibrozil-statin interaction on the STOP report. Following gemfibrozil discontinuation by protocol, 76 patients (80.9%) had TG laboratory results available and were included in the analysis. Sixty-two patients’ (82%) TG levels decreased or increased by < 100 mg/dL (Figure 4), and the TG levels of 1 patient (1.3%) increased above the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter was 6.5 (3.6) months (range, 1-17). The pharmacists spent a mean of 16 minutes per patient resolving each interaction.
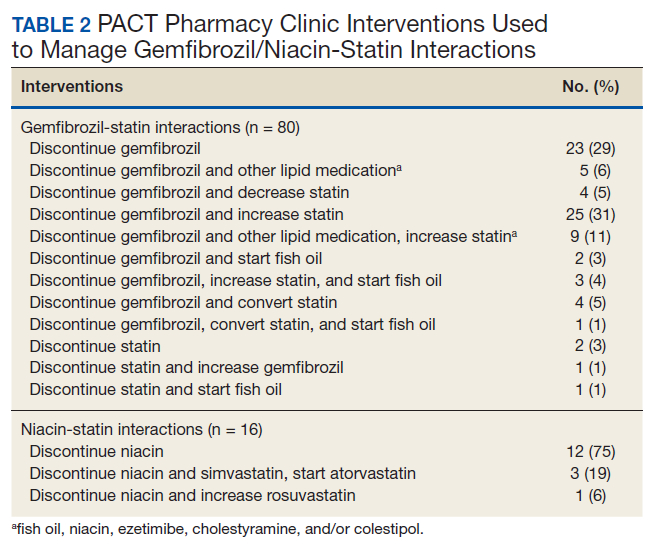
Of the 107 patients referred to the PACT Pharmacy Clinic, 80 (74.8%) had TG laboratory results available and were included in the analysis. These patients were followed by the PACT CPS until the drug interaction was resolved and confirmed to have TG levels at goal (< 500 mg/dL). Gemfibrozil doses ranged from 300 mg daily to 600 mg twice daily, with 70% (n = 56) of patients taking 600 mg twice daily. The PACT CPS made 148 interventions (Table 2). Twenty-three (29%) patients required only gemfibrozil discontinuation. The remaining 57 patients (71%) required at least 2 medication interventions. The PACT CPS generated 213 encounters for resolving drug interactions with a median of 2 encounters per patient.
Quality assurance review identified 5 patients (5.3%) who underwent gemfibrozil discontinuation by protocol, despite having criteria that would have recommended against discontinuation. In accordance with the protocol criteria, these patients were later referred to the PACT Pharmacy Clinic. None of these patients experienced a TG increase at or above the threshold of 500 mg/dL after gemfibrozil was initially discontinued but were excluded from the earlier analysis.
Niacin-Statin Interactions
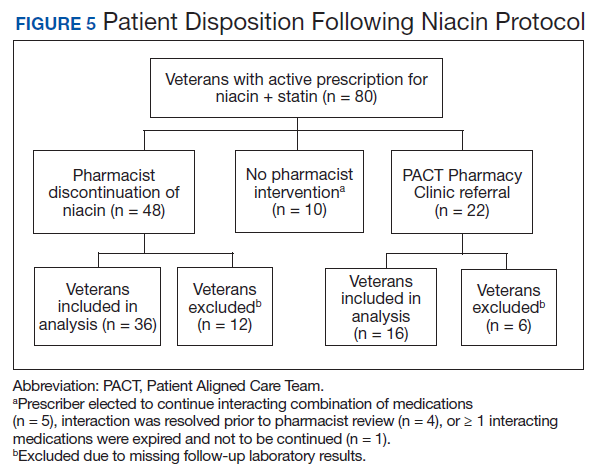
Pharmacists discontinued niacin by protocol for 48 patients (60.0%), and 22 patients (27.5%) were referred to the PACT Pharmacy Clinic (Figure 5). For the remaining 5 patients (6.3%), the interaction was either addressed outside the protocol prior to pharmacist review, or an interacting prescription was expired and not to be continued. Additionally, niacin was continued per prescriber preference in 5 patients (6.3%).
Thirty-six patients (75%) had TG laboratory results available following niacin discontinuation by protocol and were included in the analysis. Most patients’ (n = 33, 91.7%) TG levels decreased or increased by < 100 mg/dL. No patient had a TG level that increased higher than the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter, was 5.3 (2.5) months (range, 1.2-9.8). The pharmacists spent a mean of 15 minutes per patient resolving each interaction. The quality assurance review found no discrepancies in the pharmacists’ application of the protocol.
Of the 22 patients referred to the PACT Pharmacy Clinic, 16 (72.7%) patients had TG laboratory results available and were included in the analysis. As with the gemfibrozil interactions, these patients were followed by the PACT Pharmacy Clinic until the drug interaction was resolved and confirmed to have TGs at goal (< 500 mg/dL). Niacin doses ranged from 500 mg daily to 2,000 mg daily, with the majority of patients taking 1,000 mg daily. The PACT CPS made 23 interventions. The PACT CPS generated 46 encounters for resolving drug interactions with a median of 2 encounters per patient.
Discussion
Following gemfibrozil or niacin discontinuation by protocol, most patients with available laboratory results experienced either a decrease or modest TG elevation. The proportion of patients experiencing a decrease in TGs was unexpected but potentially multifactorial. Individual causes for the decrease in TGs were beyond the scope of this analysis. The retrospective design limited the ability to identify variables that could have impacted TG levels when gemfibrozil or niacin were started and discontinued. Although the treatment of TG levels is not indicated until it is ≥ 500 mg/dL, due to an increased risk of pancreatitis, both protocols excluded patients with a history of TGs ≥ 400 mg/dL.19 The lower threshold was set to compensate for anticipated increase in TG levels, following gemfibrozil or niacin discontinuation, and to minimize the number of patients with TG levels ≥ 500 mg/dL. The actual impact on patients’ TG levels supports the use of this lower threshold in the protocol.
When TG levels increased by 200 to 249 mg/dL after gemfibrozil or niacin discontinuation, patients were evaluated for possible underlying causes, which occurred for 4 gemfibrozil and 1 niacin patient. One patient started a β-blocker after gemfibrozil was initiated, and 3 patients were taking gemfibrozil prior to establishing care at the VA. The TG levels of the patient taking niacin correlated with an increased hemoglobin A1c. The TG level for only 1 patient taking gemfibrozil increased above the 500 mg/dL threshold. The patient had several comorbidities known to increase TG levels, but the comorbidities were previously well controlled. No additional medication changes were made at that time, and the TG levels on the next fasting lipid panel decreased to goal. The patient did not experience any negative clinical sequelae from the elevated TG levels.
Thirty-five patients (36%) who were referred to the PACT Pharmacy Clinic required only either gemfibrozil or niacin discontinuation. These patients were evaluated to identify whether adjustments to the protocols would have allowed for pharmacist discontinuation without referral to the PACT Pharmacy Clinic. Twenty-four of these patients (69%) had repeated TG levels ≥ 400 mg/dL prior to referral to the PACT Pharmacy Clinic. Additionally, there was no correlation between the gemfibrozil or niacin doses and the change in TG levels following discontinuation. These data indicate the protocols appropriately identified patients who did not have an indication for gemfibrozil or niacin.
In addition to drug interactions identified on the STOP report, the PACT CPS resolved 12 additional interactions involving simvastatin and gemfibrozil. Additionally, unnecessary lipid medications were deprescribed. The PACT CPS identified 13 patients who experienced myalgias, an ADR attributed to the gemfibrozil- statin interaction. Of those, 9 patients’ ADRs resolved after discontinuing gemfibrozil alone. For the remaining 4 patients, additional interventions to convert the patient to another statin were required to resolve the ADR.
Using pharmacists to address the drug interactions shifted workload from the prescribers and other primary care team members. The mean time spent to resolve both gemfibrozil and niacin interactions by protocol was 15.5 minutes. One hundred fortytwo patients (35.8%) had drug interactions resolved by protocol, saving the PACT CPS’ expertise for patients requiring individualized interventions. Drug interactions were resolved within 4 PACT CPS encounters for 93.8% of the patients taking gemfibrozil and within 3 PACT CPS encounters for 93.8% of the patients taking niacin.
The protocols allowed 12 additional pharmacists who did not have an ambulatory care scope of practice to assist the PACT CPS in mitigating the STOP drug interactions. These pharmacists otherwise would have been limited to making consultative recommendations. Simultaneously, the design allowed for the PACT pharmacists’ expertise to be allocated for patients most likely to require interventions beyond the protocols. This type of intraprofessional referral process is not well described in the medical literature. To the authors’ knowledge, the only studies described referrals from hospital pharmacists to community pharmacists during transitions of care on hospital discharge.20,21
Limitations
The results of this study are derived from a retrospective chart review at a single VA facility. The autonomous nature of PACT CPS interventions may be difficult to replicate in other settings that do not permit pharmacists the same prescriptive authority. This analysis was designed to demonstrate the impact of the pharmacist in resolving major drug interactions. Patients referred to the PACT Pharmacy Clinic who also had their lipid medications adjusted by a nonpharmacist provider were excluded. However, this may have minimized the impact of the PACT CPS on the patient care provided. As postintervention laboratory results were not available for all patients, some patients’ TG levels could have increased above the 500 mg/dL threshold but were not identified. The time investment was extensive and likely underestimates the true cost of implementing the interventions.
Because notification letters were used to instruct patients to stop gemfibrozil or niacin, several considerations need to be addressed when interpreting the follow-up laboratory results. First, we cannot confirm whether the patients received the letter or the exact date the letter was received. Additionally, we cannot confirm whether the patients followed the instructions to stop the interacting medications or the date the medications were stopped. It is possible some patients were still taking the interacting medication when the first laboratory was drawn. Should a patient have continued the interacting medication, most would have run out and been unable to obtain a refill within 90 days of receiving the letter, as this is the maximum amount dispensed at one time. The mean time to the first laboratory result for both gemfibrozil and niacin was 6.5 and 5.3 months, respectively. Approximately 85% of patients completed the first laboratory test at least 3 months after the letter was mailed.
The protocols were designed to assess whether gemfibrozil or niacin was indicated and did not assess whether the statin was indicated. Therefore, discontinuing the statin also could have resolved the interaction appropriately. However, due to characteristics of the patient population and recommendations in current lipid guidelines, it was more likely the statin would be indicated.22,23 The protocols also assumed that patients eligible for gemfibrozil or niacin discontinuation would not need additional changes to their lipid medications. The medication changes made by the PACT CPS may have gone beyond those minimally necessary to resolve the drug interaction and maintain TG goals. Patients who had gemfibrozil or niacin discontinued by protocol also may have benefited from additional optimization of their lipid medications.
Conclusions
This quality improvement analysis supports further evaluation of the complementary use of protocols and PACT CPS prescriptive authority to resolve statin drug interactions. The gemfibrozil and niacin protocols appropriately identified patients who were less likely to experience an adverse change in TG laboratory results. Patients more likely to require additional medication interventions were appropriately referred to the PACT Pharmacy Clinics for individualized care. These data support expanded roles for pharmacists, across various settings, to mitigate select drug interactions at the Truman VA.
Acknowledgments
This quality improvement project is the result of work supported with resources and use of the Harry S. Truman Memorial Veterans’ Hospital in Columbia, Missouri.
1. The top 200 drugs of 2020 Provided by the ClinCalc DrugStats Database. http://clincalc.com/DrugStats /Top200Drugs.aspx. Updated February 11, 2017. Accessed May 12, 2020.
2. Wiggins BS, Saseen JJ, Page RL 2nd, et al; American Heart Association Clinical Pharmacology Committee of the Council on Clinical Cardiology; Council on Hypertension; Council on Quality of Care and Outcomes Research; and Council on Functional Genomics and Translational Biology. Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2016;134(21):e468‐e495. doi:10.1161/CIR.0000000000000456
3. Smithburger PL, Buckley MS, Bejian S, Burenheide K, Kane-Gill SL. A critical evaluation of clinical decision support for the detection of drug-drug interactions. Expert Opin Drug Saf. 2011;10(6):871‐882. doi:10.1517/14740338.2011.583916
4. US Food and Drug Administration. FDA drug safety communication: new restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. https://www.fda.gov/Drugs/DrugSafety /ucm256581.htm. Updated December 15, 2017. Accessed May 12, 2020.
5. US Food and Drug Administration. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. https://www.fda.gov /Drugs/DrugSafety/ucm293101.htm. Updated January 19, 2016. Accessed May 12, 2020.
6. US Food and Drug Administration Federal Register. AbbVie Inc. et al; withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://www.federalregister .gov/documents/2016/04/18/2016-08887/abbvie-inc -et-al-withdrawal-of-approval-of-indications-related -to-the-coadministration-with-statins. Published April 18, 2016. Accessed May 12, 2020.
7. Lamprecht DG Jr, Todd BA, Denham AM, Ruppe LK, Stadler SL. Clinical pharmacist patient-safety initiative to reduce against-label prescribing of statins with cyclosporine. Ann Pharmacother. 2017;51(2):140‐145. doi:10.1177/1060028016675352
8. Roblek T, Deticek A, Leskovar B, et al. Clinical-pharmacist intervention reduces clinically relevant drugdrug interactions in patients with heart failure: A randomized, double-blind, controlled trial. Int J Cardiol. 2016;203:647‐652. doi:10.1016/j.ijcard.2015.10.206
9. Tuchscherer RM, Nair K, Ghushchyan V, Saseen JJ. Simvastatin prescribing patterns before and after FDA dosing restrictions: a retrospective analysis of a large healthcare claims database. Am J Cardiovasc Drugs. 2015;15(1):27‐34. doi:10.1007/s40256-014-0096-x
10. Alford JC, Saseen JJ, Allen RR, Nair KV. Persistent use of against-label statin-fibrate combinations from 2003-2009 despite United States Food and Drug Administration dose restrictions. Pharmacotherapy. 2012;32(7):623‐630. doi:10.1002/j.1875-9114.2011.01090.x
11. Leape LL, Cullen DJ, Clapp MD, et al. Pharmacist participation on physician rounds and adverse drug events in the intensive care unit [published correction appears in JAMA 2000 Mar 8;283(10):1293]. JAMA. 1999;282(3):267‐270. doi:10.1001/jama.282.3.267
12. Kucukarslan SN, Peters M, Mlynarek M, Nafziger DA. Pharmacists on rounding teams reduce preventable adverse drug events in hospital general medicine units. Arch Intern Med. 2003;163(17):2014‐2018. doi:10.1001/archinte.163.17.2014
13. Humphries TL, Carroll N, Chester EA, Magid D, Rocho B. Evaluation of an electronic critical drug interaction program coupled with active pharmacist intervention. Ann Pharmacother. 2007;41(12):1979‐1985. doi:10.1345/aph.1K349
14. Zocor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2018.
15. Lipitor [package insert]. New York, NY: Pfizer; 2017.
16. Crestor [package insert]. Wilmington, DE: AstraZeneca; 2018.
17. Mevacor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2012.
18. Wolters Kluwer Health, Lexi-Drugs, Lexicomp. Pravastatin. www.online.lexi.com. [Source not verified.]
19. Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333. doi: 10.1161/CIR.0b013e3182160726
20. Ferguson J, Seston L, Ashcroft DM. Refer-to-pharmacy: a qualitative study exploring the implementation of an electronic transfer of care initiative to improve medicines optimisation following hospital discharge. BMC Health Serv Res. 2018;18(1):424. doi:10.1186/s12913-018-3262-z
21. Ensing HT, Koster ES, Dubero DJ, van Dooren AA, Bouvy ML. Collaboration between hospital and community pharmacists to address drug-related problems: the HomeCoMe-program. Res Social Adm Pharm. 2019;15(3):267‐278. doi:10.1016/j.sapharm.2018.05.001
22. US Department of Defense, US Department of Veterans Affairs. VA/DoD clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction guideline summary. https://www.healthquality.va.gov /guidelines/CD/lipids/LipidSumOptSinglePg31Aug15.pdf. Published 2014. Accessed May 14, 2020.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines [published correction appears in Circulation. 2014 Jun 24;129(25) (suppl 2):S46-48] [published correction appears in Circulation. 2015 Dec 22;132(25):e396]. Circulation. 2014;129(25)(suppl 2): S1‐S45. doi:10.1161/01.cir.0000437738.63853.7a
Statins are one of the most common medications dispensed in the US and are associated with clinically significant drug interactions.1,2 The most common adverse drug reaction (ADR) of statin drug interactions is muscle-related toxicities.2 Despite technology advances to alert clinicians to drug interactions, updated statin manufacturer labeling, and guideline recommendations, inappropriate prescribing and dispensing of statin drug interactions continues to occur in health care systems.2-10
The medical literature has demonstrated many opportunities for pharmacists to prevent and mitigate drug interactions. At the points of prescribing and dispensing, pharmacists can reduce the number of potential drug interactions for the patient.11-13 Pharmacists also have identified and resolved drug interactions through quality assurance review after dispensing to a patient.7,8
Regardless of the time point of an intervention, the most common method pharmacists used to resolve drug interactions was through recommendations to a prescriber. The recommendations were generated through academic detailing, clinical decision support algorithms, drug conversions, or the pharmacist’s expertise. Regardless of the method the pharmacist used, the prescriber had the final authority to accept or decline the recommendation.7,8,11-13 Although these interventions were effective, pharmacists could further streamline the process by autonomously resolving drug interactions. However, these types of interventions are not well described in the medical literature.
Background
The US Department of Veterans Affairs (VA) Veterans Integrated Service Network (VISN), established the Safety Target of Polypharmacy (STOP) report in 2015. At each facility in the network, the report identified patients who were dispensed medications known to have drug interactions. The interactions were chosen by the VISN, and the severity of the interactions was based on coding parameters within the VA computerized order entry system, which uses a severity score based on First Databank data. At the Harry S. Truman Memorial Veterans’ Hospital (Truman VA) in Columbia, Missouri, > 500 drug interactions were initially active on the STOP report. The most common drug interactions were statins with gemfibrozil and statins with niacin.14-18 The Truman VA Pharmacy Service was charged with resolving the interactions for the facility.
The Truman VA employs 3 Patient Aligned Care Team (PACT) Clinical Pharmacy Specialists (CPS) practicing within primary care clinics. PACT is the patientcentered medical home model used by the VA. PACT CPS are ambulatory care pharmacists who assist providers in managing diseases using a scope of practice. Having a scope of practice would have allowed the PACT CPS to manage drug interactions with independent prescribing authority. However, due to the high volume of STOP report interactions and limited PACT CPS resources, the Pharmacy Service needed to develop an efficient, patient-centered method to resolve them. The intervention also needed to allow pharmacists, both with and without a scope of practice, to address the interactions.
Methods
The Truman VA Pharmacy Service developed protocols, approved by the Pharmacy and Therapeutics (P&T) Committee, to manage the specific gemfibrozil-statin and niacinstatin interactions chosen for the VISN 15 STOP report (Figures 1 and 2). The protocols were designed to identify patients who did not have a clear indication for gemfibrozil or niacin, were likely to maintain triglycerides (TGs) < 500 mg/dL without these medications, and would not likely require close monitoring after discontinuation.19 The protocols allowed pharmacists to autonomously discontinue gemfibrozil or niacin if patients did not have a history of pancreatitis, TGs ≥ 400 mg/dL or a nonlipid indication for niacin (eg, pellagra) after establishing care at Truman VA. Additionally, both interacting medications had to be dispensed by the VA. When pharmacists discontinued a medication, it was documented in a note in the patient electronic health record. The prescriber was notified through the note and the patient received a notification letter. Follow-up laboratory monitoring was not required as part of the protocol.
If patients met any of the exclusion criteria for discontinuation, the primary care provider (PCP) was notified to place a consult to the PACT Pharmacy Clinic for individualized interventions and close monitoring. Patients prescribed niacin for nonlipid indications were allowed to continue with their current drug regimen. At each encounter, the PACT CPS assessed for ADRs, made individualized medication changes, and arranged follow-up appointments. Once the interaction was resolved and treatment goals met, the PCP resumed monitoring of the patient’s lipid therapy.
Following all pharmacist interventions, a retrospective quality improvement analysis was conducted. The primary outcome was to evaluate the impact of discontinuing gemfibrozil and niacin by protocol on patients’ laboratory results. The coprimary endpoints were to describe the change in TG levels and the percentage of patients with TGs ≥ 500 mg/dL at least 5 weeks following the pharmacist-directed discontinuation by protocol. Secondary outcomes included the time required to resolve the interactions and a description of the PACT CPS pharmacologic interventions. Additionally, a quality assurance peer review was used to ensure the pharmacists appropriately utilized the protocols.
Data were collected from August 2016 to September 2017 for patients prescribed gemfibrozil and from May 2017 to January 2018 for patients prescribed niacin. The time spent resolving interactions was quantified based on encounter data. Descriptive statistics were used to analyze demographic information and the endpoints associated with each outcome. The project was reviewed by the University of Missouri Institutional Review Board, Truman VA privacy and information security officers, and was determined to meet guidelines for quality improvement.
Results
The original STOP report included 397 drug interactions involving statins with gemfibrozil or niacin (Table 1). The majority of patients were white and male aged 60 to 79 years. Gemfibrozil was the most common drug involved in all interactions (79.8%). The most common statins were atorvastatin (40%) and simvastatin (36.5%).
Gemfibrozil-Statin Interactions
Pharmacists discontinued gemfibrozil by protocol for 94 patients (29.6%), and 107 patients (33.8%) were referred to the PACT Pharmacy Clinic (Figure 3). For the remaining 116 patients (36.6%), the drug interaction was addressed outside of the protocol for the following reasons: the drug interaction was resolved prior to pharmacist review; an interacting prescription was expired and not to be continued; the patient self-discontinued ≥ 1 interacting medications; the patient was deceased; the patient moved; the patient was receiving ≥ 1 interacting medications outside of the VA; or the prescriber resolved the interaction following notification by the pharmacist.
Ultimately, the interaction was resolved for all patients with a gemfibrozil-statin interaction on the STOP report. Following gemfibrozil discontinuation by protocol, 76 patients (80.9%) had TG laboratory results available and were included in the analysis. Sixty-two patients’ (82%) TG levels decreased or increased by < 100 mg/dL (Figure 4), and the TG levels of 1 patient (1.3%) increased above the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter was 6.5 (3.6) months (range, 1-17). The pharmacists spent a mean of 16 minutes per patient resolving each interaction.
Of the 107 patients referred to the PACT Pharmacy Clinic, 80 (74.8%) had TG laboratory results available and were included in the analysis. These patients were followed by the PACT CPS until the drug interaction was resolved and confirmed to have TG levels at goal (< 500 mg/dL). Gemfibrozil doses ranged from 300 mg daily to 600 mg twice daily, with 70% (n = 56) of patients taking 600 mg twice daily. The PACT CPS made 148 interventions (Table 2). Twenty-three (29%) patients required only gemfibrozil discontinuation. The remaining 57 patients (71%) required at least 2 medication interventions. The PACT CPS generated 213 encounters for resolving drug interactions with a median of 2 encounters per patient.
Quality assurance review identified 5 patients (5.3%) who underwent gemfibrozil discontinuation by protocol, despite having criteria that would have recommended against discontinuation. In accordance with the protocol criteria, these patients were later referred to the PACT Pharmacy Clinic. None of these patients experienced a TG increase at or above the threshold of 500 mg/dL after gemfibrozil was initially discontinued but were excluded from the earlier analysis.
Niacin-Statin Interactions
Pharmacists discontinued niacin by protocol for 48 patients (60.0%), and 22 patients (27.5%) were referred to the PACT Pharmacy Clinic (Figure 5). For the remaining 5 patients (6.3%), the interaction was either addressed outside the protocol prior to pharmacist review, or an interacting prescription was expired and not to be continued. Additionally, niacin was continued per prescriber preference in 5 patients (6.3%).
Thirty-six patients (75%) had TG laboratory results available following niacin discontinuation by protocol and were included in the analysis. Most patients’ (n = 33, 91.7%) TG levels decreased or increased by < 100 mg/dL. No patient had a TG level that increased higher than the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter, was 5.3 (2.5) months (range, 1.2-9.8). The pharmacists spent a mean of 15 minutes per patient resolving each interaction. The quality assurance review found no discrepancies in the pharmacists’ application of the protocol.
Of the 22 patients referred to the PACT Pharmacy Clinic, 16 (72.7%) patients had TG laboratory results available and were included in the analysis. As with the gemfibrozil interactions, these patients were followed by the PACT Pharmacy Clinic until the drug interaction was resolved and confirmed to have TGs at goal (< 500 mg/dL). Niacin doses ranged from 500 mg daily to 2,000 mg daily, with the majority of patients taking 1,000 mg daily. The PACT CPS made 23 interventions. The PACT CPS generated 46 encounters for resolving drug interactions with a median of 2 encounters per patient.
