User login
Study identifies strong association between use of rotavirus vaccines, 60% reduction in infection
Two widely used rotavirus vaccines performed comparably in a meta-analysis, reducing risk of rotavirus gastroenteritis (RVGE) by more than 60% in young children. While the findings evidence a high protection level and low-risk safety profile, investigators of the study called for additional head-to-head comparisons to assess risks and benefits.
RVGE, which accounts for 28.8% of all deaths from diarrhea worldwide, is the leading cause of diarrhea in children under age 5. More than 100 countries include rotavirus vaccines in their immunization programs. Among six types of vaccines currently in use, two live-attenuated oral vaccines: the two-dose monovalent Rotarix (RV1) and three-dose pentavalent RotaTeq (RV5]) are in use worldwide.
Not much is known about their interchangeability, although a previous meta-analysis reported similarities in effectiveness of Rotarix (83%), RotaTeq (85%), and Rotarix and RotaTeq mixed series (86%) in low-mortality countries. RVGE morbidity and mortality have declined since the introduction of these vaccines, but concerns persist about their safety, Zi-Wei Sun, MSc, of Nanjing (China) Medical University and colleagues wrote in JAMA Pediatrics.
Their systematic review and meta-analysis of randomized clinical trials, case-control, and cohort studies compared benefit, risk, and immunogenicity of these vaccines and their effectiveness in reducing RVGE. Combing through databases Embase, PubMed, the Cochrane Library, and Web of Science using search terms “rotavirus” and “vaccine,” they chose 121 randomized clinical trials and cohort and case-control studies that included more than 100 children younger than 5 years. Thirty-eight of the randomized clinical trials had related data that examined the vaccines’ protection against RVGE hospitalization, study coauthor Hemant Goyal, MD, FACP, explained in an interview.
All of the studies reported on the safety and effectiveness or immunogenicity of rotavirus vaccines. The investigators used a random-effects model to calculate relative risks, odds ratios, risk differences, and 95% confidence intervals. They also stratified studies by economic development of countries, given that vaccine efficacy is often higher in middle- and high-income countries, compared with low-income countries. An adjusted indirect treatment comparison evaluated differences in vaccine protection among different subgroups, adopting P < .05 as the level of statistical significance.
Primary outcomes included RVGE, severe RVGE, and RVGE hospitalization and safety-associated outcomes such as serious adverse events, intussusception, and mortality.
Rotarix and RotaTeq reduced RVGE in children younger than 5 years by 68.4% and 63.6%, respectively. Dr. Goyal and colleagues confirmed these results in case-control studies (65.3% and 72.8%, respectively). Both vaccines significantly reduced RVGE and RVGE hospitalization risk and demonstrated higher protection against severe RVGE. In adjusted indirect comparisons, the two vaccines showed no significant differences in protection. They also found a positive correlation between immunogenicity and vaccine protection.
“RotaTeq seems to show lower protection in low-income countries, compared with Rotarix, but these estimates should be interpreted with caution as there was only one study for low-income countries and indirect comparison," said Dr. Goyal, a second-year gastroenterology fellow at the Wright Center for Graduate Medical Education, Scranton, Penn.
None of the vaccines demonstrated risk of serious adverse events. However, an Australian study in 2013 did report a small increased risk of intussusception after RV1 and RV5 vaccination. “Therefore, continuous surveillance of the benefits and adverse effects of rotavirus vaccines is required after vaccination,” the investigators noted.
Analyzing newer, less widely distributed vaccines, Rotavac, Rotasiil, and Lanzhou lamb rotavirus vaccine also showed moderate effectiveness in reducing RVGE risk.
Immunity wanes over time
Protection against rotavirus diseases seems to wane over time after vaccination. “Although our results indicated that rotavirus vaccines can provide substantial protection against RVGE during the first 2 years of life, more studies following up the vaccine efficacy for more than 2 years are required,” the investigators recommended.
Declining vaccine-induced antibodies, RVGE-acquired protection from the vaccine’s indirect effects, or exposure to unvaccinated populations may explain gradual loss of immunity.
Monitoring of rotavirus strains following vaccination should take place “to avoid population-based selection of so-called escape strains, especially fully heterotypic strains and new strains, because of the long-term pressure of vaccine immunity,” they recommended.