Discussion
Following gemfibrozil or niacin discontinuation by protocol, most patients with available laboratory results experienced either a decrease or modest TG elevation. The proportion of patients experiencing a decrease in TGs was unexpected but potentially multifactorial. Individual causes for the decrease in TGs were beyond the scope of this analysis. The retrospective design limited the ability to identify variables that could have impacted TG levels when gemfibrozil or niacin were started and discontinued. Although the treatment of TG levels is not indicated until it is ≥ 500 mg/dL, due to an increased risk of pancreatitis, both protocols excluded patients with a history of TGs ≥ 400 mg/dL.19 The lower threshold was set to compensate for anticipated increase in TG levels, following gemfibrozil or niacin discontinuation, and to minimize the number of patients with TG levels ≥ 500 mg/dL. The actual impact on patients’ TG levels supports the use of this lower threshold in the protocol.
When TG levels increased by 200 to 249 mg/dL after gemfibrozil or niacin discontinuation, patients were evaluated for possible underlying causes, which occurred for 4 gemfibrozil and 1 niacin patient. One patient started a β-blocker after gemfibrozil was initiated, and 3 patients were taking gemfibrozil prior to establishing care at the VA. The TG levels of the patient taking niacin correlated with an increased hemoglobin A1c. The TG level for only 1 patient taking gemfibrozil increased above the 500 mg/dL threshold. The patient had several comorbidities known to increase TG levels, but the comorbidities were previously well controlled. No additional medication changes were made at that time, and the TG levels on the next fasting lipid panel decreased to goal. The patient did not experience any negative clinical sequelae from the elevated TG levels.
Thirty-five patients (36%) who were referred to the PACT Pharmacy Clinic required only either gemfibrozil or niacin discontinuation. These patients were evaluated to identify whether adjustments to the protocols would have allowed for pharmacist discontinuation without referral to the PACT Pharmacy Clinic. Twenty-four of these patients (69%) had repeated TG levels ≥ 400 mg/dL prior to referral to the PACT Pharmacy Clinic. Additionally, there was no correlation between the gemfibrozil or niacin doses and the change in TG levels following discontinuation. These data indicate the protocols appropriately identified patients who did not have an indication for gemfibrozil or niacin.
In addition to drug interactions identified on the STOP report, the PACT CPS resolved 12 additional interactions involving simvastatin and gemfibrozil. Additionally, unnecessary lipid medications were deprescribed. The PACT CPS identified 13 patients who experienced myalgias, an ADR attributed to the gemfibrozil- statin interaction. Of those, 9 patients’ ADRs resolved after discontinuing gemfibrozil alone. For the remaining 4 patients, additional interventions to convert the patient to another statin were required to resolve the ADR.
Using pharmacists to address the drug interactions shifted workload from the prescribers and other primary care team members. The mean time spent to resolve both gemfibrozil and niacin interactions by protocol was 15.5 minutes. One hundred fortytwo patients (35.8%) had drug interactions resolved by protocol, saving the PACT CPS’ expertise for patients requiring individualized interventions. Drug interactions were resolved within 4 PACT CPS encounters for 93.8% of the patients taking gemfibrozil and within 3 PACT CPS encounters for 93.8% of the patients taking niacin.
The protocols allowed 12 additional pharmacists who did not have an ambulatory care scope of practice to assist the PACT CPS in mitigating the STOP drug interactions. These pharmacists otherwise would have been limited to making consultative recommendations. Simultaneously, the design allowed for the PACT pharmacists’ expertise to be allocated for patients most likely to require interventions beyond the protocols. This type of intraprofessional referral process is not well described in the medical literature. To the authors’ knowledge, the only studies described referrals from hospital pharmacists to community pharmacists during transitions of care on hospital discharge.20,21
Limitations
The results of this study are derived from a retrospective chart review at a single VA facility. The autonomous nature of PACT CPS interventions may be difficult to replicate in other settings that do not permit pharmacists the same prescriptive authority. This analysis was designed to demonstrate the impact of the pharmacist in resolving major drug interactions. Patients referred to the PACT Pharmacy Clinic who also had their lipid medications adjusted by a nonpharmacist provider were excluded. However, this may have minimized the impact of the PACT CPS on the patient care provided. As postintervention laboratory results were not available for all patients, some patients’ TG levels could have increased above the 500 mg/dL threshold but were not identified. The time investment was extensive and likely underestimates the true cost of implementing the interventions.
Because notification letters were used to instruct patients to stop gemfibrozil or niacin, several considerations need to be addressed when interpreting the follow-up laboratory results. First, we cannot confirm whether the patients received the letter or the exact date the letter was received. Additionally, we cannot confirm whether the patients followed the instructions to stop the interacting medications or the date the medications were stopped. It is possible some patients were still taking the interacting medication when the first laboratory was drawn. Should a patient have continued the interacting medication, most would have run out and been unable to obtain a refill within 90 days of receiving the letter, as this is the maximum amount dispensed at one time. The mean time to the first laboratory result for both gemfibrozil and niacin was 6.5 and 5.3 months, respectively. Approximately 85% of patients completed the first laboratory test at least 3 months after the letter was mailed.
The protocols were designed to assess whether gemfibrozil or niacin was indicated and did not assess whether the statin was indicated. Therefore, discontinuing the statin also could have resolved the interaction appropriately. However, due to characteristics of the patient population and recommendations in current lipid guidelines, it was more likely the statin would be indicated.22,23 The protocols also assumed that patients eligible for gemfibrozil or niacin discontinuation would not need additional changes to their lipid medications. The medication changes made by the PACT CPS may have gone beyond those minimally necessary to resolve the drug interaction and maintain TG goals. Patients who had gemfibrozil or niacin discontinued by protocol also may have benefited from additional optimization of their lipid medications.
Conclusions
This quality improvement analysis supports further evaluation of the complementary use of protocols and PACT CPS prescriptive authority to resolve statin drug interactions. The gemfibrozil and niacin protocols appropriately identified patients who were less likely to experience an adverse change in TG laboratory results. Patients more likely to require additional medication interventions were appropriately referred to the PACT Pharmacy Clinics for individualized care. These data support expanded roles for pharmacists, across various settings, to mitigate select drug interactions at the Truman VA.
Acknowledgments
This quality improvement project is the result of work supported with resources and use of the Harry S. Truman Memorial Veterans’ Hospital in Columbia, Missouri.
Statins are one of the most common medications dispensed in the US and are associated with clinically significant drug interactions.1,2 The most common adverse drug reaction (ADR) of statin drug interactions is muscle-related toxicities.2 Despite technology advances to alert clinicians to drug interactions, updated statin manufacturer labeling, and guideline recommendations, inappropriate prescribing and dispensing of statin drug interactions continues to occur in health care systems.2-10
The medical literature has demonstrated many opportunities for pharmacists to prevent and mitigate drug interactions. At the points of prescribing and dispensing, pharmacists can reduce the number of potential drug interactions for the patient.11-13 Pharmacists also have identified and resolved drug interactions through quality assurance review after dispensing to a patient.7,8
Regardless of the time point of an intervention, the most common method pharmacists used to resolve drug interactions was through recommendations to a prescriber. The recommendations were generated through academic detailing, clinical decision support algorithms, drug conversions, or the pharmacist’s expertise. Regardless of the method the pharmacist used, the prescriber had the final authority to accept or decline the recommendation.7,8,11-13 Although these interventions were effective, pharmacists could further streamline the process by autonomously resolving drug interactions. However, these types of interventions are not well described in the medical literature.
Background
The US Department of Veterans Affairs (VA) Veterans Integrated Service Network (VISN), established the Safety Target of Polypharmacy (STOP) report in 2015. At each facility in the network, the report identified patients who were dispensed medications known to have drug interactions. The interactions were chosen by the VISN, and the severity of the interactions was based on coding parameters within the VA computerized order entry system, which uses a severity score based on First Databank data. At the Harry S. Truman Memorial Veterans’ Hospital (Truman VA) in Columbia, Missouri, > 500 drug interactions were initially active on the STOP report. The most common drug interactions were statins with gemfibrozil and statins with niacin.14-18 The Truman VA Pharmacy Service was charged with resolving the interactions for the facility.
The Truman VA employs 3 Patient Aligned Care Team (PACT) Clinical Pharmacy Specialists (CPS) practicing within primary care clinics. PACT is the patientcentered medical home model used by the VA. PACT CPS are ambulatory care pharmacists who assist providers in managing diseases using a scope of practice. Having a scope of practice would have allowed the PACT CPS to manage drug interactions with independent prescribing authority. However, due to the high volume of STOP report interactions and limited PACT CPS resources, the Pharmacy Service needed to develop an efficient, patient-centered method to resolve them. The intervention also needed to allow pharmacists, both with and without a scope of practice, to address the interactions.
Methods
The Truman VA Pharmacy Service developed protocols, approved by the Pharmacy and Therapeutics (P&T) Committee, to manage the specific gemfibrozil-statin and niacinstatin interactions chosen for the VISN 15 STOP report (Figures 1 and 2). The protocols were designed to identify patients who did not have a clear indication for gemfibrozil or niacin, were likely to maintain triglycerides (TGs) < 500 mg/dL without these medications, and would not likely require close monitoring after discontinuation.19 The protocols allowed pharmacists to autonomously discontinue gemfibrozil or niacin if patients did not have a history of pancreatitis, TGs ≥ 400 mg/dL or a nonlipid indication for niacin (eg, pellagra) after establishing care at Truman VA. Additionally, both interacting medications had to be dispensed by the VA. When pharmacists discontinued a medication, it was documented in a note in the patient electronic health record. The prescriber was notified through the note and the patient received a notification letter. Follow-up laboratory monitoring was not required as part of the protocol.
If patients met any of the exclusion criteria for discontinuation, the primary care provider (PCP) was notified to place a consult to the PACT Pharmacy Clinic for individualized interventions and close monitoring. Patients prescribed niacin for nonlipid indications were allowed to continue with their current drug regimen. At each encounter, the PACT CPS assessed for ADRs, made individualized medication changes, and arranged follow-up appointments. Once the interaction was resolved and treatment goals met, the PCP resumed monitoring of the patient’s lipid therapy.
Following all pharmacist interventions, a retrospective quality improvement analysis was conducted. The primary outcome was to evaluate the impact of discontinuing gemfibrozil and niacin by protocol on patients’ laboratory results. The coprimary endpoints were to describe the change in TG levels and the percentage of patients with TGs ≥ 500 mg/dL at least 5 weeks following the pharmacist-directed discontinuation by protocol. Secondary outcomes included the time required to resolve the interactions and a description of the PACT CPS pharmacologic interventions. Additionally, a quality assurance peer review was used to ensure the pharmacists appropriately utilized the protocols.
Data were collected from August 2016 to September 2017 for patients prescribed gemfibrozil and from May 2017 to January 2018 for patients prescribed niacin. The time spent resolving interactions was quantified based on encounter data. Descriptive statistics were used to analyze demographic information and the endpoints associated with each outcome. The project was reviewed by the University of Missouri Institutional Review Board, Truman VA privacy and information security officers, and was determined to meet guidelines for quality improvement.
Results
The original STOP report included 397 drug interactions involving statins with gemfibrozil or niacin (Table 1). The majority of patients were white and male aged 60 to 79 years. Gemfibrozil was the most common drug involved in all interactions (79.8%). The most common statins were atorvastatin (40%) and simvastatin (36.5%).
Gemfibrozil-Statin Interactions
Pharmacists discontinued gemfibrozil by protocol for 94 patients (29.6%), and 107 patients (33.8%) were referred to the PACT Pharmacy Clinic (Figure 3). For the remaining 116 patients (36.6%), the drug interaction was addressed outside of the protocol for the following reasons: the drug interaction was resolved prior to pharmacist review; an interacting prescription was expired and not to be continued; the patient self-discontinued ≥ 1 interacting medications; the patient was deceased; the patient moved; the patient was receiving ≥ 1 interacting medications outside of the VA; or the prescriber resolved the interaction following notification by the pharmacist.
Ultimately, the interaction was resolved for all patients with a gemfibrozil-statin interaction on the STOP report. Following gemfibrozil discontinuation by protocol, 76 patients (80.9%) had TG laboratory results available and were included in the analysis. Sixty-two patients’ (82%) TG levels decreased or increased by < 100 mg/dL (Figure 4), and the TG levels of 1 patient (1.3%) increased above the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter was 6.5 (3.6) months (range, 1-17). The pharmacists spent a mean of 16 minutes per patient resolving each interaction.
Of the 107 patients referred to the PACT Pharmacy Clinic, 80 (74.8%) had TG laboratory results available and were included in the analysis. These patients were followed by the PACT CPS until the drug interaction was resolved and confirmed to have TG levels at goal (< 500 mg/dL). Gemfibrozil doses ranged from 300 mg daily to 600 mg twice daily, with 70% (n = 56) of patients taking 600 mg twice daily. The PACT CPS made 148 interventions (Table 2). Twenty-three (29%) patients required only gemfibrozil discontinuation. The remaining 57 patients (71%) required at least 2 medication interventions. The PACT CPS generated 213 encounters for resolving drug interactions with a median of 2 encounters per patient.
Quality assurance review identified 5 patients (5.3%) who underwent gemfibrozil discontinuation by protocol, despite having criteria that would have recommended against discontinuation. In accordance with the protocol criteria, these patients were later referred to the PACT Pharmacy Clinic. None of these patients experienced a TG increase at or above the threshold of 500 mg/dL after gemfibrozil was initially discontinued but were excluded from the earlier analysis.
Niacin-Statin Interactions
Pharmacists discontinued niacin by protocol for 48 patients (60.0%), and 22 patients (27.5%) were referred to the PACT Pharmacy Clinic (Figure 5). For the remaining 5 patients (6.3%), the interaction was either addressed outside the protocol prior to pharmacist review, or an interacting prescription was expired and not to be continued. Additionally, niacin was continued per prescriber preference in 5 patients (6.3%).
Thirty-six patients (75%) had TG laboratory results available following niacin discontinuation by protocol and were included in the analysis. Most patients’ (n = 33, 91.7%) TG levels decreased or increased by < 100 mg/dL. No patient had a TG level that increased higher than the threshold of 500 mg/dL. The mean (SD) time to the first laboratory result after the pharmacists mailed the notification letter, was 5.3 (2.5) months (range, 1.2-9.8). The pharmacists spent a mean of 15 minutes per patient resolving each interaction. The quality assurance review found no discrepancies in the pharmacists’ application of the protocol.
Of the 22 patients referred to the PACT Pharmacy Clinic, 16 (72.7%) patients had TG laboratory results available and were included in the analysis. As with the gemfibrozil interactions, these patients were followed by the PACT Pharmacy Clinic until the drug interaction was resolved and confirmed to have TGs at goal (< 500 mg/dL). Niacin doses ranged from 500 mg daily to 2,000 mg daily, with the majority of patients taking 1,000 mg daily. The PACT CPS made 23 interventions. The PACT CPS generated 46 encounters for resolving drug interactions with a median of 2 encounters per patient.
Discussion
Following gemfibrozil or niacin discontinuation by protocol, most patients with available laboratory results experienced either a decrease or modest TG elevation. The proportion of patients experiencing a decrease in TGs was unexpected but potentially multifactorial. Individual causes for the decrease in TGs were beyond the scope of this analysis. The retrospective design limited the ability to identify variables that could have impacted TG levels when gemfibrozil or niacin were started and discontinued. Although the treatment of TG levels is not indicated until it is ≥ 500 mg/dL, due to an increased risk of pancreatitis, both protocols excluded patients with a history of TGs ≥ 400 mg/dL.19 The lower threshold was set to compensate for anticipated increase in TG levels, following gemfibrozil or niacin discontinuation, and to minimize the number of patients with TG levels ≥ 500 mg/dL. The actual impact on patients’ TG levels supports the use of this lower threshold in the protocol.
When TG levels increased by 200 to 249 mg/dL after gemfibrozil or niacin discontinuation, patients were evaluated for possible underlying causes, which occurred for 4 gemfibrozil and 1 niacin patient. One patient started a β-blocker after gemfibrozil was initiated, and 3 patients were taking gemfibrozil prior to establishing care at the VA. The TG levels of the patient taking niacin correlated with an increased hemoglobin A1c. The TG level for only 1 patient taking gemfibrozil increased above the 500 mg/dL threshold. The patient had several comorbidities known to increase TG levels, but the comorbidities were previously well controlled. No additional medication changes were made at that time, and the TG levels on the next fasting lipid panel decreased to goal. The patient did not experience any negative clinical sequelae from the elevated TG levels.
Thirty-five patients (36%) who were referred to the PACT Pharmacy Clinic required only either gemfibrozil or niacin discontinuation. These patients were evaluated to identify whether adjustments to the protocols would have allowed for pharmacist discontinuation without referral to the PACT Pharmacy Clinic. Twenty-four of these patients (69%) had repeated TG levels ≥ 400 mg/dL prior to referral to the PACT Pharmacy Clinic. Additionally, there was no correlation between the gemfibrozil or niacin doses and the change in TG levels following discontinuation. These data indicate the protocols appropriately identified patients who did not have an indication for gemfibrozil or niacin.
In addition to drug interactions identified on the STOP report, the PACT CPS resolved 12 additional interactions involving simvastatin and gemfibrozil. Additionally, unnecessary lipid medications were deprescribed. The PACT CPS identified 13 patients who experienced myalgias, an ADR attributed to the gemfibrozil- statin interaction. Of those, 9 patients’ ADRs resolved after discontinuing gemfibrozil alone. For the remaining 4 patients, additional interventions to convert the patient to another statin were required to resolve the ADR.
Using pharmacists to address the drug interactions shifted workload from the prescribers and other primary care team members. The mean time spent to resolve both gemfibrozil and niacin interactions by protocol was 15.5 minutes. One hundred fortytwo patients (35.8%) had drug interactions resolved by protocol, saving the PACT CPS’ expertise for patients requiring individualized interventions. Drug interactions were resolved within 4 PACT CPS encounters for 93.8% of the patients taking gemfibrozil and within 3 PACT CPS encounters for 93.8% of the patients taking niacin.
The protocols allowed 12 additional pharmacists who did not have an ambulatory care scope of practice to assist the PACT CPS in mitigating the STOP drug interactions. These pharmacists otherwise would have been limited to making consultative recommendations. Simultaneously, the design allowed for the PACT pharmacists’ expertise to be allocated for patients most likely to require interventions beyond the protocols. This type of intraprofessional referral process is not well described in the medical literature. To the authors’ knowledge, the only studies described referrals from hospital pharmacists to community pharmacists during transitions of care on hospital discharge.20,21
Limitations
The results of this study are derived from a retrospective chart review at a single VA facility. The autonomous nature of PACT CPS interventions may be difficult to replicate in other settings that do not permit pharmacists the same prescriptive authority. This analysis was designed to demonstrate the impact of the pharmacist in resolving major drug interactions. Patients referred to the PACT Pharmacy Clinic who also had their lipid medications adjusted by a nonpharmacist provider were excluded. However, this may have minimized the impact of the PACT CPS on the patient care provided. As postintervention laboratory results were not available for all patients, some patients’ TG levels could have increased above the 500 mg/dL threshold but were not identified. The time investment was extensive and likely underestimates the true cost of implementing the interventions.
Because notification letters were used to instruct patients to stop gemfibrozil or niacin, several considerations need to be addressed when interpreting the follow-up laboratory results. First, we cannot confirm whether the patients received the letter or the exact date the letter was received. Additionally, we cannot confirm whether the patients followed the instructions to stop the interacting medications or the date the medications were stopped. It is possible some patients were still taking the interacting medication when the first laboratory was drawn. Should a patient have continued the interacting medication, most would have run out and been unable to obtain a refill within 90 days of receiving the letter, as this is the maximum amount dispensed at one time. The mean time to the first laboratory result for both gemfibrozil and niacin was 6.5 and 5.3 months, respectively. Approximately 85% of patients completed the first laboratory test at least 3 months after the letter was mailed.
The protocols were designed to assess whether gemfibrozil or niacin was indicated and did not assess whether the statin was indicated. Therefore, discontinuing the statin also could have resolved the interaction appropriately. However, due to characteristics of the patient population and recommendations in current lipid guidelines, it was more likely the statin would be indicated.22,23 The protocols also assumed that patients eligible for gemfibrozil or niacin discontinuation would not need additional changes to their lipid medications. The medication changes made by the PACT CPS may have gone beyond those minimally necessary to resolve the drug interaction and maintain TG goals. Patients who had gemfibrozil or niacin discontinued by protocol also may have benefited from additional optimization of their lipid medications.
Conclusions
This quality improvement analysis supports further evaluation of the complementary use of protocols and PACT CPS prescriptive authority to resolve statin drug interactions. The gemfibrozil and niacin protocols appropriately identified patients who were less likely to experience an adverse change in TG laboratory results. Patients more likely to require additional medication interventions were appropriately referred to the PACT Pharmacy Clinics for individualized care. These data support expanded roles for pharmacists, across various settings, to mitigate select drug interactions at the Truman VA.
Acknowledgments
This quality improvement project is the result of work supported with resources and use of the Harry S. Truman Memorial Veterans’ Hospital in Columbia, Missouri.
1. The top 200 drugs of 2020 Provided by the ClinCalc DrugStats Database. http://clincalc.com/DrugStats /Top200Drugs.aspx. Updated February 11, 2017. Accessed May 12, 2020.
2. Wiggins BS, Saseen JJ, Page RL 2nd, et al; American Heart Association Clinical Pharmacology Committee of the Council on Clinical Cardiology; Council on Hypertension; Council on Quality of Care and Outcomes Research; and Council on Functional Genomics and Translational Biology. Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2016;134(21):e468‐e495. doi:10.1161/CIR.0000000000000456
3. Smithburger PL, Buckley MS, Bejian S, Burenheide K, Kane-Gill SL. A critical evaluation of clinical decision support for the detection of drug-drug interactions. Expert Opin Drug Saf. 2011;10(6):871‐882. doi:10.1517/14740338.2011.583916
4. US Food and Drug Administration. FDA drug safety communication: new restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. https://www.fda.gov/Drugs/DrugSafety /ucm256581.htm. Updated December 15, 2017. Accessed May 12, 2020.
5. US Food and Drug Administration. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. https://www.fda.gov /Drugs/DrugSafety/ucm293101.htm. Updated January 19, 2016. Accessed May 12, 2020.
6. US Food and Drug Administration Federal Register. AbbVie Inc. et al; withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://www.federalregister .gov/documents/2016/04/18/2016-08887/abbvie-inc -et-al-withdrawal-of-approval-of-indications-related -to-the-coadministration-with-statins. Published April 18, 2016. Accessed May 12, 2020.
7. Lamprecht DG Jr, Todd BA, Denham AM, Ruppe LK, Stadler SL. Clinical pharmacist patient-safety initiative to reduce against-label prescribing of statins with cyclosporine. Ann Pharmacother. 2017;51(2):140‐145. doi:10.1177/1060028016675352
8. Roblek T, Deticek A, Leskovar B, et al. Clinical-pharmacist intervention reduces clinically relevant drugdrug interactions in patients with heart failure: A randomized, double-blind, controlled trial. Int J Cardiol. 2016;203:647‐652. doi:10.1016/j.ijcard.2015.10.206
9. Tuchscherer RM, Nair K, Ghushchyan V, Saseen JJ. Simvastatin prescribing patterns before and after FDA dosing restrictions: a retrospective analysis of a large healthcare claims database. Am J Cardiovasc Drugs. 2015;15(1):27‐34. doi:10.1007/s40256-014-0096-x
10. Alford JC, Saseen JJ, Allen RR, Nair KV. Persistent use of against-label statin-fibrate combinations from 2003-2009 despite United States Food and Drug Administration dose restrictions. Pharmacotherapy. 2012;32(7):623‐630. doi:10.1002/j.1875-9114.2011.01090.x
11. Leape LL, Cullen DJ, Clapp MD, et al. Pharmacist participation on physician rounds and adverse drug events in the intensive care unit [published correction appears in JAMA 2000 Mar 8;283(10):1293]. JAMA. 1999;282(3):267‐270. doi:10.1001/jama.282.3.267
12. Kucukarslan SN, Peters M, Mlynarek M, Nafziger DA. Pharmacists on rounding teams reduce preventable adverse drug events in hospital general medicine units. Arch Intern Med. 2003;163(17):2014‐2018. doi:10.1001/archinte.163.17.2014
13. Humphries TL, Carroll N, Chester EA, Magid D, Rocho B. Evaluation of an electronic critical drug interaction program coupled with active pharmacist intervention. Ann Pharmacother. 2007;41(12):1979‐1985. doi:10.1345/aph.1K349
14. Zocor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2018.
15. Lipitor [package insert]. New York, NY: Pfizer; 2017.
16. Crestor [package insert]. Wilmington, DE: AstraZeneca; 2018.
17. Mevacor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2012.
18. Wolters Kluwer Health, Lexi-Drugs, Lexicomp. Pravastatin. www.online.lexi.com. [Source not verified.]
19. Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333. doi: 10.1161/CIR.0b013e3182160726
20. Ferguson J, Seston L, Ashcroft DM. Refer-to-pharmacy: a qualitative study exploring the implementation of an electronic transfer of care initiative to improve medicines optimisation following hospital discharge. BMC Health Serv Res. 2018;18(1):424. doi:10.1186/s12913-018-3262-z
21. Ensing HT, Koster ES, Dubero DJ, van Dooren AA, Bouvy ML. Collaboration between hospital and community pharmacists to address drug-related problems: the HomeCoMe-program. Res Social Adm Pharm. 2019;15(3):267‐278. doi:10.1016/j.sapharm.2018.05.001
22. US Department of Defense, US Department of Veterans Affairs. VA/DoD clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction guideline summary. https://www.healthquality.va.gov /guidelines/CD/lipids/LipidSumOptSinglePg31Aug15.pdf. Published 2014. Accessed May 14, 2020.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines [published correction appears in Circulation. 2014 Jun 24;129(25) (suppl 2):S46-48] [published correction appears in Circulation. 2015 Dec 22;132(25):e396]. Circulation. 2014;129(25)(suppl 2): S1‐S45. doi:10.1161/01.cir.0000437738.63853.7a
1. The top 200 drugs of 2020 Provided by the ClinCalc DrugStats Database. http://clincalc.com/DrugStats /Top200Drugs.aspx. Updated February 11, 2017. Accessed May 12, 2020.
2. Wiggins BS, Saseen JJ, Page RL 2nd, et al; American Heart Association Clinical Pharmacology Committee of the Council on Clinical Cardiology; Council on Hypertension; Council on Quality of Care and Outcomes Research; and Council on Functional Genomics and Translational Biology. Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2016;134(21):e468‐e495. doi:10.1161/CIR.0000000000000456
3. Smithburger PL, Buckley MS, Bejian S, Burenheide K, Kane-Gill SL. A critical evaluation of clinical decision support for the detection of drug-drug interactions. Expert Opin Drug Saf. 2011;10(6):871‐882. doi:10.1517/14740338.2011.583916
4. US Food and Drug Administration. FDA drug safety communication: new restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. https://www.fda.gov/Drugs/DrugSafety /ucm256581.htm. Updated December 15, 2017. Accessed May 12, 2020.
5. US Food and Drug Administration. FDA drug safety communication: important safety label changes to cholesterol-lowering statin drugs. https://www.fda.gov /Drugs/DrugSafety/ucm293101.htm. Updated January 19, 2016. Accessed May 12, 2020.
6. US Food and Drug Administration Federal Register. AbbVie Inc. et al; withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://www.federalregister .gov/documents/2016/04/18/2016-08887/abbvie-inc -et-al-withdrawal-of-approval-of-indications-related -to-the-coadministration-with-statins. Published April 18, 2016. Accessed May 12, 2020.
7. Lamprecht DG Jr, Todd BA, Denham AM, Ruppe LK, Stadler SL. Clinical pharmacist patient-safety initiative to reduce against-label prescribing of statins with cyclosporine. Ann Pharmacother. 2017;51(2):140‐145. doi:10.1177/1060028016675352
8. Roblek T, Deticek A, Leskovar B, et al. Clinical-pharmacist intervention reduces clinically relevant drugdrug interactions in patients with heart failure: A randomized, double-blind, controlled trial. Int J Cardiol. 2016;203:647‐652. doi:10.1016/j.ijcard.2015.10.206
9. Tuchscherer RM, Nair K, Ghushchyan V, Saseen JJ. Simvastatin prescribing patterns before and after FDA dosing restrictions: a retrospective analysis of a large healthcare claims database. Am J Cardiovasc Drugs. 2015;15(1):27‐34. doi:10.1007/s40256-014-0096-x
10. Alford JC, Saseen JJ, Allen RR, Nair KV. Persistent use of against-label statin-fibrate combinations from 2003-2009 despite United States Food and Drug Administration dose restrictions. Pharmacotherapy. 2012;32(7):623‐630. doi:10.1002/j.1875-9114.2011.01090.x
11. Leape LL, Cullen DJ, Clapp MD, et al. Pharmacist participation on physician rounds and adverse drug events in the intensive care unit [published correction appears in JAMA 2000 Mar 8;283(10):1293]. JAMA. 1999;282(3):267‐270. doi:10.1001/jama.282.3.267
12. Kucukarslan SN, Peters M, Mlynarek M, Nafziger DA. Pharmacists on rounding teams reduce preventable adverse drug events in hospital general medicine units. Arch Intern Med. 2003;163(17):2014‐2018. doi:10.1001/archinte.163.17.2014
13. Humphries TL, Carroll N, Chester EA, Magid D, Rocho B. Evaluation of an electronic critical drug interaction program coupled with active pharmacist intervention. Ann Pharmacother. 2007;41(12):1979‐1985. doi:10.1345/aph.1K349
14. Zocor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2018.
15. Lipitor [package insert]. New York, NY: Pfizer; 2017.
16. Crestor [package insert]. Wilmington, DE: AstraZeneca; 2018.
17. Mevacor [package insert]. Whitehouse Station, NJ: Merck & Co, Inc; 2012.
18. Wolters Kluwer Health, Lexi-Drugs, Lexicomp. Pravastatin. www.online.lexi.com. [Source not verified.]
19. Miller M, Stone NJ, Ballantyne C, et al; American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333. doi: 10.1161/CIR.0b013e3182160726
20. Ferguson J, Seston L, Ashcroft DM. Refer-to-pharmacy: a qualitative study exploring the implementation of an electronic transfer of care initiative to improve medicines optimisation following hospital discharge. BMC Health Serv Res. 2018;18(1):424. doi:10.1186/s12913-018-3262-z
21. Ensing HT, Koster ES, Dubero DJ, van Dooren AA, Bouvy ML. Collaboration between hospital and community pharmacists to address drug-related problems: the HomeCoMe-program. Res Social Adm Pharm. 2019;15(3):267‐278. doi:10.1016/j.sapharm.2018.05.001
22. US Department of Defense, US Department of Veterans Affairs. VA/DoD clinical practice guideline for the management of dyslipidemia for cardiovascular risk reduction guideline summary. https://www.healthquality.va.gov /guidelines/CD/lipids/LipidSumOptSinglePg31Aug15.pdf. Published 2014. Accessed May 14, 2020.
23. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines [published correction appears in Circulation. 2014 Jun 24;129(25) (suppl 2):S46-48] [published correction appears in Circulation. 2015 Dec 22;132(25):e396]. Circulation. 2014;129(25)(suppl 2): S1‐S45. doi:10.1161/01.cir.0000437738.63853.7a
Biologics may carry melanoma risk for patients with immune-mediated inflammatory diseases
The in a systematic review and meta-analysis published in JAMA Dermatology.
The studies included in the analysis, however, had limitations, including a lack of those comparing biologic and conventional systemic therapy in psoriasis and inflammatory bowel disease (IBD), according to Shamarke Esse, MRes, of the division of musculoskeletal and dermatological sciences at the University of Manchester (England) and colleagues. “We advocate for more large, well-designed studies of this issue to be performed to help improve certainty” regarding this association, they wrote.
Previous studies that have found an increased risk of melanoma in patients on biologics for psoriasis, rheumatoid arthritis, and IBD have “typically used the general population as the comparator,” they noted. There is a large amount of evidence that has established short-term efficacy and safety of biologics, compared with conventional systemic treatments, but concerns about longer-term cancer risk associated with biologics remains a concern. Moreover, they added, “melanoma is a highly immunogenic skin cancer and therefore of concern to patients treated with TNFIs [tumor necrosis factor inhibitors] because melanoma risk increases with suppression of the immune system and TNF-alpha plays an important role in the immune surveillance of tumors.12,13
In their review, the researchers identified seven cohort studies from MEDLINE, Embase, and Cochrane Central Register of Controlled Trials (CENTRAL) databases published between January 1995 and February 2019 that evaluated melanoma risk in about 34,000 patients receiving biologics and 135,370 patients who had never been treated with biologics, and were receiving conventional systemic therapy for psoriasis, RA, or IBD. Of these, four studies were in patients with RA, two studies were in patients with IBD, and a single study was in patients with psoriasis. Six studies examined patients taking TNF inhibitors, but only one of six studies had information on specific TNF inhibitors (adalimumab, etanercept, and infliximab) in patients with RA. One study evaluated abatacept and rituximab in RA patients.
The researchers analyzed the pooled relative risk across all studies. Compared with patients who received conventional systemic therapy, there was a nonsignificant association with risk of melanoma in patients with psoriasis (hazard ratio, 1.57; 95% confidence interval, 0.61-4.09), RA (pooled relative risk, 1.20; 95% CI, 0.83-1.74), and IBD (pRR, 1.20; 95% CI, 0.60-2.40).
Among RA patients who received TNF inhibitors only, there was a slightly elevated nonsignificant risk of melanoma (pRR, 1.08; 95% CI, 0.81-1.43). Patients receiving rituximab had a pRR of 0.73 (95% CI, 0.38-1.39), and patients taking abatacept had a pRR of 1.43 (95% CI, 0.66-3.09), compared with RA patients receiving conventional systemic therapy. When excluding two major studies in the RA subgroup of patients in a sensitivity analysis, pooled risk estimates varied from 0.91 (95% CI, 0.69-1.18) to 1.95 (95% CI, 1.16- 3.30). There were no significant between-study heterogeneity or publication bias among the IBD and RA studies.
Mr. Esse and colleagues acknowledged the small number of IBD and psoriasis studies in the meta-analysis, which could affect pooled risk estimates. “Any future update of our study through the inclusion of newly published studies may produce significantly different pooled risk estimates than those reported in our meta-analysis,” they said. In addition, the use of health insurance databases, lack of risk factors for melanoma, and inconsistent information about treatment duration for patients receiving conventional systemic therapy were also limitations.
“Prospective cohort studies using an active comparator, new-user study design providing detailed information on treatment history, concomitant treatments, biologic and conventional systemic treatment duration, recreational and treatment-related UV exposure, skin color, and date of melanoma diagnosis are required to help improve certainty. These studies would also need to account for key risk factors and the latency period of melanoma,” the researchers said.
Mr. Esse disclosed being funded by a PhD studentship from the Psoriasis Association. One author disclosed receiving personal fees from Janssen, LEO Pharma, Lilly, and Novartis outside the study; another disclosed receiving grants and personal fees from those and several other pharmaceutical companies during the study, and personal fees from several pharmaceutical companies outside of the submitted work; the fourth author had no disclosures.
SOURCE: Esse S et al. JAMA Dermatol. 2020 May 20;e201300.
The in a systematic review and meta-analysis published in JAMA Dermatology.
The studies included in the analysis, however, had limitations, including a lack of those comparing biologic and conventional systemic therapy in psoriasis and inflammatory bowel disease (IBD), according to Shamarke Esse, MRes, of the division of musculoskeletal and dermatological sciences at the University of Manchester (England) and colleagues. “We advocate for more large, well-designed studies of this issue to be performed to help improve certainty” regarding this association, they wrote.
Previous studies that have found an increased risk of melanoma in patients on biologics for psoriasis, rheumatoid arthritis, and IBD have “typically used the general population as the comparator,” they noted. There is a large amount of evidence that has established short-term efficacy and safety of biologics, compared with conventional systemic treatments, but concerns about longer-term cancer risk associated with biologics remains a concern. Moreover, they added, “melanoma is a highly immunogenic skin cancer and therefore of concern to patients treated with TNFIs [tumor necrosis factor inhibitors] because melanoma risk increases with suppression of the immune system and TNF-alpha plays an important role in the immune surveillance of tumors.12,13
In their review, the researchers identified seven cohort studies from MEDLINE, Embase, and Cochrane Central Register of Controlled Trials (CENTRAL) databases published between January 1995 and February 2019 that evaluated melanoma risk in about 34,000 patients receiving biologics and 135,370 patients who had never been treated with biologics, and were receiving conventional systemic therapy for psoriasis, RA, or IBD. Of these, four studies were in patients with RA, two studies were in patients with IBD, and a single study was in patients with psoriasis. Six studies examined patients taking TNF inhibitors, but only one of six studies had information on specific TNF inhibitors (adalimumab, etanercept, and infliximab) in patients with RA. One study evaluated abatacept and rituximab in RA patients.
The researchers analyzed the pooled relative risk across all studies. Compared with patients who received conventional systemic therapy, there was a nonsignificant association with risk of melanoma in patients with psoriasis (hazard ratio, 1.57; 95% confidence interval, 0.61-4.09), RA (pooled relative risk, 1.20; 95% CI, 0.83-1.74), and IBD (pRR, 1.20; 95% CI, 0.60-2.40).
Among RA patients who received TNF inhibitors only, there was a slightly elevated nonsignificant risk of melanoma (pRR, 1.08; 95% CI, 0.81-1.43). Patients receiving rituximab had a pRR of 0.73 (95% CI, 0.38-1.39), and patients taking abatacept had a pRR of 1.43 (95% CI, 0.66-3.09), compared with RA patients receiving conventional systemic therapy. When excluding two major studies in the RA subgroup of patients in a sensitivity analysis, pooled risk estimates varied from 0.91 (95% CI, 0.69-1.18) to 1.95 (95% CI, 1.16- 3.30). There were no significant between-study heterogeneity or publication bias among the IBD and RA studies.
Mr. Esse and colleagues acknowledged the small number of IBD and psoriasis studies in the meta-analysis, which could affect pooled risk estimates. “Any future update of our study through the inclusion of newly published studies may produce significantly different pooled risk estimates than those reported in our meta-analysis,” they said. In addition, the use of health insurance databases, lack of risk factors for melanoma, and inconsistent information about treatment duration for patients receiving conventional systemic therapy were also limitations.
“Prospective cohort studies using an active comparator, new-user study design providing detailed information on treatment history, concomitant treatments, biologic and conventional systemic treatment duration, recreational and treatment-related UV exposure, skin color, and date of melanoma diagnosis are required to help improve certainty. These studies would also need to account for key risk factors and the latency period of melanoma,” the researchers said.
Mr. Esse disclosed being funded by a PhD studentship from the Psoriasis Association. One author disclosed receiving personal fees from Janssen, LEO Pharma, Lilly, and Novartis outside the study; another disclosed receiving grants and personal fees from those and several other pharmaceutical companies during the study, and personal fees from several pharmaceutical companies outside of the submitted work; the fourth author had no disclosures.
SOURCE: Esse S et al. JAMA Dermatol. 2020 May 20;e201300.
The in a systematic review and meta-analysis published in JAMA Dermatology.
The studies included in the analysis, however, had limitations, including a lack of those comparing biologic and conventional systemic therapy in psoriasis and inflammatory bowel disease (IBD), according to Shamarke Esse, MRes, of the division of musculoskeletal and dermatological sciences at the University of Manchester (England) and colleagues. “We advocate for more large, well-designed studies of this issue to be performed to help improve certainty” regarding this association, they wrote.
Previous studies that have found an increased risk of melanoma in patients on biologics for psoriasis, rheumatoid arthritis, and IBD have “typically used the general population as the comparator,” they noted. There is a large amount of evidence that has established short-term efficacy and safety of biologics, compared with conventional systemic treatments, but concerns about longer-term cancer risk associated with biologics remains a concern. Moreover, they added, “melanoma is a highly immunogenic skin cancer and therefore of concern to patients treated with TNFIs [tumor necrosis factor inhibitors] because melanoma risk increases with suppression of the immune system and TNF-alpha plays an important role in the immune surveillance of tumors.12,13
In their review, the researchers identified seven cohort studies from MEDLINE, Embase, and Cochrane Central Register of Controlled Trials (CENTRAL) databases published between January 1995 and February 2019 that evaluated melanoma risk in about 34,000 patients receiving biologics and 135,370 patients who had never been treated with biologics, and were receiving conventional systemic therapy for psoriasis, RA, or IBD. Of these, four studies were in patients with RA, two studies were in patients with IBD, and a single study was in patients with psoriasis. Six studies examined patients taking TNF inhibitors, but only one of six studies had information on specific TNF inhibitors (adalimumab, etanercept, and infliximab) in patients with RA. One study evaluated abatacept and rituximab in RA patients.
The researchers analyzed the pooled relative risk across all studies. Compared with patients who received conventional systemic therapy, there was a nonsignificant association with risk of melanoma in patients with psoriasis (hazard ratio, 1.57; 95% confidence interval, 0.61-4.09), RA (pooled relative risk, 1.20; 95% CI, 0.83-1.74), and IBD (pRR, 1.20; 95% CI, 0.60-2.40).
Among RA patients who received TNF inhibitors only, there was a slightly elevated nonsignificant risk of melanoma (pRR, 1.08; 95% CI, 0.81-1.43). Patients receiving rituximab had a pRR of 0.73 (95% CI, 0.38-1.39), and patients taking abatacept had a pRR of 1.43 (95% CI, 0.66-3.09), compared with RA patients receiving conventional systemic therapy. When excluding two major studies in the RA subgroup of patients in a sensitivity analysis, pooled risk estimates varied from 0.91 (95% CI, 0.69-1.18) to 1.95 (95% CI, 1.16- 3.30). There were no significant between-study heterogeneity or publication bias among the IBD and RA studies.
Mr. Esse and colleagues acknowledged the small number of IBD and psoriasis studies in the meta-analysis, which could affect pooled risk estimates. “Any future update of our study through the inclusion of newly published studies may produce significantly different pooled risk estimates than those reported in our meta-analysis,” they said. In addition, the use of health insurance databases, lack of risk factors for melanoma, and inconsistent information about treatment duration for patients receiving conventional systemic therapy were also limitations.
“Prospective cohort studies using an active comparator, new-user study design providing detailed information on treatment history, concomitant treatments, biologic and conventional systemic treatment duration, recreational and treatment-related UV exposure, skin color, and date of melanoma diagnosis are required to help improve certainty. These studies would also need to account for key risk factors and the latency period of melanoma,” the researchers said.
Mr. Esse disclosed being funded by a PhD studentship from the Psoriasis Association. One author disclosed receiving personal fees from Janssen, LEO Pharma, Lilly, and Novartis outside the study; another disclosed receiving grants and personal fees from those and several other pharmaceutical companies during the study, and personal fees from several pharmaceutical companies outside of the submitted work; the fourth author had no disclosures.
SOURCE: Esse S et al. JAMA Dermatol. 2020 May 20;e201300.
FROM JAMA DERMATOLOGY
Steroid-Induced Sleep Disturbance and Delirium: A Focused Review for Critically Ill Patients
Sleep disturbance in the critically ill has received much attention over recent years as this is a common result of intensive care unit (ICU) admission. Disruptions in sleep not only can, at a minimum, cause distress and lower patient satisfaction, but also inhibit recovery from illness and increase morbidity.1,2 Several studies have been conducted highlighting the altered sleep patterns of critically ill patients; although total sleep time may seem normal (7-9 hours), patients can experience multiple awakenings per hour, more time in light sleep (stages 1 and 2), and less time in restorative sleep (stages 3 and 4, [REM]rapid eye movement).2-5
There are several hypothesized physiologic detriments that contribute to slower ICU recovery with sleep deprivation. Research in noncritically ill subjects suggests that sleep deprivation contributes to hypoventilation and potentially prolonged time on the ventilator.6-9 Cardiovascular morbidity may be adversely affected by inflammatory cytokine release seen in sleep disruption.10,11 Studies of noncritically ill patients also suggest that immune response is impaired, potentially protracting infection recovery.12,13 Finally, although not directly investigated, sleep deprivation may contribute to ICU delirium, an independent adverse effect (AE) associated with increased mortality and worse long-term outcomes.14-16
The Society of Critical Care Medicine (SCCM) recently updated its consensus guidelines for the management of pain, agitation/sedation, delirium, immobility, and sleep disruption (PADIS) in adult patients.17 These guidelines offer limited interventions to promote sleep in ICU patients based on available evidence and steer the clinician toward minimizing exacerbating factors. Although factors that affect sleep patterns are multifactorial, such as noise levels, pain, mechanical ventilation, and inflammatory mediators, medication therapy is a known modifiable risk factor for sleep disturbance in critically ill patients.2 This focused review will specifically evaluate the effects of steroids on sleep deprivation, psychosis, delirium, and what is known about these effects in a critically ill population.
To include articles relevant to a critically ill population, a systematic search of MEDLINE and PubMed from 1966 to 2019 was performed using the following Medical Subject Headings (MeSH) terms: delirium/etiology, psychoses, substance-induced/etiology, sleep-wake disorders/chemically induced, neurocognitive disorders/chemically induced, dyssomnias/drug effects plus glucocorticoids/adverse effects, adrenal cortex hormones/adverse effects, prednisone/adverse effects, methylprednisolone/adverse effects, and hydrocortisone/adverse effects. The initial search produced 285 articles. Case reports, reviews, letters, and articles pertaining to primary care or palliative populations were excluded, leaving 8 relevant articles for inclusion (Table 1).18-25
ICU Steroid Use
Steroids are commonly used in the ICU and affect nearly every critically ill population. Common indications for steroids in the ICU include anaphylaxis, airway edema, septic shock, asthma and COPD exacerbations, pneumocystis pneumonia, adrenal crisis, antiemetic treatment, elevated intracranial pressure from tumors, autoimmune disorders, and stress doses needed for chronic steroid users before invasive procedures.26 Whether divided into glucocorticoid or mineralocorticoid subgroups, corticosteroids offer therapeutic benefit from their pharmacologic similarity to endogenously produced cortisol, which includes anti-inflammatory, immunosuppressive, antiproliferative, and vasoconstrictive effects.
Steroid receptors are present in most human tissue, and in varying degrees of binding affinity produce a wide variety of effects. After passive diffusion across cell membranes, steroid-receptor activation binds to various DNA sites, called glucocorticoid regulatory elements, which either stimulates or inhibits transcription of multiple nearby genes.
At the cellular level, corticosteroids inhibit the release of arachidonic acid through upstream production of lipocortin peptides and antagonism of phospholipase A2. This action decreases subsequent inflammatory mediators, including kinins, histamine, liposomal enzymes, and prostaglandins. Steroids also inhibit NF-κB, which further decreases expression of proinflammatory genes while promoting interleukin-10 and its anti-inflammatory properties. Antiproliferative effects of steroids are seen by triggering cell apoptosis and inhibition of fibroblast proliferation.27,28
By binding to mineralocorticoid receptors, steroids cause sodium retention coupled with hydrogen and potassium excretion in the distal renal tubule. Steroids also promote vasoconstriction by upregulating the production and sensitivity of β receptors in the endothelium while suppressing the production of vasodilators. Although rarely used for these physiologic effects, steroids also are involved in a number of metabolic pathways, including calcium regulation, gluconeogenesis, protein metabolism, and fat distribution. Given the similar structure to cortisol, exogenous steroids depress the hypothalamic-pituitary axis (HPA) and decrease the release of adrenocorticotropic hormone (ACTH). Tapering doses of steroid regimens is often required to allow natural androgen and cortisol synthesis and prevent steroid withdrawal.27,28
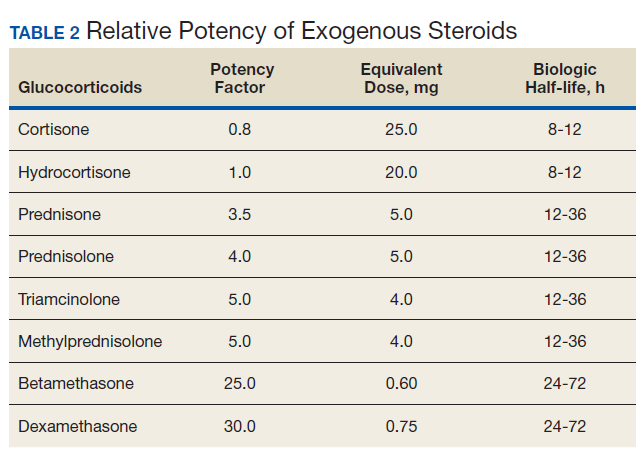

The potency of various exogenous steroids closely parallels their ability to retain sodium (Table 2). Prolonged activation of steroid receptors can have numerous systemic AEs, including unwanted neurocognitive effects (Table 3). Insomnia and psychosis are commonly described in corticosteroid clinical trials, and in one meta-analysis, both are associated with high costs per episode per year.29
Steroid-Induced Sleep Disruption and Psychosis
Sleep disruption caused by exogenous administration of steroids is thought to trigger other psychostimulant effects, such as mood swings, nervousness, psychoses, and delirium.30 Similarly, the SCCM PADIS guidelines included an ungraded statement: “although an association between sleep quality and delirium occurrence exists in critically ill adults, a cause-effect relationship has not been established.”17 For this review, these AEs will be discussed as related events.
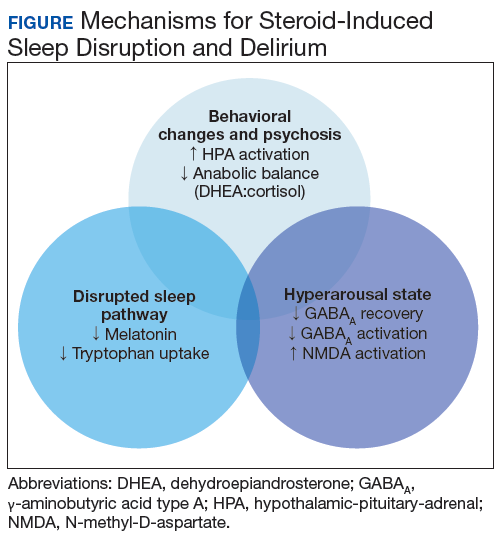
The medical literature proposes 3 pathways primarily responsible for neurocognitive AEs of steroids: behavior changes through modification of the HPA axis, changes in natural sleep-wake cycles, and hyperarousal caused by modification in neuroinhibitory pathways (Figure).