The findings emphasize the importance of introducing vaccines worldwide to reduce infection, summarized Dr. Goyal and colleagues. Given how challenging it is to treat the wide varieties of rotavirus, “It encouraging that RV1 and RV5 work well against heterotypic strains,” they added. Similar performance between Rotarix and RotaTeq also makes it easier for clinicians to choose a vaccine.
Increasing the availability and efficacy of these vaccines in low-income countries with high mortality rates is a high priority,
David I. Bernstein, MD, MA, wrote in a related editorial: “A clear gradient in vaccine protections was noted by country income level in the analysis presented, and much effort has been spent to understand this discrepancy.”
Overall, the study confirmed the efficacy of these two vaccines and their equivalence, noted Dr. Bernstein.
The study’s literature search process had some limitations. “Especially in stratified analyses, sparse data in some subgroups limit generalizability. ... The most accurate method, head-to-head comparisons, to evaluate the comparative efficacy of different vaccines is required in further studies,” the study investigators wrote.
Such studies would directly compare Rotarix and RotaTeq from multiple perspectives: efficacy, cost-effectiveness, strain-specific protection, the duration of protection, safety, and immunogenicity, said Dr. Goyal.
*This story was updated on May 24, 2021.
Two widely used rotavirus vaccines performed comparably in a meta-analysis, reducing risk of rotavirus gastroenteritis (RVGE) by more than 60% in young children. While the findings evidence a high protection level and low-risk safety profile, investigators of the study called for additional head-to-head comparisons to assess risks and benefits.
RVGE, which accounts for 28.8% of all deaths from diarrhea worldwide, is the leading cause of diarrhea in children under age 5. More than 100 countries include rotavirus vaccines in their immunization programs. Among six types of vaccines currently in use, two live-attenuated oral vaccines: the two-dose monovalent Rotarix (RV1) and three-dose pentavalent RotaTeq (RV5]) are in use worldwide.
Not much is known about their interchangeability, although a previous meta-analysis reported similarities in effectiveness of Rotarix (83%), RotaTeq (85%), and Rotarix and RotaTeq mixed series (86%) in low-mortality countries. RVGE morbidity and mortality have declined since the introduction of these vaccines, but concerns persist about their safety, Zi-Wei Sun, MSc, of Nanjing (China) Medical University and colleagues wrote in JAMA Pediatrics.
Their systematic review and meta-analysis of randomized clinical trials, case-control, and cohort studies compared benefit, risk, and immunogenicity of these vaccines and their effectiveness in reducing RVGE. Combing through databases Embase, PubMed, the Cochrane Library, and Web of Science using search terms “rotavirus” and “vaccine,” they chose 121 randomized clinical trials and cohort and case-control studies that included more than 100 children younger than 5 years. Thirty-eight of the randomized clinical trials had related data that examined the vaccines’ protection against RVGE hospitalization, study coauthor Hemant Goyal, MD, FACP, explained in an interview.
All of the studies reported on the safety and effectiveness or immunogenicity of rotavirus vaccines. The investigators used a random-effects model to calculate relative risks, odds ratios, risk differences, and 95% confidence intervals. They also stratified studies by economic development of countries, given that vaccine efficacy is often higher in middle- and high-income countries, compared with low-income countries. An adjusted indirect treatment comparison evaluated differences in vaccine protection among different subgroups, adopting P < .05 as the level of statistical significance.
Primary outcomes included RVGE, severe RVGE, and RVGE hospitalization and safety-associated outcomes such as serious adverse events, intussusception, and mortality.
Rotarix and RotaTeq reduced RVGE in children younger than 5 years by 68.4% and 63.6%, respectively. Dr. Goyal and colleagues confirmed these results in case-control studies (65.3% and 72.8%, respectively). Both vaccines significantly reduced RVGE and RVGE hospitalization risk and demonstrated higher protection against severe RVGE. In adjusted indirect comparisons, the two vaccines showed no significant differences in protection. They also found a positive correlation between immunogenicity and vaccine protection.
“RotaTeq seems to show lower protection in low-income countries, compared with Rotarix, but these estimates should be interpreted with caution as there was only one study for low-income countries and indirect comparison," said Dr. Goyal, a second-year gastroenterology fellow at the Wright Center for Graduate Medical Education, Scranton, Penn.
None of the vaccines demonstrated risk of serious adverse events. However, an Australian study in 2013 did report a small increased risk of intussusception after RV1 and RV5 vaccination. “Therefore, continuous surveillance of the benefits and adverse effects of rotavirus vaccines is required after vaccination,” the investigators noted.