HPA Axis Modification
Under either physical or psychological stress, neural circuits in the brain release corticotropin-releasing hormone (CRH), dehydroepiandrosterone (DHEA), and arginine vasopressin, which go on to activate the sympathetic nervous system and the HPA axis. CRH from the hypothalamus goes on to stimulate ACTH release from the pituitary. ACTH then stimulates cortisol secretion from the adrenal glands. Circulating cortisol feeds into several structures of the brain, including the pituitary, hippocampus, and amygdala. Steroid-receptor complexes alter gene transcription in the central nervous system (CNS), affecting the production of neurotransmitters (eg, dopamine, serotonin) and neuropeptides (eg, somatostatin, β-endorphin). Feedback inhibition ensues, with downregulation of the HPA axis, which prevents depletion of endogenous production of steroids.31 DHEA has protective effects against excessive cortisol activity, but DHEA secretion declines with prolonged cortisol exposure. Exogenous steroids may have different effects than endogenous steroids, and neurocognitive sequelae stem from disruption and imbalance of these physiologic mechanisms.32,33
Steroid receptors are densely located in behavior centers in the brain: the amygdala, septum, and hippocampus. Pharmacologic changes in gene expression alter norepinephrine and serotonin levels in the brain as well as their receptors.32 Prolonged exposure to exogenous steroids has been shown to decrease amygdala and hippocampal volumes.34,35 Furthermore, prolonged corticosteroid exposure has been shown to decrease the number of steroid receptors in the hippocampus, pituitary gland, and amygdala.36 In a somewhat paradoxical finding, the production of CNS proinflammatory cytokines like interleuken-1β and tumor necrosis factor α has been seen after steroid administration, suggesting alternate gene signaling in the CNS.37 Although not proven conclusively, it is felt that these physiologic changes and hyperactivity of the HPA axis are predominantly responsible for changes in behavior, mood, memory, and eventually psychosis in steroid-treated patients.33,38
Finally, alterations in cognition and behavior may be related to steroid-induced changes in CNS carbohydrate, protein, and lipid metabolism with subsequent cellular neurotoxicity.32,38 Glucose uptake into the hippocampus is decreased with steroid exposure. Additionally, breakdown of metabolic compounds to produce energy can be destructive if left unchecked for prolonged periods. DHEA, growth hormone, and testosterone work to repair catabolic damage produced by cortisol, known as anabolic balance. A low anabolic balance (low DHEA levels to high cortisol levels) leads to a cascade of dysregulation in brain activity.39
Changes in Natural Sleep-Wake Cycles
Natural sleep pathways are also affected by steroids. The sleep-wake cycle is primarily regulated in the hypothalamus with circadian release of melatonin from the pineal gland. Melatonin release is highest at night, where it promotes sleep onset and continuity. Upstream, tryptophan is an amino acid that serves as a precursor to serotonin and melatonin.40 Both endogenous and exogenous corticosteroids decrease serum melatonin levels with a markedly diminished circadian rhythm secretion.41,42Demish and colleagues found a significant decrease in mean (SD) nocturnal melatonin plasma levels after the evening administration of oral dexamethasone 1 mg in 11 healthy volunteers: 127 (42) pg/mL before vs 73 (38) pg/mL after; P < .01.42 This result is likely due to decreased cellular metabolism and melatonin synthesis in the pineal gland. Of note, melatonin has neuroprotective affects, and the administration of melatonin has been shown to reverse some steroid-induced neurotoxicities in animal models.43
Steroids also reduce the uptake of tryptophan into the brain.33 Additionally, in animal models, dexamethasone administration caused a significant decrease in the gene expression of tryptophan hydroxylase, which is part of the multistep pathway in synthesizing serotonin from L-tryptophan. These effects upstream could inhibit the biosynthetic capacity of both melatonin and serotonin.44
A third pathway investigated in sleep regulation are the orexin neuropeptides. Orexins are produced in the hypothalamus and stimulate daytime wake activity in monoaminergic and cholinergic neurons. Subsequently, orexin receptor antagonists are a newer class of drugs aimed at mitigating nighttime hyperarousal and sleep disruption. Orexin overexpression may be a causal factor in steroid-induced sleep disturbance. However, this effect was specifically evaluated in a recent study in children with acute lymphoblastic leukemia, which showed that cerebral spinal fluid orexin levels (SD) were not significantly different from baseline after dexamethasone administration: 574 (26.6) pg/mL vs 580 (126.1) pg/mL; P = .8.45
Hyperarousal State
Finally, a hyperarousal state is thought to be produced by nongenomic changes to natural neuroinhibitory regulation seen with nonclassical steroid production called neurosteroids. Animal studies revealed that high levels of steroids were found in the CNS long after adrenalectomy, suggesting CNS de novo synthesis.46 In addition to altering gene expression at classic intercellular steroid receptors, neurosteroids can alter neurotransmission by direct interaction on ion-gated membranes and other receptors on the cell surface. Restlessness and insomnia could be due to γ-aminobutyric acid type A (GABAA) receptor modulation in the CNS where neuroactive steroids slow the rate of recovery of GABAA and potentially inhibit postsynaptic GABAergic transmission. It also is hypothesized that neuroactive steroids have excitatory action at nicotinic acetylcholine, 5HT3 receptors, and through increasing the fractional open time of the N-methyl-D-aspartate -activated channels.47 Allopregnanolone and DHEA are neurosteroids that act as GABAA agonists and have neuroprotective effects with anxiolytic, antidepressant, and antiaggressive properties.
Neurosteroids are synthesized from cholesterol in the hippocampus. Neurosteroids are upregulated in response to stress by CNS cortisol effects on various enzyme expressions.47 Whether exogenous steroid administration affects this biosynthesis vs the stress response in the HPA axis itself is not fully elucidated. Monteleone and colleagues found that dexamethasone 1 mg given orally significantly reduced cortisol and DHEA and allopregnanolone levels in both healthy volunteers and anorexia nervosa patients.48 Similarly, Genazzani and colleagues demonstrated that oral dexamethasone administration (0.5 mg every 6 hours) caused significant reductions in both serum allopregnanolone and DHEA levels.49
Outcomes Studies
The majority of reported data in steroid-induced insomnia and psychosis is in noncritically ill populations. In a randomized, prospective crossover study of healthy volunteers, dexamethasone administration (3 mg every 8 hours for 48 hours) resulted in significant changes in sleep patterns measured with polysomnography. Compared with placebo, steroid treatment showed significantly longer percentage (SD) of stage 0/awake times (11.7% [11.4] vs 2.9% [1.8]; P < .05); longer percentage (SD) of REM sleep latency (363.8 [74.5] minutes vs 202.8 [79.6] minutes; P < .01), and a reduced number (SD) of REM periods (3.8 [2.6] vs 9.7 [3.6]; P < .01).50 Insomnia was one of the most commonly self-reported AEs (> 60%) in a survey of 2,446 chronic steroid users, and the incidence increased as steroid doses increased.51
A prospective, open-label study of 240 patients with cancer demonstrated significant sleep disruptions using the Pittsburgh Sleep Quality Index with the use of high-dose steroids in chemotherapy.52 Naber and colleagues evaluated 50 previously healthy patients taking methylprednisolone 119 mg (41 mg/d) for retinitis and uveitis.53 They reported 26% to 34% of subjects experienced hypomanic syndrome based on a semistructured interview examination. Symptoms developed within 3 days and persisted for the 8-day course of therapy. Brown and colleagues prospectively evaluated 32 asthmatic patients prescribed bursts of prednisone > 40 mg daily. They observed significantly increased scores in the Young Mania Rating Scale within 3 to 7 days of starting therapy, which dissipated to baseline after stopping therapy.54
Despite a high reported incidence of neurologic AEs, outcomes in critically ill populations are mixed. Study methods are varied, and many were largely observational. No prospective, randomized studies exist to date specifically aimed and powered to evaluate the effects of steroids on sleep disturbances or delirium in a critically ill population. Furthermore, sleep quality is difficult to measure in this population, and self-reporting often is not an option. In critical care trials, if AEs such as insomnia, delirium, or psychosis are recorded at all, there is heterogeneity in the definitions, and these AEs are generally poorly defined (eg, psychiatric or neurologic disorder not otherwise specified), making pooled analysis of this outcome difficult.55
One of the largest observational studies in hospitalized patients was through the Boston Collaborative Drug Surveillance Program. A total of 718 consecutively enrolled inpatients who received prednisone were monitored for acute reactions. Psychiatric AEs were rare (1.3%) with low doses (< 40 mg/d), more prevalent (4.6%) with higher doses (41-80 mg/d), and most prevalent (18.4%) with the highest doses (> 80 mg/d), suggesting CNS AEs are dose dependent.18 A single-center, retrospective review of 755 psychiatric consults in hospitalized patients revealed that 54% of manic patients were due to corticosteroid administration.19 In a prospective observational study of 206 consecutive ICU admissions, steroid administration was an independent risk factor for development of ICU delirium, using the Confusion Assessment Method-ICU (CAM-ICU) at a single center (odds ratio [OR], 2.8; 95% CI, 1.05-7.28).25
Two studies in hospitalized oncology patients found conflicting results using the Nursing Delirium Screening Scale (Nu-DESC). One did not find a significant association between delirium and dexamethasone equivalent doses > 15 mg, while the second found an increased hazard ratio (HR) for a positive Nu-DESC score (HR, 2.67; 95% CI, 1.18-6.03).20,21 Similarly, conflicting results were found in 2 studies using first-order Markov models. In one prospective cohort study, 520 consecutive mechanically ventilated patients in 13 ICUs were monitored for the transition to delirium (CAM-ICU positive) from nondelirium states. Steroid administration was significantly associated with transitioning to delirium (OR, 1.52; 95% CI, 1.05-2.21).22 This conflicts with a similar study by Wolters and colleagues, which monitored 1,112 ICU patients who were given a median prednisone equivalent of 50 mg (interquartile range, 25-75 mg). Steroid administration was not significantly associated with the transition to delirium from an awake without delirium state (OR, 1.08; 95% CI, 0.89-1.32; adjusted OR, 1.00; 95% CI, 0.99-1.01 per 10-mg increase in prednisone equivalent).23
Mitigating Effects
Although steroid therapy often cannot be altered in the critically ill population, research showed that steroid overuse is common in ICUs.56,57 Minimizing dosage and duration are important ways clinicians can mitigate unwanted effects. CNS AEs seen with steroids often can be reversed once therapy is discontinued. Avoiding split-dose administration has been proposed given the natural diurnal production of cortisol.58 A review by Flaherty discusses the importance of avoiding pharmacologic agents in hospitalized older patients if possible due to known risks (falls, dependency, hip fractures, rebound insomnia, and risk of delirium) and provides a HELP ME SLEEP nomogram for nonpharmacologic interventions in hospitalized patients (Table 4).59
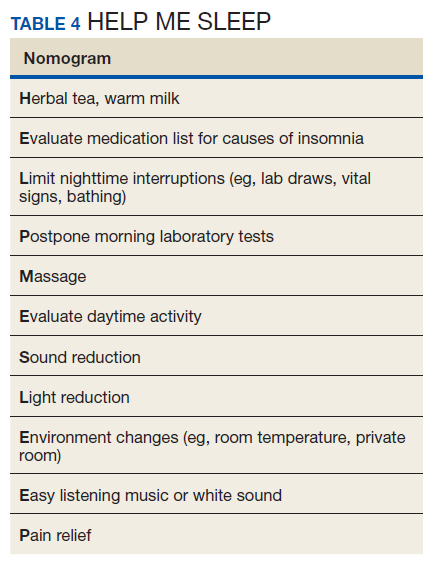
Historically, lithium has been recommended for steroid-induced mania with chronic steroid use; however, given the large volume and electrolyte shifts seen in critically ill patients, this may not be a viable option. Antidepressants, especially tricyclics, should generally be avoided in steroid-induced psychosis as these may exacerbate symptoms. If symptoms are severe, either typical (haloperidol) or atypical (olanzapine, quetiapine, risperidone) antipsychotics have been used with success.60 Given the known depletion of serum melatonin levels, melatonin supplements are an attractive and relatively safe option for steroid-induced insomnia; however, there are no robust studies specifically aimed at this intervention for this population.
Conclusions
With known, multimodal foci driving sleep impairment in ICU patients, PADIS guidelines recommend myriad interventions for improvement. Recommendations include noise and light reduction with earplugs and/or eyeshades to improve sleep quality. Nocturnal assist-control ventilation may improve sleep quality in ventilated patients. Finally, the development of institutional protocols for promoting sleep quality in ICU patients is recommended.17
1. Simini B. Patients’ perceptions of intensive care. Lancet. 1999;354(9178):571-572. doi: 10.1016/S0140-6736(99)02728-2
2. Delaney LJ, Van Haren F, Lopez V. Sleeping on a problem: the impact of sleep disturbance on intensive care patients—a clinical review. Ann Intensive Care. 2015;15:3. doi: 10.1186/s13613-015-0043-2
3. Friese RS, Diaz-Arrastia R, McBride D, Frankel H, Gentilello LM. Quality and quantity of sleep in the surgical intensive care unit; are our patients sleeping? J Trauma. 2007;63(6):1210-1214. doi: 10.1097/TA.0b013e31815b83d7
4. Elliott R, McKinley S, Cistulli P, Fien M. Characterisation of sleep in intensive care using 24-hour polysomnography: an observational study. Crit Care 2013;17(2):R46.
5. Aurell J, Elmqvist D. Sleep in the surgical intensive care unit: continuous polygraphic recording of sleep in patients receiving postoperative care. BJM (Clin Res Ed). 1985;290(6474)1029-1032. doi: 10.1136/bmj.290.6474.1029
6. White DP, Douglas NJ, Pickett CK, Zwillich CW, Weil JV. Sleep deprivation and the control of ventilation. Am Rev Respir Dis. 1983;128(6):984-986. doi: 10.1164/arrd.1983.128.6.984
7. Series F, Roy N, Marc I. Effects of sleep deprivation and sleep fragmentation on upper airway collapsibility in normal subjects. Am J Respir Crit Care Med. 1994;150(2):481-485. doi: 10.1164/ajrccm.150.2.8049833
8. Tadjalli A, Peever J. Sleep loss reduces respiratory motor plasticity. Adv Exp Med Biol. 2010;669:289-292.
doi: 10.1007/978-1-4419-5692-7_59
9. Roche Campo F, Drouot X, Thille AW, et al. Poor sleep quality is associated with late noninvasive ventilation failure in patients with acute hypercapnic respiratory failure. Crit Care Med. 2010;38(2):447-485. doi: 10.1097/CCM.0b013e3181bc8243
10. Sauvet F, Leftheriotis G, Gomez-Merino D, et al. Effect of acute sleep deprivation on vascular function in healthy subjects. J Appl Physiol (1985). 2010;108(1):68-75. doi: 10.1152/japplphysiol.00851.2009
11. Frey DJ, Fleshner M, Wright KP Jr. The effects of 40 hours of total sleep deprivation on inflammatory markers in healthy young adults. Brain Behav Immun. 2007;21(8):1050-1057. doi: 10.1016/j.bbi.2007.04.003
12. Spiegel K, Sheridan JF, Van Cauter E. Effect of sleep deprivation on response to immunization. JAMA 2002;288(12):1471-1472. doi: 10.1001/jama.288.12.1471-a
13. Dinges DF, Douglas SD, Zuagg L, et al. Leukocytosis and natural killer cell function parallel neurobehavioral fatigue induced by 64 hours of sleep deprivation. J Clin Invest. 1994;93(5):1930-1939. doi: 10.1172/JCI117184
14. Weinhouse GL, Schwab RJ, Watson PL, et al. Bench-to-bedside review: delirium in ICU patients— importance of sleep deprivation. Crit Care. 2009;13(6):234. doi: 10.1186/cc8131
15. Ely EW, Shintani A, Truman B, et al. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA. 2004;291(14):1753-1762. doi: 10.1001/jama.291.14.1753
16. Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513-1520. doi: 10.1097/CCM.0b013e3181e47be1
17. Devlin JW, Skrobik Y, Gelinas C, et al. Clinical practice guidelines for the prevention and management of pain, agitation/sedation, delirium, immobility, and sleep disruption in adult patients in the ICU. Crit Care Med. 2018;46(9):e825-e873
18. The Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther. 1972;13(5):694-698. doi: 10.1002/cpt1972135part1694
19. Rundell JR, Wise MG. Causes of organic mood disorder. J Neuropsychiatry Clin Neurosci. 1989;1(4):398-400. doi: 10.1176/jnp.1.4.398
20. Gaudreau JD, Gagnon P, Harel F, Roy MA, Tremblay A. Psychoactive medications and risk of delirium in hospitalized cancer patients. J Clin Oncol. 2005;23(27):6712-6718. doi: 10.1200/JCO.2005.05.140
21. Gaudreau JD, Gagnon P, Roy MA, Harel F, Tremblay A. Opioid medications and longitudinal risk of delirium in hospitalized cancer patients. Cancer. 2007;109(11):2365-2373.
doi: 10.1002/cncr.22665
22. Schreiber MP, Colantuoni E, Bienvenu OJ, et al. Corticosteroids and transition to delirium in patients with acute lung injury. Crit Care Med. 2014;42(6):1480-1486. doi: 10.1097/CCM.0000000000000247
23. Wolters AE, Veldhuijzen DS, Zaal IJ, et al. Systemic corticosteroids and transition to delirium in critically ill patients. Crit Care Med. 2015;43(12):e585-e588. doi: 10.1097/CCM.0000000000001302
24. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naïve adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol. 2016;136(2):101-107. doi: 10.1159/000445420
25. Tilouche N, Hassen M, Ali HBS, Jaoued AHO, Gharbi R, Atrous SS. Delirium in the intensive care unit: incidence, risk factors, and impact on outcome. Indian J Crit Care Med. 2018;22:144-149. doi: 10.4103/ijccm.IJCCM_244_17
26. Young A, Marsh S. Steroid use in critical care. BJA Education. 2018;18(5):129-134. doi: 10.1016/j.bjae.2018.01.005
27. DiPiro J, Talbert R, Yee G, Matzke GR, Wells BG, Posey M. Pharmacotherapy: A Pathophysiologic Approach. 4th ed. New York: McGraw-Hill; 1999:1277-1278.
28. Schimmer
29. Sarnes E, Crofford L, Watson M, Dennis G, Kan H, Bass D. Incidence of US costs of corticosteroid-associated adverse events: a systematic literature review. Clin Ther. 2011;33(10):1413-1432.
30. Idzikowsi C, Shapiro CM. ABC of sleep disorders, non-psychotropic drugs and sleep. BMJ. 1993;306(6885):1118-1120. doi: 10.1136/bmj.306.6885.1118

31. Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14(4):398-406.
doi: 10.3109/10253890.2011.586446
32. Wolkowitz OM, Reus VI, Weingartner H, et al. Cognitive effects of corticosteroids. Am J Psychiatry 1990;147(10):1297-1303. doi: 10.1176/ajp.147.10.1297
33. McEwen BS, Davis PG, Parsons B, Pfaff DW. The brain as a target for steroid hormone action. Ann Rev Neurosci. 1979;2:65-112. doi: 10.1146/annurev.ne.02.030179.000433
34. Brown ES, Woolston DJ, Frol AM. Amygdala volume in patients receiving chronic corticosteroid therapy. Biol Psychiatry. 2008;63(7):705-709.
doi: 10.1016/j.biopsych.2007.09.014
35. Brown ES, Woolston D, Frol A, et al. Hippocampal volume, spectroscopy, cognition, and mood in patients receiving corticosteroid. Biol Psychiatry. 2004;55(5):538-545.
36. Sapolsky RM, McEwen BS. Down-regulation of neural corticosterone receptors by corticosterone and dexamethasone. Brain Res. 1985;339(1):161-165.
doi: 10.1016/0006-8993(85)90638-9
37. Sorrells SF, Caso JR, Munhoz CD, Spolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64(1):33-39.
doi: 10.1016/j.neuron.2009.09.032
38. Wolkowitz OM, Burke H, Epel ES, Reus VI. Glucocorticoids: mood, memory, and mechanisms. Ann NY Acad Sci. 2009;1179:19-40. doi: 10.1111/j.1749-6632.2009.04980.x
39. Wolkowitz OM, Epel ES, Reus VI. Stress hormone-related psychopathology: pathophysiological and treatment implications. World J Biol Psychiatry. 2001;2(3):115-143. doi: 10.3109/15622970109026799
40. Paredes S, Barriga C, Reiter R, Rodrigues A. Assessment of the potential role of tryptophan as the precursor of serotonin and melatonin for the aged sleep-wake cycle and immune function: Streptopelia Risoria as a model. Int J Tryptophan Res. 2009;2:23-36. doi: 10.4137/ijtr.s1129
41. Soszyński P, Stowińska-Srzednicka J, Kasperlik-Zatuska A, Zgliczyński S. Decreased melatonin concentration in Cushing’s Syndrome. Horm Metab Res. 1989;21(12):673-674. doi: 10.1055/s-2007-1009317
42. Demish L, Demish K, Neckelsen T. Influence of dexamethasone on nocturnal melatonin production in healthy adult subjects. J Pineal Res. 1988;5(3):317-321. doi: 10.1111/j.1600-079x.1988.tb00657.x
43. Assaf N, Shalby AB, Khalil WK, Ahmed HH. Biochemical and genetic alterations of oxidant/antioxidant status of the brain in rats treated with dexamethasone: protective roles of melatonin and acetyl-L-carnitine. J Physiol Biochem. 2012;68(1):77-90. doi: 10.1007/s13105-011-0121-3
44. Clark MS, Russo AF. Tissue-specific glucocorticoid regulation of tryptophan hydroxylase mRNA levels. Brain Res Mol Brain Res. 1997;48(2):346-54. doi: 10.1016/s0169-328x(97)00106-x
45. Kram DE, Krasnow SM, Levasseur PR, Zhu X, Stork LC, Marks DL. Dexamethasone chemotherapy does not disrupt orexin signaling. PLoS One. 2016;11(12):e0168731. doi: 10.1371/journal.pone.0168731
46. Mellon S. Neurosteroids: biochemistry, modes of action, and clinical relevance. J Clin Endocrinol Metab. 1994;78(5):1003-1008. doi: 10.1210/jcem.78.5.8175951
47. Zorumski C, Paul SM, Izumi Y, Covey DF, Mennerick S . Neurosteroids, stress and depression: potential therapeutic opportunities. Neurosci Biobehav Rev. 2013;37(1):109-122. doi: 10.1016/j.neubiorev.2012.10.005
48. Monteleone P, Luisi M, Martiadis V, et al. Impaired reduction of enhanced levels of dehydroepiandrosterone by oral dexamethasone in anorexia nervosa. Psychoneuroendocrinology. 2006;31(4):537-542. doi: 10.1016/j.psyneuen.2005.08.015
49. Genazzani AR, Petraglia F, Bernardi F, et al. Circulating levels of allopregnanolone in humans: gender, age, and endocrine influences. J Clin Endocrinol Metab. 1998;83(6):2099-3103. doi: 10.1210/jcem.83.6.4905
50. Moser NJ, Phillips BA, Guthrie G, Barnett G. Effects of dexamethasone on sleep. Pharmacol Toxicol. 1996;79(2):100-102. doi: 10.1111/j.1600-0773.1996.tb00249.x
51. Curtis J, Westfall A, Allison J, et al. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum. 2006;55(3):420-426. doi: 10.1002/art.21984
52. Zhao J, Dai YH, Xi QS, Yu SY. A clinical study on insomnia in patients with cancer during chemotherapy containing high-dose glucocorticoids. Pharmazie. 2013;68(6):421-427
53. Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days corticosteroid treatment. A prospective study. Psychoneuroendocrinology. 1996;21(1):25-31. doi: 10.1016/0306-4530(95)00031-3
54. Brown ES, Suppes T, Khan DA, Carmody TJ 3rd. Mood changes during prednisone bursts in outpatients with asthma. J Clin Psychopharmacol. 2002;22(1):55-61.
doi: 10.1097/00004714-200202000-00009
55. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367. doi: 10.4065/81.10.1361
56. Britt RC, Devine A, Swallen KC et al. Corticosteroid use in the intensive care unit: at what cost? Arch Surg. 2006;141(2):145-159. doi:10.1001/archsurg.141.2.145
57. Kiser TH, Allen RR, Valuck RJ, Moss M, Vanivier RW. Outcomes associated with corticosteroid dosage in critically ill patients in acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(9):1052-1064. doi: 10.1164/rccm.201401-0058OC
58. Bourne RS, Mills GH. Sleep disruption in critically ill patients—pharmacological considerations. Anaesthesia. 2004;59(4):374-384. doi: 10.1111/j. 1365-2044.2004.03664.x
59. Flaherty JH. Insomnia among hospitalized older persons. Clin Geriatr Med. 2008;24(1):51-67. doi: 10.1016/j.cger.2007.08.012
60. Sirios F. Steroid psychosis: a review. Gen Hosp Psychiatry. 2003;25(1):27-33. doi: 10.1016/s0163-8343(02)00241-4
Sleep disturbance in the critically ill has received much attention over recent years as this is a common result of intensive care unit (ICU) admission. Disruptions in sleep not only can, at a minimum, cause distress and lower patient satisfaction, but also inhibit recovery from illness and increase morbidity.1,2 Several studies have been conducted highlighting the altered sleep patterns of critically ill patients; although total sleep time may seem normal (7-9 hours), patients can experience multiple awakenings per hour, more time in light sleep (stages 1 and 2), and less time in restorative sleep (stages 3 and 4, [REM]rapid eye movement).2-5
There are several hypothesized physiologic detriments that contribute to slower ICU recovery with sleep deprivation. Research in noncritically ill subjects suggests that sleep deprivation contributes to hypoventilation and potentially prolonged time on the ventilator.6-9 Cardiovascular morbidity may be adversely affected by inflammatory cytokine release seen in sleep disruption.10,11 Studies of noncritically ill patients also suggest that immune response is impaired, potentially protracting infection recovery.12,13 Finally, although not directly investigated, sleep deprivation may contribute to ICU delirium, an independent adverse effect (AE) associated with increased mortality and worse long-term outcomes.14-16
The Society of Critical Care Medicine (SCCM) recently updated its consensus guidelines for the management of pain, agitation/sedation, delirium, immobility, and sleep disruption (PADIS) in adult patients.17 These guidelines offer limited interventions to promote sleep in ICU patients based on available evidence and steer the clinician toward minimizing exacerbating factors. Although factors that affect sleep patterns are multifactorial, such as noise levels, pain, mechanical ventilation, and inflammatory mediators, medication therapy is a known modifiable risk factor for sleep disturbance in critically ill patients.2 This focused review will specifically evaluate the effects of steroids on sleep deprivation, psychosis, delirium, and what is known about these effects in a critically ill population.