Analyzing newer, less widely distributed vaccines, Rotavac, Rotasiil, and Lanzhou lamb rotavirus vaccine also showed moderate effectiveness in reducing RVGE risk.
Immunity wanes over time
Protection against rotavirus diseases seems to wane over time after vaccination. “Although our results indicated that rotavirus vaccines can provide substantial protection against RVGE during the first 2 years of life, more studies following up the vaccine efficacy for more than 2 years are required,” the investigators recommended.
Declining vaccine-induced antibodies, RVGE-acquired protection from the vaccine’s indirect effects, or exposure to unvaccinated populations may explain gradual loss of immunity.
Monitoring of rotavirus strains following vaccination should take place “to avoid population-based selection of so-called escape strains, especially fully heterotypic strains and new strains, because of the long-term pressure of vaccine immunity,” they recommended.
The findings emphasize the importance of introducing vaccines worldwide to reduce infection, summarized Dr. Goyal and colleagues. Given how challenging it is to treat the wide varieties of rotavirus, “It encouraging that RV1 and RV5 work well against heterotypic strains,” they added. Similar performance between Rotarix and RotaTeq also makes it easier for clinicians to choose a vaccine.
Increasing the availability and efficacy of these vaccines in low-income countries with high mortality rates is a high priority,
David I. Bernstein, MD, MA, wrote in a related editorial: “A clear gradient in vaccine protections was noted by country income level in the analysis presented, and much effort has been spent to understand this discrepancy.”
Overall, the study confirmed the efficacy of these two vaccines and their equivalence, noted Dr. Bernstein.
The study’s literature search process had some limitations. “Especially in stratified analyses, sparse data in some subgroups limit generalizability. ... The most accurate method, head-to-head comparisons, to evaluate the comparative efficacy of different vaccines is required in further studies,” the study investigators wrote.
Such studies would directly compare Rotarix and RotaTeq from multiple perspectives: efficacy, cost-effectiveness, strain-specific protection, the duration of protection, safety, and immunogenicity, said Dr. Goyal.
*This story was updated on May 24, 2021.
Two widely used rotavirus vaccines performed comparably in a meta-analysis, reducing risk of rotavirus gastroenteritis (RVGE) by more than 60% in young children. While the findings evidence a high protection level and low-risk safety profile, investigators of the study called for additional head-to-head comparisons to assess risks and benefits.
RVGE, which accounts for 28.8% of all deaths from diarrhea worldwide, is the leading cause of diarrhea in children under age 5. More than 100 countries include rotavirus vaccines in their immunization programs. Among six types of vaccines currently in use, two live-attenuated oral vaccines: the two-dose monovalent Rotarix (RV1) and three-dose pentavalent RotaTeq (RV5]) are in use worldwide.
Not much is known about their interchangeability, although a previous meta-analysis reported similarities in effectiveness of Rotarix (83%), RotaTeq (85%), and Rotarix and RotaTeq mixed series (86%) in low-mortality countries. RVGE morbidity and mortality have declined since the introduction of these vaccines, but concerns persist about their safety, Zi-Wei Sun, MSc, of Nanjing (China) Medical University and colleagues wrote in JAMA Pediatrics.
Their systematic review and meta-analysis of randomized clinical trials, case-control, and cohort studies compared benefit, risk, and immunogenicity of these vaccines and their effectiveness in reducing RVGE. Combing through databases Embase, PubMed, the Cochrane Library, and Web of Science using search terms “rotavirus” and “vaccine,” they chose 121 randomized clinical trials and cohort and case-control studies that included more than 100 children younger than 5 years. Thirty-eight of the randomized clinical trials had related data that examined the vaccines’ protection against RVGE hospitalization, study coauthor Hemant Goyal, MD, FACP, explained in an interview.
All of the studies reported on the safety and effectiveness or immunogenicity of rotavirus vaccines. The investigators used a random-effects model to calculate relative risks, odds ratios, risk differences, and 95% confidence intervals. They also stratified studies by economic development of countries, given that vaccine efficacy is often higher in middle- and high-income countries, compared with low-income countries. An adjusted indirect treatment comparison evaluated differences in vaccine protection among different subgroups, adopting P < .05 as the level of statistical significance.