To include articles relevant to a critically ill population, a systematic search of MEDLINE and PubMed from 1966 to 2019 was performed using the following Medical Subject Headings (MeSH) terms: delirium/etiology, psychoses, substance-induced/etiology, sleep-wake disorders/chemically induced, neurocognitive disorders/chemically induced, dyssomnias/drug effects plus glucocorticoids/adverse effects, adrenal cortex hormones/adverse effects, prednisone/adverse effects, methylprednisolone/adverse effects, and hydrocortisone/adverse effects. The initial search produced 285 articles. Case reports, reviews, letters, and articles pertaining to primary care or palliative populations were excluded, leaving 8 relevant articles for inclusion (Table 1).18-25
ICU Steroid Use
Steroids are commonly used in the ICU and affect nearly every critically ill population. Common indications for steroids in the ICU include anaphylaxis, airway edema, septic shock, asthma and COPD exacerbations, pneumocystis pneumonia, adrenal crisis, antiemetic treatment, elevated intracranial pressure from tumors, autoimmune disorders, and stress doses needed for chronic steroid users before invasive procedures.26 Whether divided into glucocorticoid or mineralocorticoid subgroups, corticosteroids offer therapeutic benefit from their pharmacologic similarity to endogenously produced cortisol, which includes anti-inflammatory, immunosuppressive, antiproliferative, and vasoconstrictive effects.
Steroid receptors are present in most human tissue, and in varying degrees of binding affinity produce a wide variety of effects. After passive diffusion across cell membranes, steroid-receptor activation binds to various DNA sites, called glucocorticoid regulatory elements, which either stimulates or inhibits transcription of multiple nearby genes.
At the cellular level, corticosteroids inhibit the release of arachidonic acid through upstream production of lipocortin peptides and antagonism of phospholipase A2. This action decreases subsequent inflammatory mediators, including kinins, histamine, liposomal enzymes, and prostaglandins. Steroids also inhibit NF-κB, which further decreases expression of proinflammatory genes while promoting interleukin-10 and its anti-inflammatory properties. Antiproliferative effects of steroids are seen by triggering cell apoptosis and inhibition of fibroblast proliferation.27,28
By binding to mineralocorticoid receptors, steroids cause sodium retention coupled with hydrogen and potassium excretion in the distal renal tubule. Steroids also promote vasoconstriction by upregulating the production and sensitivity of β receptors in the endothelium while suppressing the production of vasodilators. Although rarely used for these physiologic effects, steroids also are involved in a number of metabolic pathways, including calcium regulation, gluconeogenesis, protein metabolism, and fat distribution. Given the similar structure to cortisol, exogenous steroids depress the hypothalamic-pituitary axis (HPA) and decrease the release of adrenocorticotropic hormone (ACTH). Tapering doses of steroid regimens is often required to allow natural androgen and cortisol synthesis and prevent steroid withdrawal.27,28
The potency of various exogenous steroids closely parallels their ability to retain sodium (Table 2). Prolonged activation of steroid receptors can have numerous systemic AEs, including unwanted neurocognitive effects (Table 3). Insomnia and psychosis are commonly described in corticosteroid clinical trials, and in one meta-analysis, both are associated with high costs per episode per year.29
Steroid-Induced Sleep Disruption and Psychosis
Sleep disruption caused by exogenous administration of steroids is thought to trigger other psychostimulant effects, such as mood swings, nervousness, psychoses, and delirium.30 Similarly, the SCCM PADIS guidelines included an ungraded statement: “although an association between sleep quality and delirium occurrence exists in critically ill adults, a cause-effect relationship has not been established.”17 For this review, these AEs will be discussed as related events.
The medical literature proposes 3 pathways primarily responsible for neurocognitive AEs of steroids: behavior changes through modification of the HPA axis, changes in natural sleep-wake cycles, and hyperarousal caused by modification in neuroinhibitory pathways (Figure).
HPA Axis Modification
Under either physical or psychological stress, neural circuits in the brain release corticotropin-releasing hormone (CRH), dehydroepiandrosterone (DHEA), and arginine vasopressin, which go on to activate the sympathetic nervous system and the HPA axis. CRH from the hypothalamus goes on to stimulate ACTH release from the pituitary. ACTH then stimulates cortisol secretion from the adrenal glands. Circulating cortisol feeds into several structures of the brain, including the pituitary, hippocampus, and amygdala. Steroid-receptor complexes alter gene transcription in the central nervous system (CNS), affecting the production of neurotransmitters (eg, dopamine, serotonin) and neuropeptides (eg, somatostatin, β-endorphin). Feedback inhibition ensues, with downregulation of the HPA axis, which prevents depletion of endogenous production of steroids.31 DHEA has protective effects against excessive cortisol activity, but DHEA secretion declines with prolonged cortisol exposure. Exogenous steroids may have different effects than endogenous steroids, and neurocognitive sequelae stem from disruption and imbalance of these physiologic mechanisms.32,33
Steroid receptors are densely located in behavior centers in the brain: the amygdala, septum, and hippocampus. Pharmacologic changes in gene expression alter norepinephrine and serotonin levels in the brain as well as their receptors.32 Prolonged exposure to exogenous steroids has been shown to decrease amygdala and hippocampal volumes.34,35 Furthermore, prolonged corticosteroid exposure has been shown to decrease the number of steroid receptors in the hippocampus, pituitary gland, and amygdala.36 In a somewhat paradoxical finding, the production of CNS proinflammatory cytokines like interleuken-1β and tumor necrosis factor α has been seen after steroid administration, suggesting alternate gene signaling in the CNS.37 Although not proven conclusively, it is felt that these physiologic changes and hyperactivity of the HPA axis are predominantly responsible for changes in behavior, mood, memory, and eventually psychosis in steroid-treated patients.33,38
Finally, alterations in cognition and behavior may be related to steroid-induced changes in CNS carbohydrate, protein, and lipid metabolism with subsequent cellular neurotoxicity.32,38 Glucose uptake into the hippocampus is decreased with steroid exposure. Additionally, breakdown of metabolic compounds to produce energy can be destructive if left unchecked for prolonged periods. DHEA, growth hormone, and testosterone work to repair catabolic damage produced by cortisol, known as anabolic balance. A low anabolic balance (low DHEA levels to high cortisol levels) leads to a cascade of dysregulation in brain activity.39
Changes in Natural Sleep-Wake Cycles
Natural sleep pathways are also affected by steroids. The sleep-wake cycle is primarily regulated in the hypothalamus with circadian release of melatonin from the pineal gland. Melatonin release is highest at night, where it promotes sleep onset and continuity. Upstream, tryptophan is an amino acid that serves as a precursor to serotonin and melatonin.40 Both endogenous and exogenous corticosteroids decrease serum melatonin levels with a markedly diminished circadian rhythm secretion.41,42Demish and colleagues found a significant decrease in mean (SD) nocturnal melatonin plasma levels after the evening administration of oral dexamethasone 1 mg in 11 healthy volunteers: 127 (42) pg/mL before vs 73 (38) pg/mL after; P < .01.42 This result is likely due to decreased cellular metabolism and melatonin synthesis in the pineal gland. Of note, melatonin has neuroprotective affects, and the administration of melatonin has been shown to reverse some steroid-induced neurotoxicities in animal models.43
Steroids also reduce the uptake of tryptophan into the brain.33 Additionally, in animal models, dexamethasone administration caused a significant decrease in the gene expression of tryptophan hydroxylase, which is part of the multistep pathway in synthesizing serotonin from L-tryptophan. These effects upstream could inhibit the biosynthetic capacity of both melatonin and serotonin.44
A third pathway investigated in sleep regulation are the orexin neuropeptides. Orexins are produced in the hypothalamus and stimulate daytime wake activity in monoaminergic and cholinergic neurons. Subsequently, orexin receptor antagonists are a newer class of drugs aimed at mitigating nighttime hyperarousal and sleep disruption. Orexin overexpression may be a causal factor in steroid-induced sleep disturbance. However, this effect was specifically evaluated in a recent study in children with acute lymphoblastic leukemia, which showed that cerebral spinal fluid orexin levels (SD) were not significantly different from baseline after dexamethasone administration: 574 (26.6) pg/mL vs 580 (126.1) pg/mL; P = .8.45
Hyperarousal State
Finally, a hyperarousal state is thought to be produced by nongenomic changes to natural neuroinhibitory regulation seen with nonclassical steroid production called neurosteroids. Animal studies revealed that high levels of steroids were found in the CNS long after adrenalectomy, suggesting CNS de novo synthesis.46 In addition to altering gene expression at classic intercellular steroid receptors, neurosteroids can alter neurotransmission by direct interaction on ion-gated membranes and other receptors on the cell surface. Restlessness and insomnia could be due to γ-aminobutyric acid type A (GABAA) receptor modulation in the CNS where neuroactive steroids slow the rate of recovery of GABAA and potentially inhibit postsynaptic GABAergic transmission. It also is hypothesized that neuroactive steroids have excitatory action at nicotinic acetylcholine, 5HT3 receptors, and through increasing the fractional open time of the N-methyl-D-aspartate -activated channels.47 Allopregnanolone and DHEA are neurosteroids that act as GABAA agonists and have neuroprotective effects with anxiolytic, antidepressant, and antiaggressive properties.
Neurosteroids are synthesized from cholesterol in the hippocampus. Neurosteroids are upregulated in response to stress by CNS cortisol effects on various enzyme expressions.47 Whether exogenous steroid administration affects this biosynthesis vs the stress response in the HPA axis itself is not fully elucidated. Monteleone and colleagues found that dexamethasone 1 mg given orally significantly reduced cortisol and DHEA and allopregnanolone levels in both healthy volunteers and anorexia nervosa patients.48 Similarly, Genazzani and colleagues demonstrated that oral dexamethasone administration (0.5 mg every 6 hours) caused significant reductions in both serum allopregnanolone and DHEA levels.49
Outcomes Studies
The majority of reported data in steroid-induced insomnia and psychosis is in noncritically ill populations. In a randomized, prospective crossover study of healthy volunteers, dexamethasone administration (3 mg every 8 hours for 48 hours) resulted in significant changes in sleep patterns measured with polysomnography. Compared with placebo, steroid treatment showed significantly longer percentage (SD) of stage 0/awake times (11.7% [11.4] vs 2.9% [1.8]; P < .05); longer percentage (SD) of REM sleep latency (363.8 [74.5] minutes vs 202.8 [79.6] minutes; P < .01), and a reduced number (SD) of REM periods (3.8 [2.6] vs 9.7 [3.6]; P < .01).50 Insomnia was one of the most commonly self-reported AEs (> 60%) in a survey of 2,446 chronic steroid users, and the incidence increased as steroid doses increased.51
A prospective, open-label study of 240 patients with cancer demonstrated significant sleep disruptions using the Pittsburgh Sleep Quality Index with the use of high-dose steroids in chemotherapy.52 Naber and colleagues evaluated 50 previously healthy patients taking methylprednisolone 119 mg (41 mg/d) for retinitis and uveitis.53 They reported 26% to 34% of subjects experienced hypomanic syndrome based on a semistructured interview examination. Symptoms developed within 3 days and persisted for the 8-day course of therapy. Brown and colleagues prospectively evaluated 32 asthmatic patients prescribed bursts of prednisone > 40 mg daily. They observed significantly increased scores in the Young Mania Rating Scale within 3 to 7 days of starting therapy, which dissipated to baseline after stopping therapy.54
Despite a high reported incidence of neurologic AEs, outcomes in critically ill populations are mixed. Study methods are varied, and many were largely observational. No prospective, randomized studies exist to date specifically aimed and powered to evaluate the effects of steroids on sleep disturbances or delirium in a critically ill population. Furthermore, sleep quality is difficult to measure in this population, and self-reporting often is not an option. In critical care trials, if AEs such as insomnia, delirium, or psychosis are recorded at all, there is heterogeneity in the definitions, and these AEs are generally poorly defined (eg, psychiatric or neurologic disorder not otherwise specified), making pooled analysis of this outcome difficult.55
One of the largest observational studies in hospitalized patients was through the Boston Collaborative Drug Surveillance Program. A total of 718 consecutively enrolled inpatients who received prednisone were monitored for acute reactions. Psychiatric AEs were rare (1.3%) with low doses (< 40 mg/d), more prevalent (4.6%) with higher doses (41-80 mg/d), and most prevalent (18.4%) with the highest doses (> 80 mg/d), suggesting CNS AEs are dose dependent.18 A single-center, retrospective review of 755 psychiatric consults in hospitalized patients revealed that 54% of manic patients were due to corticosteroid administration.19 In a prospective observational study of 206 consecutive ICU admissions, steroid administration was an independent risk factor for development of ICU delirium, using the Confusion Assessment Method-ICU (CAM-ICU) at a single center (odds ratio [OR], 2.8; 95% CI, 1.05-7.28).25
Two studies in hospitalized oncology patients found conflicting results using the Nursing Delirium Screening Scale (Nu-DESC). One did not find a significant association between delirium and dexamethasone equivalent doses > 15 mg, while the second found an increased hazard ratio (HR) for a positive Nu-DESC score (HR, 2.67; 95% CI, 1.18-6.03).20,21 Similarly, conflicting results were found in 2 studies using first-order Markov models. In one prospective cohort study, 520 consecutive mechanically ventilated patients in 13 ICUs were monitored for the transition to delirium (CAM-ICU positive) from nondelirium states. Steroid administration was significantly associated with transitioning to delirium (OR, 1.52; 95% CI, 1.05-2.21).22 This conflicts with a similar study by Wolters and colleagues, which monitored 1,112 ICU patients who were given a median prednisone equivalent of 50 mg (interquartile range, 25-75 mg). Steroid administration was not significantly associated with the transition to delirium from an awake without delirium state (OR, 1.08; 95% CI, 0.89-1.32; adjusted OR, 1.00; 95% CI, 0.99-1.01 per 10-mg increase in prednisone equivalent).23
Mitigating Effects
Although steroid therapy often cannot be altered in the critically ill population, research showed that steroid overuse is common in ICUs.56,57 Minimizing dosage and duration are important ways clinicians can mitigate unwanted effects. CNS AEs seen with steroids often can be reversed once therapy is discontinued. Avoiding split-dose administration has been proposed given the natural diurnal production of cortisol.58 A review by Flaherty discusses the importance of avoiding pharmacologic agents in hospitalized older patients if possible due to known risks (falls, dependency, hip fractures, rebound insomnia, and risk of delirium) and provides a HELP ME SLEEP nomogram for nonpharmacologic interventions in hospitalized patients (Table 4).59
Historically, lithium has been recommended for steroid-induced mania with chronic steroid use; however, given the large volume and electrolyte shifts seen in critically ill patients, this may not be a viable option. Antidepressants, especially tricyclics, should generally be avoided in steroid-induced psychosis as these may exacerbate symptoms. If symptoms are severe, either typical (haloperidol) or atypical (olanzapine, quetiapine, risperidone) antipsychotics have been used with success.60 Given the known depletion of serum melatonin levels, melatonin supplements are an attractive and relatively safe option for steroid-induced insomnia; however, there are no robust studies specifically aimed at this intervention for this population.
Conclusions
With known, multimodal foci driving sleep impairment in ICU patients, PADIS guidelines recommend myriad interventions for improvement. Recommendations include noise and light reduction with earplugs and/or eyeshades to improve sleep quality. Nocturnal assist-control ventilation may improve sleep quality in ventilated patients. Finally, the development of institutional protocols for promoting sleep quality in ICU patients is recommended.17
Sleep disturbance in the critically ill has received much attention over recent years as this is a common result of intensive care unit (ICU) admission. Disruptions in sleep not only can, at a minimum, cause distress and lower patient satisfaction, but also inhibit recovery from illness and increase morbidity.1,2 Several studies have been conducted highlighting the altered sleep patterns of critically ill patients; although total sleep time may seem normal (7-9 hours), patients can experience multiple awakenings per hour, more time in light sleep (stages 1 and 2), and less time in restorative sleep (stages 3 and 4, [REM]rapid eye movement).2-5
There are several hypothesized physiologic detriments that contribute to slower ICU recovery with sleep deprivation. Research in noncritically ill subjects suggests that sleep deprivation contributes to hypoventilation and potentially prolonged time on the ventilator.6-9 Cardiovascular morbidity may be adversely affected by inflammatory cytokine release seen in sleep disruption.10,11 Studies of noncritically ill patients also suggest that immune response is impaired, potentially protracting infection recovery.12,13 Finally, although not directly investigated, sleep deprivation may contribute to ICU delirium, an independent adverse effect (AE) associated with increased mortality and worse long-term outcomes.14-16
The Society of Critical Care Medicine (SCCM) recently updated its consensus guidelines for the management of pain, agitation/sedation, delirium, immobility, and sleep disruption (PADIS) in adult patients.17 These guidelines offer limited interventions to promote sleep in ICU patients based on available evidence and steer the clinician toward minimizing exacerbating factors. Although factors that affect sleep patterns are multifactorial, such as noise levels, pain, mechanical ventilation, and inflammatory mediators, medication therapy is a known modifiable risk factor for sleep disturbance in critically ill patients.2 This focused review will specifically evaluate the effects of steroids on sleep deprivation, psychosis, delirium, and what is known about these effects in a critically ill population.
To include articles relevant to a critically ill population, a systematic search of MEDLINE and PubMed from 1966 to 2019 was performed using the following Medical Subject Headings (MeSH) terms: delirium/etiology, psychoses, substance-induced/etiology, sleep-wake disorders/chemically induced, neurocognitive disorders/chemically induced, dyssomnias/drug effects plus glucocorticoids/adverse effects, adrenal cortex hormones/adverse effects, prednisone/adverse effects, methylprednisolone/adverse effects, and hydrocortisone/adverse effects. The initial search produced 285 articles. Case reports, reviews, letters, and articles pertaining to primary care or palliative populations were excluded, leaving 8 relevant articles for inclusion (Table 1).18-25
ICU Steroid Use
Steroids are commonly used in the ICU and affect nearly every critically ill population. Common indications for steroids in the ICU include anaphylaxis, airway edema, septic shock, asthma and COPD exacerbations, pneumocystis pneumonia, adrenal crisis, antiemetic treatment, elevated intracranial pressure from tumors, autoimmune disorders, and stress doses needed for chronic steroid users before invasive procedures.26 Whether divided into glucocorticoid or mineralocorticoid subgroups, corticosteroids offer therapeutic benefit from their pharmacologic similarity to endogenously produced cortisol, which includes anti-inflammatory, immunosuppressive, antiproliferative, and vasoconstrictive effects.
Steroid receptors are present in most human tissue, and in varying degrees of binding affinity produce a wide variety of effects. After passive diffusion across cell membranes, steroid-receptor activation binds to various DNA sites, called glucocorticoid regulatory elements, which either stimulates or inhibits transcription of multiple nearby genes.
At the cellular level, corticosteroids inhibit the release of arachidonic acid through upstream production of lipocortin peptides and antagonism of phospholipase A2. This action decreases subsequent inflammatory mediators, including kinins, histamine, liposomal enzymes, and prostaglandins. Steroids also inhibit NF-κB, which further decreases expression of proinflammatory genes while promoting interleukin-10 and its anti-inflammatory properties. Antiproliferative effects of steroids are seen by triggering cell apoptosis and inhibition of fibroblast proliferation.27,28
By binding to mineralocorticoid receptors, steroids cause sodium retention coupled with hydrogen and potassium excretion in the distal renal tubule. Steroids also promote vasoconstriction by upregulating the production and sensitivity of β receptors in the endothelium while suppressing the production of vasodilators. Although rarely used for these physiologic effects, steroids also are involved in a number of metabolic pathways, including calcium regulation, gluconeogenesis, protein metabolism, and fat distribution. Given the similar structure to cortisol, exogenous steroids depress the hypothalamic-pituitary axis (HPA) and decrease the release of adrenocorticotropic hormone (ACTH). Tapering doses of steroid regimens is often required to allow natural androgen and cortisol synthesis and prevent steroid withdrawal.27,28
The potency of various exogenous steroids closely parallels their ability to retain sodium (Table 2). Prolonged activation of steroid receptors can have numerous systemic AEs, including unwanted neurocognitive effects (Table 3). Insomnia and psychosis are commonly described in corticosteroid clinical trials, and in one meta-analysis, both are associated with high costs per episode per year.29
Steroid-Induced Sleep Disruption and Psychosis
Sleep disruption caused by exogenous administration of steroids is thought to trigger other psychostimulant effects, such as mood swings, nervousness, psychoses, and delirium.30 Similarly, the SCCM PADIS guidelines included an ungraded statement: “although an association between sleep quality and delirium occurrence exists in critically ill adults, a cause-effect relationship has not been established.”17 For this review, these AEs will be discussed as related events.
The medical literature proposes 3 pathways primarily responsible for neurocognitive AEs of steroids: behavior changes through modification of the HPA axis, changes in natural sleep-wake cycles, and hyperarousal caused by modification in neuroinhibitory pathways (Figure).
HPA Axis Modification
Under either physical or psychological stress, neural circuits in the brain release corticotropin-releasing hormone (CRH), dehydroepiandrosterone (DHEA), and arginine vasopressin, which go on to activate the sympathetic nervous system and the HPA axis. CRH from the hypothalamus goes on to stimulate ACTH release from the pituitary. ACTH then stimulates cortisol secretion from the adrenal glands. Circulating cortisol feeds into several structures of the brain, including the pituitary, hippocampus, and amygdala. Steroid-receptor complexes alter gene transcription in the central nervous system (CNS), affecting the production of neurotransmitters (eg, dopamine, serotonin) and neuropeptides (eg, somatostatin, β-endorphin). Feedback inhibition ensues, with downregulation of the HPA axis, which prevents depletion of endogenous production of steroids.31 DHEA has protective effects against excessive cortisol activity, but DHEA secretion declines with prolonged cortisol exposure. Exogenous steroids may have different effects than endogenous steroids, and neurocognitive sequelae stem from disruption and imbalance of these physiologic mechanisms.32,33
Steroid receptors are densely located in behavior centers in the brain: the amygdala, septum, and hippocampus. Pharmacologic changes in gene expression alter norepinephrine and serotonin levels in the brain as well as their receptors.32 Prolonged exposure to exogenous steroids has been shown to decrease amygdala and hippocampal volumes.34,35 Furthermore, prolonged corticosteroid exposure has been shown to decrease the number of steroid receptors in the hippocampus, pituitary gland, and amygdala.36 In a somewhat paradoxical finding, the production of CNS proinflammatory cytokines like interleuken-1β and tumor necrosis factor α has been seen after steroid administration, suggesting alternate gene signaling in the CNS.37 Although not proven conclusively, it is felt that these physiologic changes and hyperactivity of the HPA axis are predominantly responsible for changes in behavior, mood, memory, and eventually psychosis in steroid-treated patients.33,38
Finally, alterations in cognition and behavior may be related to steroid-induced changes in CNS carbohydrate, protein, and lipid metabolism with subsequent cellular neurotoxicity.32,38 Glucose uptake into the hippocampus is decreased with steroid exposure. Additionally, breakdown of metabolic compounds to produce energy can be destructive if left unchecked for prolonged periods. DHEA, growth hormone, and testosterone work to repair catabolic damage produced by cortisol, known as anabolic balance. A low anabolic balance (low DHEA levels to high cortisol levels) leads to a cascade of dysregulation in brain activity.39
Changes in Natural Sleep-Wake Cycles
Natural sleep pathways are also affected by steroids. The sleep-wake cycle is primarily regulated in the hypothalamus with circadian release of melatonin from the pineal gland. Melatonin release is highest at night, where it promotes sleep onset and continuity. Upstream, tryptophan is an amino acid that serves as a precursor to serotonin and melatonin.40 Both endogenous and exogenous corticosteroids decrease serum melatonin levels with a markedly diminished circadian rhythm secretion.41,42Demish and colleagues found a significant decrease in mean (SD) nocturnal melatonin plasma levels after the evening administration of oral dexamethasone 1 mg in 11 healthy volunteers: 127 (42) pg/mL before vs 73 (38) pg/mL after; P < .01.42 This result is likely due to decreased cellular metabolism and melatonin synthesis in the pineal gland. Of note, melatonin has neuroprotective affects, and the administration of melatonin has been shown to reverse some steroid-induced neurotoxicities in animal models.43
Steroids also reduce the uptake of tryptophan into the brain.33 Additionally, in animal models, dexamethasone administration caused a significant decrease in the gene expression of tryptophan hydroxylase, which is part of the multistep pathway in synthesizing serotonin from L-tryptophan. These effects upstream could inhibit the biosynthetic capacity of both melatonin and serotonin.44
A third pathway investigated in sleep regulation are the orexin neuropeptides. Orexins are produced in the hypothalamus and stimulate daytime wake activity in monoaminergic and cholinergic neurons. Subsequently, orexin receptor antagonists are a newer class of drugs aimed at mitigating nighttime hyperarousal and sleep disruption. Orexin overexpression may be a causal factor in steroid-induced sleep disturbance. However, this effect was specifically evaluated in a recent study in children with acute lymphoblastic leukemia, which showed that cerebral spinal fluid orexin levels (SD) were not significantly different from baseline after dexamethasone administration: 574 (26.6) pg/mL vs 580 (126.1) pg/mL; P = .8.45
Hyperarousal State
Finally, a hyperarousal state is thought to be produced by nongenomic changes to natural neuroinhibitory regulation seen with nonclassical steroid production called neurosteroids. Animal studies revealed that high levels of steroids were found in the CNS long after adrenalectomy, suggesting CNS de novo synthesis.46 In addition to altering gene expression at classic intercellular steroid receptors, neurosteroids can alter neurotransmission by direct interaction on ion-gated membranes and other receptors on the cell surface. Restlessness and insomnia could be due to γ-aminobutyric acid type A (GABAA) receptor modulation in the CNS where neuroactive steroids slow the rate of recovery of GABAA and potentially inhibit postsynaptic GABAergic transmission. It also is hypothesized that neuroactive steroids have excitatory action at nicotinic acetylcholine, 5HT3 receptors, and through increasing the fractional open time of the N-methyl-D-aspartate -activated channels.47 Allopregnanolone and DHEA are neurosteroids that act as GABAA agonists and have neuroprotective effects with anxiolytic, antidepressant, and antiaggressive properties.
Neurosteroids are synthesized from cholesterol in the hippocampus. Neurosteroids are upregulated in response to stress by CNS cortisol effects on various enzyme expressions.47 Whether exogenous steroid administration affects this biosynthesis vs the stress response in the HPA axis itself is not fully elucidated. Monteleone and colleagues found that dexamethasone 1 mg given orally significantly reduced cortisol and DHEA and allopregnanolone levels in both healthy volunteers and anorexia nervosa patients.48 Similarly, Genazzani and colleagues demonstrated that oral dexamethasone administration (0.5 mg every 6 hours) caused significant reductions in both serum allopregnanolone and DHEA levels.49
Outcomes Studies
The majority of reported data in steroid-induced insomnia and psychosis is in noncritically ill populations. In a randomized, prospective crossover study of healthy volunteers, dexamethasone administration (3 mg every 8 hours for 48 hours) resulted in significant changes in sleep patterns measured with polysomnography. Compared with placebo, steroid treatment showed significantly longer percentage (SD) of stage 0/awake times (11.7% [11.4] vs 2.9% [1.8]; P < .05); longer percentage (SD) of REM sleep latency (363.8 [74.5] minutes vs 202.8 [79.6] minutes; P < .01), and a reduced number (SD) of REM periods (3.8 [2.6] vs 9.7 [3.6]; P < .01).50 Insomnia was one of the most commonly self-reported AEs (> 60%) in a survey of 2,446 chronic steroid users, and the incidence increased as steroid doses increased.51
A prospective, open-label study of 240 patients with cancer demonstrated significant sleep disruptions using the Pittsburgh Sleep Quality Index with the use of high-dose steroids in chemotherapy.52 Naber and colleagues evaluated 50 previously healthy patients taking methylprednisolone 119 mg (41 mg/d) for retinitis and uveitis.53 They reported 26% to 34% of subjects experienced hypomanic syndrome based on a semistructured interview examination. Symptoms developed within 3 days and persisted for the 8-day course of therapy. Brown and colleagues prospectively evaluated 32 asthmatic patients prescribed bursts of prednisone > 40 mg daily. They observed significantly increased scores in the Young Mania Rating Scale within 3 to 7 days of starting therapy, which dissipated to baseline after stopping therapy.54
Despite a high reported incidence of neurologic AEs, outcomes in critically ill populations are mixed. Study methods are varied, and many were largely observational. No prospective, randomized studies exist to date specifically aimed and powered to evaluate the effects of steroids on sleep disturbances or delirium in a critically ill population. Furthermore, sleep quality is difficult to measure in this population, and self-reporting often is not an option. In critical care trials, if AEs such as insomnia, delirium, or psychosis are recorded at all, there is heterogeneity in the definitions, and these AEs are generally poorly defined (eg, psychiatric or neurologic disorder not otherwise specified), making pooled analysis of this outcome difficult.55
One of the largest observational studies in hospitalized patients was through the Boston Collaborative Drug Surveillance Program. A total of 718 consecutively enrolled inpatients who received prednisone were monitored for acute reactions. Psychiatric AEs were rare (1.3%) with low doses (< 40 mg/d), more prevalent (4.6%) with higher doses (41-80 mg/d), and most prevalent (18.4%) with the highest doses (> 80 mg/d), suggesting CNS AEs are dose dependent.18 A single-center, retrospective review of 755 psychiatric consults in hospitalized patients revealed that 54% of manic patients were due to corticosteroid administration.19 In a prospective observational study of 206 consecutive ICU admissions, steroid administration was an independent risk factor for development of ICU delirium, using the Confusion Assessment Method-ICU (CAM-ICU) at a single center (odds ratio [OR], 2.8; 95% CI, 1.05-7.28).25
Two studies in hospitalized oncology patients found conflicting results using the Nursing Delirium Screening Scale (Nu-DESC). One did not find a significant association between delirium and dexamethasone equivalent doses > 15 mg, while the second found an increased hazard ratio (HR) for a positive Nu-DESC score (HR, 2.67; 95% CI, 1.18-6.03).20,21 Similarly, conflicting results were found in 2 studies using first-order Markov models. In one prospective cohort study, 520 consecutive mechanically ventilated patients in 13 ICUs were monitored for the transition to delirium (CAM-ICU positive) from nondelirium states. Steroid administration was significantly associated with transitioning to delirium (OR, 1.52; 95% CI, 1.05-2.21).22 This conflicts with a similar study by Wolters and colleagues, which monitored 1,112 ICU patients who were given a median prednisone equivalent of 50 mg (interquartile range, 25-75 mg). Steroid administration was not significantly associated with the transition to delirium from an awake without delirium state (OR, 1.08; 95% CI, 0.89-1.32; adjusted OR, 1.00; 95% CI, 0.99-1.01 per 10-mg increase in prednisone equivalent).23
Mitigating Effects
Although steroid therapy often cannot be altered in the critically ill population, research showed that steroid overuse is common in ICUs.56,57 Minimizing dosage and duration are important ways clinicians can mitigate unwanted effects. CNS AEs seen with steroids often can be reversed once therapy is discontinued. Avoiding split-dose administration has been proposed given the natural diurnal production of cortisol.58 A review by Flaherty discusses the importance of avoiding pharmacologic agents in hospitalized older patients if possible due to known risks (falls, dependency, hip fractures, rebound insomnia, and risk of delirium) and provides a HELP ME SLEEP nomogram for nonpharmacologic interventions in hospitalized patients (Table 4).59
Historically, lithium has been recommended for steroid-induced mania with chronic steroid use; however, given the large volume and electrolyte shifts seen in critically ill patients, this may not be a viable option. Antidepressants, especially tricyclics, should generally be avoided in steroid-induced psychosis as these may exacerbate symptoms. If symptoms are severe, either typical (haloperidol) or atypical (olanzapine, quetiapine, risperidone) antipsychotics have been used with success.60 Given the known depletion of serum melatonin levels, melatonin supplements are an attractive and relatively safe option for steroid-induced insomnia; however, there are no robust studies specifically aimed at this intervention for this population.
Conclusions
With known, multimodal foci driving sleep impairment in ICU patients, PADIS guidelines recommend myriad interventions for improvement. Recommendations include noise and light reduction with earplugs and/or eyeshades to improve sleep quality. Nocturnal assist-control ventilation may improve sleep quality in ventilated patients. Finally, the development of institutional protocols for promoting sleep quality in ICU patients is recommended.17
1. Simini B. Patients’ perceptions of intensive care. Lancet. 1999;354(9178):571-572. doi: 10.1016/S0140-6736(99)02728-2
2. Delaney LJ, Van Haren F, Lopez V. Sleeping on a problem: the impact of sleep disturbance on intensive care patients—a clinical review. Ann Intensive Care. 2015;15:3. doi: 10.1186/s13613-015-0043-2
3. Friese RS, Diaz-Arrastia R, McBride D, Frankel H, Gentilello LM. Quality and quantity of sleep in the surgical intensive care unit; are our patients sleeping? J Trauma. 2007;63(6):1210-1214. doi: 10.1097/TA.0b013e31815b83d7
4. Elliott R, McKinley S, Cistulli P, Fien M. Characterisation of sleep in intensive care using 24-hour polysomnography: an observational study. Crit Care 2013;17(2):R46.
5. Aurell J, Elmqvist D. Sleep in the surgical intensive care unit: continuous polygraphic recording of sleep in patients receiving postoperative care. BJM (Clin Res Ed). 1985;290(6474)1029-1032. doi: 10.1136/bmj.290.6474.1029
6. White DP, Douglas NJ, Pickett CK, Zwillich CW, Weil JV. Sleep deprivation and the control of ventilation. Am Rev Respir Dis. 1983;128(6):984-986. doi: 10.1164/arrd.1983.128.6.984
7. Series F, Roy N, Marc I. Effects of sleep deprivation and sleep fragmentation on upper airway collapsibility in normal subjects. Am J Respir Crit Care Med. 1994;150(2):481-485. doi: 10.1164/ajrccm.150.2.8049833
8. Tadjalli A, Peever J. Sleep loss reduces respiratory motor plasticity. Adv Exp Med Biol. 2010;669:289-292.
doi: 10.1007/978-1-4419-5692-7_59
9. Roche Campo F, Drouot X, Thille AW, et al. Poor sleep quality is associated with late noninvasive ventilation failure in patients with acute hypercapnic respiratory failure. Crit Care Med. 2010;38(2):447-485. doi: 10.1097/CCM.0b013e3181bc8243
10. Sauvet F, Leftheriotis G, Gomez-Merino D, et al. Effect of acute sleep deprivation on vascular function in healthy subjects. J Appl Physiol (1985). 2010;108(1):68-75. doi: 10.1152/japplphysiol.00851.2009
11. Frey DJ, Fleshner M, Wright KP Jr. The effects of 40 hours of total sleep deprivation on inflammatory markers in healthy young adults. Brain Behav Immun. 2007;21(8):1050-1057. doi: 10.1016/j.bbi.2007.04.003
12. Spiegel K, Sheridan JF, Van Cauter E. Effect of sleep deprivation on response to immunization. JAMA 2002;288(12):1471-1472. doi: 10.1001/jama.288.12.1471-a
13. Dinges DF, Douglas SD, Zuagg L, et al. Leukocytosis and natural killer cell function parallel neurobehavioral fatigue induced by 64 hours of sleep deprivation. J Clin Invest. 1994;93(5):1930-1939. doi: 10.1172/JCI117184
14. Weinhouse GL, Schwab RJ, Watson PL, et al. Bench-to-bedside review: delirium in ICU patients— importance of sleep deprivation. Crit Care. 2009;13(6):234. doi: 10.1186/cc8131
15. Ely EW, Shintani A, Truman B, et al. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA. 2004;291(14):1753-1762. doi: 10.1001/jama.291.14.1753
16. Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513-1520. doi: 10.1097/CCM.0b013e3181e47be1
17. Devlin JW, Skrobik Y, Gelinas C, et al. Clinical practice guidelines for the prevention and management of pain, agitation/sedation, delirium, immobility, and sleep disruption in adult patients in the ICU. Crit Care Med. 2018;46(9):e825-e873
18. The Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther. 1972;13(5):694-698. doi: 10.1002/cpt1972135part1694
19. Rundell JR, Wise MG. Causes of organic mood disorder. J Neuropsychiatry Clin Neurosci. 1989;1(4):398-400. doi: 10.1176/jnp.1.4.398
20. Gaudreau JD, Gagnon P, Harel F, Roy MA, Tremblay A. Psychoactive medications and risk of delirium in hospitalized cancer patients. J Clin Oncol. 2005;23(27):6712-6718. doi: 10.1200/JCO.2005.05.140
21. Gaudreau JD, Gagnon P, Roy MA, Harel F, Tremblay A. Opioid medications and longitudinal risk of delirium in hospitalized cancer patients. Cancer. 2007;109(11):2365-2373.
doi: 10.1002/cncr.22665
22. Schreiber MP, Colantuoni E, Bienvenu OJ, et al. Corticosteroids and transition to delirium in patients with acute lung injury. Crit Care Med. 2014;42(6):1480-1486. doi: 10.1097/CCM.0000000000000247
23. Wolters AE, Veldhuijzen DS, Zaal IJ, et al. Systemic corticosteroids and transition to delirium in critically ill patients. Crit Care Med. 2015;43(12):e585-e588. doi: 10.1097/CCM.0000000000001302
24. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naïve adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol. 2016;136(2):101-107. doi: 10.1159/000445420
25. Tilouche N, Hassen M, Ali HBS, Jaoued AHO, Gharbi R, Atrous SS. Delirium in the intensive care unit: incidence, risk factors, and impact on outcome. Indian J Crit Care Med. 2018;22:144-149. doi: 10.4103/ijccm.IJCCM_244_17
26. Young A, Marsh S. Steroid use in critical care. BJA Education. 2018;18(5):129-134. doi: 10.1016/j.bjae.2018.01.005
27. DiPiro J, Talbert R, Yee G, Matzke GR, Wells BG, Posey M. Pharmacotherapy: A Pathophysiologic Approach. 4th ed. New York: McGraw-Hill; 1999:1277-1278.
28. Schimmer
29. Sarnes E, Crofford L, Watson M, Dennis G, Kan H, Bass D. Incidence of US costs of corticosteroid-associated adverse events: a systematic literature review. Clin Ther. 2011;33(10):1413-1432.
30. Idzikowsi C, Shapiro CM. ABC of sleep disorders, non-psychotropic drugs and sleep. BMJ. 1993;306(6885):1118-1120. doi: 10.1136/bmj.306.6885.1118
31. Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14(4):398-406.
doi: 10.3109/10253890.2011.586446
32. Wolkowitz OM, Reus VI, Weingartner H, et al. Cognitive effects of corticosteroids. Am J Psychiatry 1990;147(10):1297-1303. doi: 10.1176/ajp.147.10.1297
33. McEwen BS, Davis PG, Parsons B, Pfaff DW. The brain as a target for steroid hormone action. Ann Rev Neurosci. 1979;2:65-112. doi: 10.1146/annurev.ne.02.030179.000433
34. Brown ES, Woolston DJ, Frol AM. Amygdala volume in patients receiving chronic corticosteroid therapy. Biol Psychiatry. 2008;63(7):705-709.
doi: 10.1016/j.biopsych.2007.09.014
35. Brown ES, Woolston D, Frol A, et al. Hippocampal volume, spectroscopy, cognition, and mood in patients receiving corticosteroid. Biol Psychiatry. 2004;55(5):538-545.
36. Sapolsky RM, McEwen BS. Down-regulation of neural corticosterone receptors by corticosterone and dexamethasone. Brain Res. 1985;339(1):161-165.
doi: 10.1016/0006-8993(85)90638-9
37. Sorrells SF, Caso JR, Munhoz CD, Spolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64(1):33-39.
doi: 10.1016/j.neuron.2009.09.032
38. Wolkowitz OM, Burke H, Epel ES, Reus VI. Glucocorticoids: mood, memory, and mechanisms. Ann NY Acad Sci. 2009;1179:19-40. doi: 10.1111/j.1749-6632.2009.04980.x
39. Wolkowitz OM, Epel ES, Reus VI. Stress hormone-related psychopathology: pathophysiological and treatment implications. World J Biol Psychiatry. 2001;2(3):115-143. doi: 10.3109/15622970109026799
40. Paredes S, Barriga C, Reiter R, Rodrigues A. Assessment of the potential role of tryptophan as the precursor of serotonin and melatonin for the aged sleep-wake cycle and immune function: Streptopelia Risoria as a model. Int J Tryptophan Res. 2009;2:23-36. doi: 10.4137/ijtr.s1129
41. Soszyński P, Stowińska-Srzednicka J, Kasperlik-Zatuska A, Zgliczyński S. Decreased melatonin concentration in Cushing’s Syndrome. Horm Metab Res. 1989;21(12):673-674. doi: 10.1055/s-2007-1009317
42. Demish L, Demish K, Neckelsen T. Influence of dexamethasone on nocturnal melatonin production in healthy adult subjects. J Pineal Res. 1988;5(3):317-321. doi: 10.1111/j.1600-079x.1988.tb00657.x
43. Assaf N, Shalby AB, Khalil WK, Ahmed HH. Biochemical and genetic alterations of oxidant/antioxidant status of the brain in rats treated with dexamethasone: protective roles of melatonin and acetyl-L-carnitine. J Physiol Biochem. 2012;68(1):77-90. doi: 10.1007/s13105-011-0121-3
44. Clark MS, Russo AF. Tissue-specific glucocorticoid regulation of tryptophan hydroxylase mRNA levels. Brain Res Mol Brain Res. 1997;48(2):346-54. doi: 10.1016/s0169-328x(97)00106-x
45. Kram DE, Krasnow SM, Levasseur PR, Zhu X, Stork LC, Marks DL. Dexamethasone chemotherapy does not disrupt orexin signaling. PLoS One. 2016;11(12):e0168731. doi: 10.1371/journal.pone.0168731
46. Mellon S. Neurosteroids: biochemistry, modes of action, and clinical relevance. J Clin Endocrinol Metab. 1994;78(5):1003-1008. doi: 10.1210/jcem.78.5.8175951
47. Zorumski C, Paul SM, Izumi Y, Covey DF, Mennerick S . Neurosteroids, stress and depression: potential therapeutic opportunities. Neurosci Biobehav Rev. 2013;37(1):109-122. doi: 10.1016/j.neubiorev.2012.10.005
48. Monteleone P, Luisi M, Martiadis V, et al. Impaired reduction of enhanced levels of dehydroepiandrosterone by oral dexamethasone in anorexia nervosa. Psychoneuroendocrinology. 2006;31(4):537-542. doi: 10.1016/j.psyneuen.2005.08.015
49. Genazzani AR, Petraglia F, Bernardi F, et al. Circulating levels of allopregnanolone in humans: gender, age, and endocrine influences. J Clin Endocrinol Metab. 1998;83(6):2099-3103. doi: 10.1210/jcem.83.6.4905
50. Moser NJ, Phillips BA, Guthrie G, Barnett G. Effects of dexamethasone on sleep. Pharmacol Toxicol. 1996;79(2):100-102. doi: 10.1111/j.1600-0773.1996.tb00249.x
51. Curtis J, Westfall A, Allison J, et al. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum. 2006;55(3):420-426. doi: 10.1002/art.21984
52. Zhao J, Dai YH, Xi QS, Yu SY. A clinical study on insomnia in patients with cancer during chemotherapy containing high-dose glucocorticoids. Pharmazie. 2013;68(6):421-427
53. Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days corticosteroid treatment. A prospective study. Psychoneuroendocrinology. 1996;21(1):25-31. doi: 10.1016/0306-4530(95)00031-3
54. Brown ES, Suppes T, Khan DA, Carmody TJ 3rd. Mood changes during prednisone bursts in outpatients with asthma. J Clin Psychopharmacol. 2002;22(1):55-61.
doi: 10.1097/00004714-200202000-00009
55. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367. doi: 10.4065/81.10.1361
56. Britt RC, Devine A, Swallen KC et al. Corticosteroid use in the intensive care unit: at what cost? Arch Surg. 2006;141(2):145-159. doi:10.1001/archsurg.141.2.145
57. Kiser TH, Allen RR, Valuck RJ, Moss M, Vanivier RW. Outcomes associated with corticosteroid dosage in critically ill patients in acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(9):1052-1064. doi: 10.1164/rccm.201401-0058OC
58. Bourne RS, Mills GH. Sleep disruption in critically ill patients—pharmacological considerations. Anaesthesia. 2004;59(4):374-384. doi: 10.1111/j. 1365-2044.2004.03664.x
59. Flaherty JH. Insomnia among hospitalized older persons. Clin Geriatr Med. 2008;24(1):51-67. doi: 10.1016/j.cger.2007.08.012
60. Sirios F. Steroid psychosis: a review. Gen Hosp Psychiatry. 2003;25(1):27-33. doi: 10.1016/s0163-8343(02)00241-4
1. Simini B. Patients’ perceptions of intensive care. Lancet. 1999;354(9178):571-572. doi: 10.1016/S0140-6736(99)02728-2
2. Delaney LJ, Van Haren F, Lopez V. Sleeping on a problem: the impact of sleep disturbance on intensive care patients—a clinical review. Ann Intensive Care. 2015;15:3. doi: 10.1186/s13613-015-0043-2
3. Friese RS, Diaz-Arrastia R, McBride D, Frankel H, Gentilello LM. Quality and quantity of sleep in the surgical intensive care unit; are our patients sleeping? J Trauma. 2007;63(6):1210-1214. doi: 10.1097/TA.0b013e31815b83d7
4. Elliott R, McKinley S, Cistulli P, Fien M. Characterisation of sleep in intensive care using 24-hour polysomnography: an observational study. Crit Care 2013;17(2):R46.
5. Aurell J, Elmqvist D. Sleep in the surgical intensive care unit: continuous polygraphic recording of sleep in patients receiving postoperative care. BJM (Clin Res Ed). 1985;290(6474)1029-1032. doi: 10.1136/bmj.290.6474.1029
6. White DP, Douglas NJ, Pickett CK, Zwillich CW, Weil JV. Sleep deprivation and the control of ventilation. Am Rev Respir Dis. 1983;128(6):984-986. doi: 10.1164/arrd.1983.128.6.984
7. Series F, Roy N, Marc I. Effects of sleep deprivation and sleep fragmentation on upper airway collapsibility in normal subjects. Am J Respir Crit Care Med. 1994;150(2):481-485. doi: 10.1164/ajrccm.150.2.8049833
8. Tadjalli A, Peever J. Sleep loss reduces respiratory motor plasticity. Adv Exp Med Biol. 2010;669:289-292.
doi: 10.1007/978-1-4419-5692-7_59
9. Roche Campo F, Drouot X, Thille AW, et al. Poor sleep quality is associated with late noninvasive ventilation failure in patients with acute hypercapnic respiratory failure. Crit Care Med. 2010;38(2):447-485. doi: 10.1097/CCM.0b013e3181bc8243
10. Sauvet F, Leftheriotis G, Gomez-Merino D, et al. Effect of acute sleep deprivation on vascular function in healthy subjects. J Appl Physiol (1985). 2010;108(1):68-75. doi: 10.1152/japplphysiol.00851.2009
11. Frey DJ, Fleshner M, Wright KP Jr. The effects of 40 hours of total sleep deprivation on inflammatory markers in healthy young adults. Brain Behav Immun. 2007;21(8):1050-1057. doi: 10.1016/j.bbi.2007.04.003
12. Spiegel K, Sheridan JF, Van Cauter E. Effect of sleep deprivation on response to immunization. JAMA 2002;288(12):1471-1472. doi: 10.1001/jama.288.12.1471-a
13. Dinges DF, Douglas SD, Zuagg L, et al. Leukocytosis and natural killer cell function parallel neurobehavioral fatigue induced by 64 hours of sleep deprivation. J Clin Invest. 1994;93(5):1930-1939. doi: 10.1172/JCI117184
14. Weinhouse GL, Schwab RJ, Watson PL, et al. Bench-to-bedside review: delirium in ICU patients— importance of sleep deprivation. Crit Care. 2009;13(6):234. doi: 10.1186/cc8131
15. Ely EW, Shintani A, Truman B, et al. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA. 2004;291(14):1753-1762. doi: 10.1001/jama.291.14.1753
16. Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513-1520. doi: 10.1097/CCM.0b013e3181e47be1
17. Devlin JW, Skrobik Y, Gelinas C, et al. Clinical practice guidelines for the prevention and management of pain, agitation/sedation, delirium, immobility, and sleep disruption in adult patients in the ICU. Crit Care Med. 2018;46(9):e825-e873
18. The Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther. 1972;13(5):694-698. doi: 10.1002/cpt1972135part1694
19. Rundell JR, Wise MG. Causes of organic mood disorder. J Neuropsychiatry Clin Neurosci. 1989;1(4):398-400. doi: 10.1176/jnp.1.4.398
20. Gaudreau JD, Gagnon P, Harel F, Roy MA, Tremblay A. Psychoactive medications and risk of delirium in hospitalized cancer patients. J Clin Oncol. 2005;23(27):6712-6718. doi: 10.1200/JCO.2005.05.140
21. Gaudreau JD, Gagnon P, Roy MA, Harel F, Tremblay A. Opioid medications and longitudinal risk of delirium in hospitalized cancer patients. Cancer. 2007;109(11):2365-2373.
doi: 10.1002/cncr.22665
22. Schreiber MP, Colantuoni E, Bienvenu OJ, et al. Corticosteroids and transition to delirium in patients with acute lung injury. Crit Care Med. 2014;42(6):1480-1486. doi: 10.1097/CCM.0000000000000247
23. Wolters AE, Veldhuijzen DS, Zaal IJ, et al. Systemic corticosteroids and transition to delirium in critically ill patients. Crit Care Med. 2015;43(12):e585-e588. doi: 10.1097/CCM.0000000000001302
24. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naïve adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol. 2016;136(2):101-107. doi: 10.1159/000445420
25. Tilouche N, Hassen M, Ali HBS, Jaoued AHO, Gharbi R, Atrous SS. Delirium in the intensive care unit: incidence, risk factors, and impact on outcome. Indian J Crit Care Med. 2018;22:144-149. doi: 10.4103/ijccm.IJCCM_244_17
26. Young A, Marsh S. Steroid use in critical care. BJA Education. 2018;18(5):129-134. doi: 10.1016/j.bjae.2018.01.005
27. DiPiro J, Talbert R, Yee G, Matzke GR, Wells BG, Posey M. Pharmacotherapy: A Pathophysiologic Approach. 4th ed. New York: McGraw-Hill; 1999:1277-1278.
28. Schimmer
29. Sarnes E, Crofford L, Watson M, Dennis G, Kan H, Bass D. Incidence of US costs of corticosteroid-associated adverse events: a systematic literature review. Clin Ther. 2011;33(10):1413-1432.
30. Idzikowsi C, Shapiro CM. ABC of sleep disorders, non-psychotropic drugs and sleep. BMJ. 1993;306(6885):1118-1120. doi: 10.1136/bmj.306.6885.1118
31. Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14(4):398-406.
doi: 10.3109/10253890.2011.586446
32. Wolkowitz OM, Reus VI, Weingartner H, et al. Cognitive effects of corticosteroids. Am J Psychiatry 1990;147(10):1297-1303. doi: 10.1176/ajp.147.10.1297
33. McEwen BS, Davis PG, Parsons B, Pfaff DW. The brain as a target for steroid hormone action. Ann Rev Neurosci. 1979;2:65-112. doi: 10.1146/annurev.ne.02.030179.000433
34. Brown ES, Woolston DJ, Frol AM. Amygdala volume in patients receiving chronic corticosteroid therapy. Biol Psychiatry. 2008;63(7):705-709.
doi: 10.1016/j.biopsych.2007.09.014
35. Brown ES, Woolston D, Frol A, et al. Hippocampal volume, spectroscopy, cognition, and mood in patients receiving corticosteroid. Biol Psychiatry. 2004;55(5):538-545.
36. Sapolsky RM, McEwen BS. Down-regulation of neural corticosterone receptors by corticosterone and dexamethasone. Brain Res. 1985;339(1):161-165.
doi: 10.1016/0006-8993(85)90638-9
37. Sorrells SF, Caso JR, Munhoz CD, Spolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64(1):33-39.
doi: 10.1016/j.neuron.2009.09.032
38. Wolkowitz OM, Burke H, Epel ES, Reus VI. Glucocorticoids: mood, memory, and mechanisms. Ann NY Acad Sci. 2009;1179:19-40. doi: 10.1111/j.1749-6632.2009.04980.x
39. Wolkowitz OM, Epel ES, Reus VI. Stress hormone-related psychopathology: pathophysiological and treatment implications. World J Biol Psychiatry. 2001;2(3):115-143. doi: 10.3109/15622970109026799
40. Paredes S, Barriga C, Reiter R, Rodrigues A. Assessment of the potential role of tryptophan as the precursor of serotonin and melatonin for the aged sleep-wake cycle and immune function: Streptopelia Risoria as a model. Int J Tryptophan Res. 2009;2:23-36. doi: 10.4137/ijtr.s1129
41. Soszyński P, Stowińska-Srzednicka J, Kasperlik-Zatuska A, Zgliczyński S. Decreased melatonin concentration in Cushing’s Syndrome. Horm Metab Res. 1989;21(12):673-674. doi: 10.1055/s-2007-1009317
42. Demish L, Demish K, Neckelsen T. Influence of dexamethasone on nocturnal melatonin production in healthy adult subjects. J Pineal Res. 1988;5(3):317-321. doi: 10.1111/j.1600-079x.1988.tb00657.x
43. Assaf N, Shalby AB, Khalil WK, Ahmed HH. Biochemical and genetic alterations of oxidant/antioxidant status of the brain in rats treated with dexamethasone: protective roles of melatonin and acetyl-L-carnitine. J Physiol Biochem. 2012;68(1):77-90. doi: 10.1007/s13105-011-0121-3
44. Clark MS, Russo AF. Tissue-specific glucocorticoid regulation of tryptophan hydroxylase mRNA levels. Brain Res Mol Brain Res. 1997;48(2):346-54. doi: 10.1016/s0169-328x(97)00106-x
45. Kram DE, Krasnow SM, Levasseur PR, Zhu X, Stork LC, Marks DL. Dexamethasone chemotherapy does not disrupt orexin signaling. PLoS One. 2016;11(12):e0168731. doi: 10.1371/journal.pone.0168731
46. Mellon S. Neurosteroids: biochemistry, modes of action, and clinical relevance. J Clin Endocrinol Metab. 1994;78(5):1003-1008. doi: 10.1210/jcem.78.5.8175951
47. Zorumski C, Paul SM, Izumi Y, Covey DF, Mennerick S . Neurosteroids, stress and depression: potential therapeutic opportunities. Neurosci Biobehav Rev. 2013;37(1):109-122. doi: 10.1016/j.neubiorev.2012.10.005
48. Monteleone P, Luisi M, Martiadis V, et al. Impaired reduction of enhanced levels of dehydroepiandrosterone by oral dexamethasone in anorexia nervosa. Psychoneuroendocrinology. 2006;31(4):537-542. doi: 10.1016/j.psyneuen.2005.08.015
49. Genazzani AR, Petraglia F, Bernardi F, et al. Circulating levels of allopregnanolone in humans: gender, age, and endocrine influences. J Clin Endocrinol Metab. 1998;83(6):2099-3103. doi: 10.1210/jcem.83.6.4905
50. Moser NJ, Phillips BA, Guthrie G, Barnett G. Effects of dexamethasone on sleep. Pharmacol Toxicol. 1996;79(2):100-102. doi: 10.1111/j.1600-0773.1996.tb00249.x
51. Curtis J, Westfall A, Allison J, et al. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum. 2006;55(3):420-426. doi: 10.1002/art.21984
52. Zhao J, Dai YH, Xi QS, Yu SY. A clinical study on insomnia in patients with cancer during chemotherapy containing high-dose glucocorticoids. Pharmazie. 2013;68(6):421-427
53. Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days corticosteroid treatment. A prospective study. Psychoneuroendocrinology. 1996;21(1):25-31. doi: 10.1016/0306-4530(95)00031-3
54. Brown ES, Suppes T, Khan DA, Carmody TJ 3rd. Mood changes during prednisone bursts in outpatients with asthma. J Clin Psychopharmacol. 2002;22(1):55-61.
doi: 10.1097/00004714-200202000-00009
55. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367. doi: 10.4065/81.10.1361
56. Britt RC, Devine A, Swallen KC et al. Corticosteroid use in the intensive care unit: at what cost? Arch Surg. 2006;141(2):145-159. doi:10.1001/archsurg.141.2.145
57. Kiser TH, Allen RR, Valuck RJ, Moss M, Vanivier RW. Outcomes associated with corticosteroid dosage in critically ill patients in acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(9):1052-1064. doi: 10.1164/rccm.201401-0058OC
58. Bourne RS, Mills GH. Sleep disruption in critically ill patients—pharmacological considerations. Anaesthesia. 2004;59(4):374-384. doi: 10.1111/j. 1365-2044.2004.03664.x
59. Flaherty JH. Insomnia among hospitalized older persons. Clin Geriatr Med. 2008;24(1):51-67. doi: 10.1016/j.cger.2007.08.012
60. Sirios F. Steroid psychosis: a review. Gen Hosp Psychiatry. 2003;25(1):27-33. doi: 10.1016/s0163-8343(02)00241-4
TNF inhibitor plus methotrexate surpassed methotrexate monotherapy in PsA
Adding a tumor necrosis factor inhibitor to the treatment regimen of patients with psoriatic arthritis who failed to reach minimal disease activity on methotrexate monotherapy after 4 or more weeks had more than triple the rate of minimal disease activity after 16 weeks, compared with patients who had their methotrexate dosage escalated but received no second drug, in a multicenter, randomized study with 245 patients.

After 16 weeks, 42% of 123 patients with psoriatic arthritis (PsA) treated with methotrexate and the tumor necrosis factor (TNF) inhibitor adalimumab achieved minimal disease activity, compared with 13% of 122 patients randomized to receive escalated methotrexate monotherapy to their maximally tolerated dosage or to a maximum of 25 mg/week, Laura C. Coates, MBChB, PhD, reported at the annual European Congress of Rheumatology, held online this year due to COVID-19.
The findings are “supportive of the EULAR recommendations” for managing patients with PsA, said Dr. Coates, a rheumatologist at the University of Oxford (England). The EULAR recommendations call for starting a biologic disease-modifying antirheumatic drug (bDMARD) in patients with PsA and peripheral arthritis and “inadequate response to at least one [conventional synthetic] DMARD,” such as methotrexate (Ann Rheum Dis. 2019 Jun;79[6]:700-12). “A proportion of patients treated with methotrexate do well, but for those struggling on methotrexate, these results support use of a TNF inhibitor. It’s a balance of cost and benefit. If TNF inhibitors were as cheap as methotrexate, I suspect that would be first line more frequently,” Dr. Coates said in an interview. In contrast, the PsA management recommendations from the American College of Rheumatology make treatment with a TNF inhibitor first line, before starting with what these guidelines call an oral small molecule, the same as a conventional synthetic DMARD such as methotrexate (Arthritis Rheumatol. 2019 Jan;71[1]:5-32).

“It’s a well-known fact that adalimumab is more effective than methotrexate in [PsA] patients who do not respond sufficiently well to methotrexate. Patients failing on methotrexate have been escalated to a TNF inhibitor for years,” commented Robert B.M. Landewé, MD, a rheumatologist and professor of medicine at the University of Amsterdam, and a coauthor of the EULAR PsA treatment recommendations. “In the Netherlands and in my practice, every [PsA] patient starts on methotrexate until a dosage of at least 15 mg/week, but if they don’t have sufficient response we escalate to adding a TNF inhibitor,” he said in an interview. “A significant proportion of patients with PsA respond well to moderate to higher dosages of methotrexate,” and this monotherapy with escalation of methotrexate can be safely continued for more than 3 months in many patients without the risk of “losing too much time by waiting” to start a bDMARD.
Dr. Coates said that her practice was to look for some level of response to methotrexate by 12 weeks on treatment and for achievement of minimal disease activity within 24 weeks of treatment. If these targets are not reached, she then adds a TNF inhibitor.
The CONTROL study ran at 60 sites in the United States and in 12 other countries and enrolled patients with active PsA despite treatment with methotrexate for at least 4 weeks and no history of treatment with a bDMARD. Patients received either 40 mg adalimumab every other week plus 15 mg of methotrexate weekly, or maximum-tolerated methotrexate up to 25 mg/week. The results also showed that the primary endpoint of the rate of achieved minimal disease activity seen overall in each of the two study arms was consistent in both the roughly half of patients who had been on methotrexate monotherapy for 3 months or less before entering the study as well as those who had been on initial methotrexate monotherapy for a longer period. Other secondary endpoints examined also showed significantly better responses to adding adalimumab, including a tripling of the rate at which patients achieved complete resolution of their Psoriasis Area and Severity Index score, which occurred in 30% of patients on the TNF inhibitor plus methotrexate and in 9% of those on methotrexate monotherapy.
The results seen in the CONTROL study with adalimumab would likely be similar using a different TNF inhibitor or an agent that’s an adalimumab biosimilar, Dr. Coates said. The only patients with PsA and not achieving minimal disease activity on methotrexate monotherapy who should not then receive add-on treatment with a TNF inhibitor are those known to have a safety exclusion for this drug class or patients for whom the incremental cost poses a barrier, she added. In addition, patients with more substantial skin involvement may get greater benefit from a different class of bDMARD, such as a drug that inhibits interleukin-17 or IL-12 and -23 as recommended by the EULAR panel.
“We still get very good results with a TNF inhibitor for psoriasis, but in patients with severe psoriasis there is an argument to use a different drug,” Dr. Coates acknowledged. Skin responses with an IL-17 inhibitor or an IL-12/23 inhibitor “are far better” than with a TNF inhibitor, said Dr. Landewé. He also added the caution that longer-term use of adalimumab “may induce aggravation of PsA in a significant number of patients.”
CONTROL was sponsored by AbbVie, the company that markets adalimumab (Humira). Dr. Coates has been a consultant to AbbVie, as well as to Amgen, Biogen, Boehringer Ingelheim, Celgene, Jansen, Novartis, Pfizer, and UCB. Dr. Landewé has been a consultant to AbbVie, as well as to Eli Lilly, Novartis, Pfizer, and UCB.
SOURCE: Coates LC et al. Ann Rheum Dis. 2020 Jun;79[suppl 1]:33, Abstract OP0050.
Adding a tumor necrosis factor inhibitor to the treatment regimen of patients with psoriatic arthritis who failed to reach minimal disease activity on methotrexate monotherapy after 4 or more weeks had more than triple the rate of minimal disease activity after 16 weeks, compared with patients who had their methotrexate dosage escalated but received no second drug, in a multicenter, randomized study with 245 patients.
After 16 weeks, 42% of 123 patients with psoriatic arthritis (PsA) treated with methotrexate and the tumor necrosis factor (TNF) inhibitor adalimumab achieved minimal disease activity, compared with 13% of 122 patients randomized to receive escalated methotrexate monotherapy to their maximally tolerated dosage or to a maximum of 25 mg/week, Laura C. Coates, MBChB, PhD, reported at the annual European Congress of Rheumatology, held online this year due to COVID-19.
The findings are “supportive of the EULAR recommendations” for managing patients with PsA, said Dr. Coates, a rheumatologist at the University of Oxford (England). The EULAR recommendations call for starting a biologic disease-modifying antirheumatic drug (bDMARD) in patients with PsA and peripheral arthritis and “inadequate response to at least one [conventional synthetic] DMARD,” such as methotrexate (Ann Rheum Dis. 2019 Jun;79[6]:700-12). “A proportion of patients treated with methotrexate do well, but for those struggling on methotrexate, these results support use of a TNF inhibitor. It’s a balance of cost and benefit. If TNF inhibitors were as cheap as methotrexate, I suspect that would be first line more frequently,” Dr. Coates said in an interview. In contrast, the PsA management recommendations from the American College of Rheumatology make treatment with a TNF inhibitor first line, before starting with what these guidelines call an oral small molecule, the same as a conventional synthetic DMARD such as methotrexate (Arthritis Rheumatol. 2019 Jan;71[1]:5-32).
“It’s a well-known fact that adalimumab is more effective than methotrexate in [PsA] patients who do not respond sufficiently well to methotrexate. Patients failing on methotrexate have been escalated to a TNF inhibitor for years,” commented Robert B.M. Landewé, MD, a rheumatologist and professor of medicine at the University of Amsterdam, and a coauthor of the EULAR PsA treatment recommendations. “In the Netherlands and in my practice, every [PsA] patient starts on methotrexate until a dosage of at least 15 mg/week, but if they don’t have sufficient response we escalate to adding a TNF inhibitor,” he said in an interview. “A significant proportion of patients with PsA respond well to moderate to higher dosages of methotrexate,” and this monotherapy with escalation of methotrexate can be safely continued for more than 3 months in many patients without the risk of “losing too much time by waiting” to start a bDMARD.
Dr. Coates said that her practice was to look for some level of response to methotrexate by 12 weeks on treatment and for achievement of minimal disease activity within 24 weeks of treatment. If these targets are not reached, she then adds a TNF inhibitor.
The CONTROL study ran at 60 sites in the United States and in 12 other countries and enrolled patients with active PsA despite treatment with methotrexate for at least 4 weeks and no history of treatment with a bDMARD. Patients received either 40 mg adalimumab every other week plus 15 mg of methotrexate weekly, or maximum-tolerated methotrexate up to 25 mg/week. The results also showed that the primary endpoint of the rate of achieved minimal disease activity seen overall in each of the two study arms was consistent in both the roughly half of patients who had been on methotrexate monotherapy for 3 months or less before entering the study as well as those who had been on initial methotrexate monotherapy for a longer period. Other secondary endpoints examined also showed significantly better responses to adding adalimumab, including a tripling of the rate at which patients achieved complete resolution of their Psoriasis Area and Severity Index score, which occurred in 30% of patients on the TNF inhibitor plus methotrexate and in 9% of those on methotrexate monotherapy.
The results seen in the CONTROL study with adalimumab would likely be similar using a different TNF inhibitor or an agent that’s an adalimumab biosimilar, Dr. Coates said. The only patients with PsA and not achieving minimal disease activity on methotrexate monotherapy who should not then receive add-on treatment with a TNF inhibitor are those known to have a safety exclusion for this drug class or patients for whom the incremental cost poses a barrier, she added. In addition, patients with more substantial skin involvement may get greater benefit from a different class of bDMARD, such as a drug that inhibits interleukin-17 or IL-12 and -23 as recommended by the EULAR panel.
“We still get very good results with a TNF inhibitor for psoriasis, but in patients with severe psoriasis there is an argument to use a different drug,” Dr. Coates acknowledged. Skin responses with an IL-17 inhibitor or an IL-12/23 inhibitor “are far better” than with a TNF inhibitor, said Dr. Landewé. He also added the caution that longer-term use of adalimumab “may induce aggravation of PsA in a significant number of patients.”
CONTROL was sponsored by AbbVie, the company that markets adalimumab (Humira). Dr. Coates has been a consultant to AbbVie, as well as to Amgen, Biogen, Boehringer Ingelheim, Celgene, Jansen, Novartis, Pfizer, and UCB. Dr. Landewé has been a consultant to AbbVie, as well as to Eli Lilly, Novartis, Pfizer, and UCB.
SOURCE: Coates LC et al. Ann Rheum Dis. 2020 Jun;79[suppl 1]:33, Abstract OP0050.
Adding a tumor necrosis factor inhibitor to the treatment regimen of patients with psoriatic arthritis who failed to reach minimal disease activity on methotrexate monotherapy after 4 or more weeks had more than triple the rate of minimal disease activity after 16 weeks, compared with patients who had their methotrexate dosage escalated but received no second drug, in a multicenter, randomized study with 245 patients.
After 16 weeks, 42% of 123 patients with psoriatic arthritis (PsA) treated with methotrexate and the tumor necrosis factor (TNF) inhibitor adalimumab achieved minimal disease activity, compared with 13% of 122 patients randomized to receive escalated methotrexate monotherapy to their maximally tolerated dosage or to a maximum of 25 mg/week, Laura C. Coates, MBChB, PhD, reported at the annual European Congress of Rheumatology, held online this year due to COVID-19.
The findings are “supportive of the EULAR recommendations” for managing patients with PsA, said Dr. Coates, a rheumatologist at the University of Oxford (England). The EULAR recommendations call for starting a biologic disease-modifying antirheumatic drug (bDMARD) in patients with PsA and peripheral arthritis and “inadequate response to at least one [conventional synthetic] DMARD,” such as methotrexate (Ann Rheum Dis. 2019 Jun;79[6]:700-12). “A proportion of patients treated with methotrexate do well, but for those struggling on methotrexate, these results support use of a TNF inhibitor. It’s a balance of cost and benefit. If TNF inhibitors were as cheap as methotrexate, I suspect that would be first line more frequently,” Dr. Coates said in an interview. In contrast, the PsA management recommendations from the American College of Rheumatology make treatment with a TNF inhibitor first line, before starting with what these guidelines call an oral small molecule, the same as a conventional synthetic DMARD such as methotrexate (Arthritis Rheumatol. 2019 Jan;71[1]:5-32).
“It’s a well-known fact that adalimumab is more effective than methotrexate in [PsA] patients who do not respond sufficiently well to methotrexate. Patients failing on methotrexate have been escalated to a TNF inhibitor for years,” commented Robert B.M. Landewé, MD, a rheumatologist and professor of medicine at the University of Amsterdam, and a coauthor of the EULAR PsA treatment recommendations. “In the Netherlands and in my practice, every [PsA] patient starts on methotrexate until a dosage of at least 15 mg/week, but if they don’t have sufficient response we escalate to adding a TNF inhibitor,” he said in an interview. “A significant proportion of patients with PsA respond well to moderate to higher dosages of methotrexate,” and this monotherapy with escalation of methotrexate can be safely continued for more than 3 months in many patients without the risk of “losing too much time by waiting” to start a bDMARD.
Dr. Coates said that her practice was to look for some level of response to methotrexate by 12 weeks on treatment and for achievement of minimal disease activity within 24 weeks of treatment. If these targets are not reached, she then adds a TNF inhibitor.
The CONTROL study ran at 60 sites in the United States and in 12 other countries and enrolled patients with active PsA despite treatment with methotrexate for at least 4 weeks and no history of treatment with a bDMARD. Patients received either 40 mg adalimumab every other week plus 15 mg of methotrexate weekly, or maximum-tolerated methotrexate up to 25 mg/week. The results also showed that the primary endpoint of the rate of achieved minimal disease activity seen overall in each of the two study arms was consistent in both the roughly half of patients who had been on methotrexate monotherapy for 3 months or less before entering the study as well as those who had been on initial methotrexate monotherapy for a longer period. Other secondary endpoints examined also showed significantly better responses to adding adalimumab, including a tripling of the rate at which patients achieved complete resolution of their Psoriasis Area and Severity Index score, which occurred in 30% of patients on the TNF inhibitor plus methotrexate and in 9% of those on methotrexate monotherapy.
The results seen in the CONTROL study with adalimumab would likely be similar using a different TNF inhibitor or an agent that’s an adalimumab biosimilar, Dr. Coates said. The only patients with PsA and not achieving minimal disease activity on methotrexate monotherapy who should not then receive add-on treatment with a TNF inhibitor are those known to have a safety exclusion for this drug class or patients for whom the incremental cost poses a barrier, she added. In addition, patients with more substantial skin involvement may get greater benefit from a different class of bDMARD, such as a drug that inhibits interleukin-17 or IL-12 and -23 as recommended by the EULAR panel.
“We still get very good results with a TNF inhibitor for psoriasis, but in patients with severe psoriasis there is an argument to use a different drug,” Dr. Coates acknowledged. Skin responses with an IL-17 inhibitor or an IL-12/23 inhibitor “are far better” than with a TNF inhibitor, said Dr. Landewé. He also added the caution that longer-term use of adalimumab “may induce aggravation of PsA in a significant number of patients.”
CONTROL was sponsored by AbbVie, the company that markets adalimumab (Humira). Dr. Coates has been a consultant to AbbVie, as well as to Amgen, Biogen, Boehringer Ingelheim, Celgene, Jansen, Novartis, Pfizer, and UCB. Dr. Landewé has been a consultant to AbbVie, as well as to Eli Lilly, Novartis, Pfizer, and UCB.
SOURCE: Coates LC et al. Ann Rheum Dis. 2020 Jun;79[suppl 1]:33, Abstract OP0050.
FROM EULAR 2020 E-CONGRESS
FDA approves new antibiotic for HABP/VABP treatment
in people aged 18 years and older.

Approval for Recarbrio was based on results of a randomized, controlled clinical trial of 535 hospitalized adults with hospital-acquired and ventilator-associated bacterial pneumonia who received either Recarbrio or piperacillin-tazobactam. After 28 days, 16% of patients who received Recarbrio and 21% of patients who received piperacillin-tazobactam had died.
The most common adverse events associated with Recarbrio are increased alanine aminotransferase/ aspartate aminotransferase, anemia, diarrhea, hypokalemia, and hyponatremia. Recarbrio was previously approved by the FDA to treat patients with complicated urinary tract infections and complicated intra-abdominal infections who have limited or no alternative treatment options, according to an FDA press release.
“As a public health agency, the FDA addresses the threat of antimicrobial-resistant infections by facilitating the development of safe and effective new treatments. These efforts provide more options to fight serious bacterial infections and get new, safe and effective therapies to patients as soon as possible,” said Sumathi Nambiar, MD, MPH, director of the division of anti-infectives within the office of infectious disease at the Center for Drug Evaluation and Research.
in people aged 18 years and older.
Approval for Recarbrio was based on results of a randomized, controlled clinical trial of 535 hospitalized adults with hospital-acquired and ventilator-associated bacterial pneumonia who received either Recarbrio or piperacillin-tazobactam. After 28 days, 16% of patients who received Recarbrio and 21% of patients who received piperacillin-tazobactam had died.
The most common adverse events associated with Recarbrio are increased alanine aminotransferase/ aspartate aminotransferase, anemia, diarrhea, hypokalemia, and hyponatremia. Recarbrio was previously approved by the FDA to treat patients with complicated urinary tract infections and complicated intra-abdominal infections who have limited or no alternative treatment options, according to an FDA press release.
“As a public health agency, the FDA addresses the threat of antimicrobial-resistant infections by facilitating the development of safe and effective new treatments. These efforts provide more options to fight serious bacterial infections and get new, safe and effective therapies to patients as soon as possible,” said Sumathi Nambiar, MD, MPH, director of the division of anti-infectives within the office of infectious disease at the Center for Drug Evaluation and Research.
in people aged 18 years and older.
Approval for Recarbrio was based on results of a randomized, controlled clinical trial of 535 hospitalized adults with hospital-acquired and ventilator-associated bacterial pneumonia who received either Recarbrio or piperacillin-tazobactam. After 28 days, 16% of patients who received Recarbrio and 21% of patients who received piperacillin-tazobactam had died.
The most common adverse events associated with Recarbrio are increased alanine aminotransferase/ aspartate aminotransferase, anemia, diarrhea, hypokalemia, and hyponatremia. Recarbrio was previously approved by the FDA to treat patients with complicated urinary tract infections and complicated intra-abdominal infections who have limited or no alternative treatment options, according to an FDA press release.
“As a public health agency, the FDA addresses the threat of antimicrobial-resistant infections by facilitating the development of safe and effective new treatments. These efforts provide more options to fight serious bacterial infections and get new, safe and effective therapies to patients as soon as possible,” said Sumathi Nambiar, MD, MPH, director of the division of anti-infectives within the office of infectious disease at the Center for Drug Evaluation and Research.
Lancet, NEJM retract studies on hydroxychloroquine for COVID-19
The Lancet announced today that it has retracted a highly cited study that suggested hydroxychloroquine may cause more harm than benefit in patients with COVID-19. Hours later, the New England Journal of Medicine announced that it had retracted a second article by some of the same authors, also on heart disease and COVID-19.
The Lancet article, titled “Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: A multinational registry analysis” was originally published online May 22. The NEJM article, “Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19” was initially published May 1.
Three authors of the Lancet article, Mandeep R. Mehra, MD, Frank Ruschitzka, MD, and Amit N. Patel, MD, wrote in a letter that the action came after concerns were raised about the integrity of the data, and about how the analysis was conducted by Chicago-based Surgisphere Corp and study coauthor Sapan Desai, MD, Surgisphere’s founder and CEO.
The authors asked for an independent third-party review of Surgisphere to evaluate the integrity of the trial elements and to replicate the analyses in the article.
“Our independent peer reviewers informed us that Surgisphere would not transfer the full dataset, client contracts, and the full ISO audit report to their servers for analysis, as such transfer would violate client agreements and confidentiality requirements,” the authors wrote.
Therefore, reviewers were not able to conduct the review and notified the authors they would withdraw from the peer-review process.
The Lancet said in a statement: “The Lancet takes issues of scientific integrity extremely seriously, and there are many outstanding questions about Surgisphere and the data that were allegedly included in this study. Following guidelines from the Committee on Publication Ethics and International Committee of Medical Journal Editors, institutional reviews of Surgisphere’s research collaborations are urgently needed.”
The authors wrote, “We can never forget the responsibility we have as researchers to scrupulously ensure that we rely on data sources that adhere to our high standards. Based on this development, we can no longer vouch for the veracity of the primary data sources. Due to this unfortunate development, the authors request that the paper be retracted.
“We all entered this collaboration to contribute in good faith and at a time of great need during the COVID-19 pandemic. We deeply apologize to you, the editors, and the journal readership for any embarrassment or inconvenience that this may have caused.”
In a similar, if briefer, note, the authors requested that the New England Journal of Medicine retract the earlier article as well. The retraction notice on the website reads: “Because all the authors were not granted access to the raw data and the raw data could not be made available to a third-party auditor, we are unable to validate the primary data sources underlying our article, ‘Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19.’ We therefore request that the article be retracted. We apologize to the editors and to readers of the Journal for the difficulties that this has caused.”
Both journals had already published “Expression of Concern” notices about the articles. The expression of concern followed an open letter, endorsed by more than 200 scientists, ethicists, and clinicians and posted on May 28, questioning the data and ethics of the study.
A version of this article originally appeared on Medscape.com.
The Lancet announced today that it has retracted a highly cited study that suggested hydroxychloroquine may cause more harm than benefit in patients with COVID-19. Hours later, the New England Journal of Medicine announced that it had retracted a second article by some of the same authors, also on heart disease and COVID-19.
The Lancet article, titled “Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: A multinational registry analysis” was originally published online May 22. The NEJM article, “Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19” was initially published May 1.
Three authors of the Lancet article, Mandeep R. Mehra, MD, Frank Ruschitzka, MD, and Amit N. Patel, MD, wrote in a letter that the action came after concerns were raised about the integrity of the data, and about how the analysis was conducted by Chicago-based Surgisphere Corp and study coauthor Sapan Desai, MD, Surgisphere’s founder and CEO.
The authors asked for an independent third-party review of Surgisphere to evaluate the integrity of the trial elements and to replicate the analyses in the article.
“Our independent peer reviewers informed us that Surgisphere would not transfer the full dataset, client contracts, and the full ISO audit report to their servers for analysis, as such transfer would violate client agreements and confidentiality requirements,” the authors wrote.
Therefore, reviewers were not able to conduct the review and notified the authors they would withdraw from the peer-review process.
The Lancet said in a statement: “The Lancet takes issues of scientific integrity extremely seriously, and there are many outstanding questions about Surgisphere and the data that were allegedly included in this study. Following guidelines from the Committee on Publication Ethics and International Committee of Medical Journal Editors, institutional reviews of Surgisphere’s research collaborations are urgently needed.”
The authors wrote, “We can never forget the responsibility we have as researchers to scrupulously ensure that we rely on data sources that adhere to our high standards. Based on this development, we can no longer vouch for the veracity of the primary data sources. Due to this unfortunate development, the authors request that the paper be retracted.
“We all entered this collaboration to contribute in good faith and at a time of great need during the COVID-19 pandemic. We deeply apologize to you, the editors, and the journal readership for any embarrassment or inconvenience that this may have caused.”
In a similar, if briefer, note, the authors requested that the New England Journal of Medicine retract the earlier article as well. The retraction notice on the website reads: “Because all the authors were not granted access to the raw data and the raw data could not be made available to a third-party auditor, we are unable to validate the primary data sources underlying our article, ‘Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19.’ We therefore request that the article be retracted. We apologize to the editors and to readers of the Journal for the difficulties that this has caused.”
Both journals had already published “Expression of Concern” notices about the articles. The expression of concern followed an open letter, endorsed by more than 200 scientists, ethicists, and clinicians and posted on May 28, questioning the data and ethics of the study.
A version of this article originally appeared on Medscape.com.
The Lancet announced today that it has retracted a highly cited study that suggested hydroxychloroquine may cause more harm than benefit in patients with COVID-19. Hours later, the New England Journal of Medicine announced that it had retracted a second article by some of the same authors, also on heart disease and COVID-19.
The Lancet article, titled “Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: A multinational registry analysis” was originally published online May 22. The NEJM article, “Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19” was initially published May 1.
Three authors of the Lancet article, Mandeep R. Mehra, MD, Frank Ruschitzka, MD, and Amit N. Patel, MD, wrote in a letter that the action came after concerns were raised about the integrity of the data, and about how the analysis was conducted by Chicago-based Surgisphere Corp and study coauthor Sapan Desai, MD, Surgisphere’s founder and CEO.
The authors asked for an independent third-party review of Surgisphere to evaluate the integrity of the trial elements and to replicate the analyses in the article.
“Our independent peer reviewers informed us that Surgisphere would not transfer the full dataset, client contracts, and the full ISO audit report to their servers for analysis, as such transfer would violate client agreements and confidentiality requirements,” the authors wrote.
Therefore, reviewers were not able to conduct the review and notified the authors they would withdraw from the peer-review process.
The Lancet said in a statement: “The Lancet takes issues of scientific integrity extremely seriously, and there are many outstanding questions about Surgisphere and the data that were allegedly included in this study. Following guidelines from the Committee on Publication Ethics and International Committee of Medical Journal Editors, institutional reviews of Surgisphere’s research collaborations are urgently needed.”
The authors wrote, “We can never forget the responsibility we have as researchers to scrupulously ensure that we rely on data sources that adhere to our high standards. Based on this development, we can no longer vouch for the veracity of the primary data sources. Due to this unfortunate development, the authors request that the paper be retracted.
“We all entered this collaboration to contribute in good faith and at a time of great need during the COVID-19 pandemic. We deeply apologize to you, the editors, and the journal readership for any embarrassment or inconvenience that this may have caused.”
In a similar, if briefer, note, the authors requested that the New England Journal of Medicine retract the earlier article as well. The retraction notice on the website reads: “Because all the authors were not granted access to the raw data and the raw data could not be made available to a third-party auditor, we are unable to validate the primary data sources underlying our article, ‘Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19.’ We therefore request that the article be retracted. We apologize to the editors and to readers of the Journal for the difficulties that this has caused.”
Both journals had already published “Expression of Concern” notices about the articles. The expression of concern followed an open letter, endorsed by more than 200 scientists, ethicists, and clinicians and posted on May 28, questioning the data and ethics of the study.
A version of this article originally appeared on Medscape.com.
‘Promising’ durvalumab results spark phase 3 trial in mesothelioma
Adding durvalumab to first-line pemetrexed and cisplatin improved survival in patients with unresectable malignant pleural mesothelioma (MPM) in a phase 2 trial, compared with historical controls who received only pemetrexed and cisplatin.
The median overall survival was 20.4 months in patients who received durvalumab plus pemetrexed-cisplatin. This is significantly longer than the median overall survival of 12.1 months (P = .0014) observed with pemetrexed-cisplatin in a prior phase 3 study (J Clin Oncol. 2003 Jul 15;21[14]:2636-44).
The new phase 2 results are “promising,” said lead investigator Patrick Forde, MBBCh, director of the thoracic cancer clinical research program at Johns Hopkins University in Baltimore.
He presented the results as part of the American Society of Clinical Oncology virtual scientific program.
Dr. Forde noted that a phase 3 trial directly comparing pemetrexed-cisplatin plus durvalumab to pemetrexed-cisplatin will begin recruiting this year. The trial is a collaboration between U.S. investigators and Australian researchers who reported their own phase 2 results with durvalumab plus pemetrexed-cisplatin in 2018 (J Thorac Oncol. 2018 Oct;13[10]:S338-339).
Study details
Dr. Forde’s phase 2 study enrolled 55 patients with treatment-naive, unresectable MPM. Their median age was 68 years (range, 35-83 years), and 45 (82%) were men. All had an Eastern Cooperative Oncology Group performance status of 0-1.
Epithelioid mesothelioma was the histologic subtype in three-quarters of patients. “It was a fairly typical mesothelioma population,” Dr. Forde said.
The patients received durvalumab at 1,120 mg plus pemetrexed at 500 mg/m2 and cisplatin at 75 mg/m2 every 3 weeks for up to six cycles. Carboplatin was substituted when cisplatin was contraindicated or patients developed toxicities.
All but one patient had stable or responding disease on radiography and went on to durvalumab maintenance, also given at 1,120 mg every 3 weeks, for up to 1 year from study entry.
Results
Dr. Forde said this study had 90% power to detect a 58% improvement in median overall survival, from the 12.1 months seen in historical controls to 19 months, which was the goal of this study.
It was a positive study, he said, as the median overall survival was 20.4 months (P = .0014).
The overall survival rate was 87.2% at 6 months, 70.4% at 12 months, and 44.2% at 24 months. The progression-free survival rate was 69.1% at 6 months, 16.4% at 12 months, and 10.9% at 24 months.
The overall response rate was 56.4%, which comprised 31 partial responses. Forty percent of patients (n = 22) had stable disease. One patient had progressive disease, and one was not evaluable (1.8% each).
To help with future patient selection, the researchers looked for baseline biomarkers that predicted response. Tumor PD-L1 expression, tumor mutation burden, and other potential candidates haven’t worked out so far, but the work continues, Dr. Forde said.
He noted that many of the adverse events in this trial are those typically seen with platinum-based chemotherapy.
Grade 3/4 treatment-emergent adverse events included anemia (n = 14), fatigue (n = 4), decreased appetite (n = 1), and hypomagnesemia (n = 1).
The most common grade 1/2 adverse events of special interest were hypothyroidism (n = 7), rash (n = 5), pruritus (n = 3), AST elevation (n = 3), and hyperthyroidism (n = 3).
Putting the results in context
Given the role of inflammation in MPM, durvalumab is among several immunotherapies under investigation for the disease.
A phase 3 French trial showed MPM patients had a median overall survival of 18.8 months with pemetrexed-cisplatin plus bevacizumab versus 16.1 months with pemetrexed-cisplatin only (Lancet. 2016 Apr 2;387[10026]:1405-1414).
The higher overall survival in the French study’s pemetrexed-cisplatin arm, compared with the 2003 trial results, is likely due to the use of modern second-line options, said Marjorie Zauderer, MD, codirector of the mesothelioma program at Memorial Sloan Kettering Cancer Center in New York, who was the discussant for Dr. Forde’s presentation.
“I think the improvement in overall survival presented by Dr. Forde is potentially clinically meaningful,” she said, but it was “well within the 95% confidence interval” of the bevacizumab trial. Even so, “I look forward” to the phase 3 results, she said.
Dr. Zauderer also pointed out an April press release from Bristol Myers Squibb that reported improved survival over pemetrexed-cisplatin with two of the company’s immunotherapies, nivolumab and ipilimumab, not as additions but as replacement first-line therapy. However, the randomized trial data haven’t been released yet. “We are all eager to evaluate this option further,” she said.
AstraZeneca, maker of durvalumab, funded the current study. Dr. Forde is an adviser for the company and reported research funding. Dr. Zauderer reported a relationship with Roche, which markets bevacizumab through its subsidiary, Genentech. She also disclosed research funding from Bristol Myers Squibb.
SOURCE: Forde PM et al. ASCO 2020, Abstract 9003.
Adding durvalumab to first-line pemetrexed and cisplatin improved survival in patients with unresectable malignant pleural mesothelioma (MPM) in a phase 2 trial, compared with historical controls who received only pemetrexed and cisplatin.
The median overall survival was 20.4 months in patients who received durvalumab plus pemetrexed-cisplatin. This is significantly longer than the median overall survival of 12.1 months (P = .0014) observed with pemetrexed-cisplatin in a prior phase 3 study (J Clin Oncol. 2003 Jul 15;21[14]:2636-44).
The new phase 2 results are “promising,” said lead investigator Patrick Forde, MBBCh, director of the thoracic cancer clinical research program at Johns Hopkins University in Baltimore.
He presented the results as part of the American Society of Clinical Oncology virtual scientific program.
Dr. Forde noted that a phase 3 trial directly comparing pemetrexed-cisplatin plus durvalumab to pemetrexed-cisplatin will begin recruiting this year. The trial is a collaboration between U.S. investigators and Australian researchers who reported their own phase 2 results with durvalumab plus pemetrexed-cisplatin in 2018 (J Thorac Oncol. 2018 Oct;13[10]:S338-339).
Study details
Dr. Forde’s phase 2 study enrolled 55 patients with treatment-naive, unresectable MPM. Their median age was 68 years (range, 35-83 years), and 45 (82%) were men. All had an Eastern Cooperative Oncology Group performance status of 0-1.
Epithelioid mesothelioma was the histologic subtype in three-quarters of patients. “It was a fairly typical mesothelioma population,” Dr. Forde said.
The patients received durvalumab at 1,120 mg plus pemetrexed at 500 mg/m2 and cisplatin at 75 mg/m2 every 3 weeks for up to six cycles. Carboplatin was substituted when cisplatin was contraindicated or patients developed toxicities.
All but one patient had stable or responding disease on radiography and went on to durvalumab maintenance, also given at 1,120 mg every 3 weeks, for up to 1 year from study entry.
Results
Dr. Forde said this study had 90% power to detect a 58% improvement in median overall survival, from the 12.1 months seen in historical controls to 19 months, which was the goal of this study.
It was a positive study, he said, as the median overall survival was 20.4 months (P = .0014).
The overall survival rate was 87.2% at 6 months, 70.4% at 12 months, and 44.2% at 24 months. The progression-free survival rate was 69.1% at 6 months, 16.4% at 12 months, and 10.9% at 24 months.
The overall response rate was 56.4%, which comprised 31 partial responses. Forty percent of patients (n = 22) had stable disease. One patient had progressive disease, and one was not evaluable (1.8% each).
To help with future patient selection, the researchers looked for baseline biomarkers that predicted response. Tumor PD-L1 expression, tumor mutation burden, and other potential candidates haven’t worked out so far, but the work continues, Dr. Forde said.
He noted that many of the adverse events in this trial are those typically seen with platinum-based chemotherapy.
Grade 3/4 treatment-emergent adverse events included anemia (n = 14), fatigue (n = 4), decreased appetite (n = 1), and hypomagnesemia (n = 1).
The most common grade 1/2 adverse events of special interest were hypothyroidism (n = 7), rash (n = 5), pruritus (n = 3), AST elevation (n = 3), and hyperthyroidism (n = 3).
Putting the results in context
Given the role of inflammation in MPM, durvalumab is among several immunotherapies under investigation for the disease.
A phase 3 French trial showed MPM patients had a median overall survival of 18.8 months with pemetrexed-cisplatin plus bevacizumab versus 16.1 months with pemetrexed-cisplatin only (Lancet. 2016 Apr 2;387[10026]:1405-1414).
The higher overall survival in the French study’s pemetrexed-cisplatin arm, compared with the 2003 trial results, is likely due to the use of modern second-line options, said Marjorie Zauderer, MD, codirector of the mesothelioma program at Memorial Sloan Kettering Cancer Center in New York, who was the discussant for Dr. Forde’s presentation.
“I think the improvement in overall survival presented by Dr. Forde is potentially clinically meaningful,” she said, but it was “well within the 95% confidence interval” of the bevacizumab trial. Even so, “I look forward” to the phase 3 results, she said.
Dr. Zauderer also pointed out an April press release from Bristol Myers Squibb that reported improved survival over pemetrexed-cisplatin with two of the company’s immunotherapies, nivolumab and ipilimumab, not as additions but as replacement first-line therapy. However, the randomized trial data haven’t been released yet. “We are all eager to evaluate this option further,” she said.
AstraZeneca, maker of durvalumab, funded the current study. Dr. Forde is an adviser for the company and reported research funding. Dr. Zauderer reported a relationship with Roche, which markets bevacizumab through its subsidiary, Genentech. She also disclosed research funding from Bristol Myers Squibb.
SOURCE: Forde PM et al. ASCO 2020, Abstract 9003.
Adding durvalumab to first-line pemetrexed and cisplatin improved survival in patients with unresectable malignant pleural mesothelioma (MPM) in a phase 2 trial, compared with historical controls who received only pemetrexed and cisplatin.
The median overall survival was 20.4 months in patients who received durvalumab plus pemetrexed-cisplatin. This is significantly longer than the median overall survival of 12.1 months (P = .0014) observed with pemetrexed-cisplatin in a prior phase 3 study (J Clin Oncol. 2003 Jul 15;21[14]:2636-44).
The new phase 2 results are “promising,” said lead investigator Patrick Forde, MBBCh, director of the thoracic cancer clinical research program at Johns Hopkins University in Baltimore.
He presented the results as part of the American Society of Clinical Oncology virtual scientific program.
Dr. Forde noted that a phase 3 trial directly comparing pemetrexed-cisplatin plus durvalumab to pemetrexed-cisplatin will begin recruiting this year. The trial is a collaboration between U.S. investigators and Australian researchers who reported their own phase 2 results with durvalumab plus pemetrexed-cisplatin in 2018 (J Thorac Oncol. 2018 Oct;13[10]:S338-339).
Study details
Dr. Forde’s phase 2 study enrolled 55 patients with treatment-naive, unresectable MPM. Their median age was 68 years (range, 35-83 years), and 45 (82%) were men. All had an Eastern Cooperative Oncology Group performance status of 0-1.
Epithelioid mesothelioma was the histologic subtype in three-quarters of patients. “It was a fairly typical mesothelioma population,” Dr. Forde said.
The patients received durvalumab at 1,120 mg plus pemetrexed at 500 mg/m2 and cisplatin at 75 mg/m2 every 3 weeks for up to six cycles. Carboplatin was substituted when cisplatin was contraindicated or patients developed toxicities.
All but one patient had stable or responding disease on radiography and went on to durvalumab maintenance, also given at 1,120 mg every 3 weeks, for up to 1 year from study entry.
Results
Dr. Forde said this study had 90% power to detect a 58% improvement in median overall survival, from the 12.1 months seen in historical controls to 19 months, which was the goal of this study.
It was a positive study, he said, as the median overall survival was 20.4 months (P = .0014).
The overall survival rate was 87.2% at 6 months, 70.4% at 12 months, and 44.2% at 24 months. The progression-free survival rate was 69.1% at 6 months, 16.4% at 12 months, and 10.9% at 24 months.
The overall response rate was 56.4%, which comprised 31 partial responses. Forty percent of patients (n = 22) had stable disease. One patient had progressive disease, and one was not evaluable (1.8% each).
To help with future patient selection, the researchers looked for baseline biomarkers that predicted response. Tumor PD-L1 expression, tumor mutation burden, and other potential candidates haven’t worked out so far, but the work continues, Dr. Forde said.
He noted that many of the adverse events in this trial are those typically seen with platinum-based chemotherapy.
Grade 3/4 treatment-emergent adverse events included anemia (n = 14), fatigue (n = 4), decreased appetite (n = 1), and hypomagnesemia (n = 1).
The most common grade 1/2 adverse events of special interest were hypothyroidism (n = 7), rash (n = 5), pruritus (n = 3), AST elevation (n = 3), and hyperthyroidism (n = 3).
Putting the results in context
Given the role of inflammation in MPM, durvalumab is among several immunotherapies under investigation for the disease.
A phase 3 French trial showed MPM patients had a median overall survival of 18.8 months with pemetrexed-cisplatin plus bevacizumab versus 16.1 months with pemetrexed-cisplatin only (Lancet. 2016 Apr 2;387[10026]:1405-1414).
The higher overall survival in the French study’s pemetrexed-cisplatin arm, compared with the 2003 trial results, is likely due to the use of modern second-line options, said Marjorie Zauderer, MD, codirector of the mesothelioma program at Memorial Sloan Kettering Cancer Center in New York, who was the discussant for Dr. Forde’s presentation.
“I think the improvement in overall survival presented by Dr. Forde is potentially clinically meaningful,” she said, but it was “well within the 95% confidence interval” of the bevacizumab trial. Even so, “I look forward” to the phase 3 results, she said.
Dr. Zauderer also pointed out an April press release from Bristol Myers Squibb that reported improved survival over pemetrexed-cisplatin with two of the company’s immunotherapies, nivolumab and ipilimumab, not as additions but as replacement first-line therapy. However, the randomized trial data haven’t been released yet. “We are all eager to evaluate this option further,” she said.
AstraZeneca, maker of durvalumab, funded the current study. Dr. Forde is an adviser for the company and reported research funding. Dr. Zauderer reported a relationship with Roche, which markets bevacizumab through its subsidiary, Genentech. She also disclosed research funding from Bristol Myers Squibb.
SOURCE: Forde PM et al. ASCO 2020, Abstract 9003.
FROM ASCO 2020