Primary outcomes included RVGE, severe RVGE, and RVGE hospitalization and safety-associated outcomes such as serious adverse events, intussusception, and mortality.
Rotarix and RotaTeq reduced RVGE in children younger than 5 years by 68.4% and 63.6%, respectively. Dr. Goyal and colleagues confirmed these results in case-control studies (65.3% and 72.8%, respectively). Both vaccines significantly reduced RVGE and RVGE hospitalization risk and demonstrated higher protection against severe RVGE. In adjusted indirect comparisons, the two vaccines showed no significant differences in protection. They also found a positive correlation between immunogenicity and vaccine protection.
“RotaTeq seems to show lower protection in low-income countries, compared with Rotarix, but these estimates should be interpreted with caution as there was only one study for low-income countries and indirect comparison," said Dr. Goyal, a second-year gastroenterology fellow at the Wright Center for Graduate Medical Education, Scranton, Penn.
None of the vaccines demonstrated risk of serious adverse events. However, an Australian study in 2013 did report a small increased risk of intussusception after RV1 and RV5 vaccination. “Therefore, continuous surveillance of the benefits and adverse effects of rotavirus vaccines is required after vaccination,” the investigators noted.
Analyzing newer, less widely distributed vaccines, Rotavac, Rotasiil, and Lanzhou lamb rotavirus vaccine also showed moderate effectiveness in reducing RVGE risk.
Immunity wanes over time
Protection against rotavirus diseases seems to wane over time after vaccination. “Although our results indicated that rotavirus vaccines can provide substantial protection against RVGE during the first 2 years of life, more studies following up the vaccine efficacy for more than 2 years are required,” the investigators recommended.
Declining vaccine-induced antibodies, RVGE-acquired protection from the vaccine’s indirect effects, or exposure to unvaccinated populations may explain gradual loss of immunity.
Monitoring of rotavirus strains following vaccination should take place “to avoid population-based selection of so-called escape strains, especially fully heterotypic strains and new strains, because of the long-term pressure of vaccine immunity,” they recommended.
The findings emphasize the importance of introducing vaccines worldwide to reduce infection, summarized Dr. Goyal and colleagues. Given how challenging it is to treat the wide varieties of rotavirus, “It encouraging that RV1 and RV5 work well against heterotypic strains,” they added. Similar performance between Rotarix and RotaTeq also makes it easier for clinicians to choose a vaccine.
Increasing the availability and efficacy of these vaccines in low-income countries with high mortality rates is a high priority,
David I. Bernstein, MD, MA, wrote in a related editorial: “A clear gradient in vaccine protections was noted by country income level in the analysis presented, and much effort has been spent to understand this discrepancy.”
Overall, the study confirmed the efficacy of these two vaccines and their equivalence, noted Dr. Bernstein.
The study’s literature search process had some limitations. “Especially in stratified analyses, sparse data in some subgroups limit generalizability. ... The most accurate method, head-to-head comparisons, to evaluate the comparative efficacy of different vaccines is required in further studies,” the study investigators wrote.
Such studies would directly compare Rotarix and RotaTeq from multiple perspectives: efficacy, cost-effectiveness, strain-specific protection, the duration of protection, safety, and immunogenicity, said Dr. Goyal.
*This story was updated on May 24, 2021.
FROM JAMA PEDIATRICS
Hospital outcomes for children with MIS-C unaffected by initial presentation site
Length of hospital stay and the need for intensive care for pediatric COVID-19 patients with multisystem inflammatory syndrome in children was not significantly different for those who presented first as outpatients or emergency patients, based on data from 34 children.

Multisystem inflammatory syndrome in children (MIS-C) can be challenging to diagnose, as the key characteristics of fever, elevated inflammatory markers, and involvement of at least two organ systems often overlap with other illnesses, said Erin B. Treemarcki, DO, of the University of Utah, Salt Lake City, and colleagues.
“Primary care and urgent care providers are often the first point of health care for children with symptoms of MIS-C,” the researchers wrote. In a study (Poster 142) presented at the annual meeting of the Pediatric Academic Societies, held virtually, the researchers conducted a retrospective review of 34 patients younger than 21 years who were hospitalized with MIS-C at a single center between April 2020 and December 2020. The average age of the patients was 7.9 years, 68% were male, 82% were White, and 53% first presented to an outpatient clinic.
Sixteen patients presented to an emergency department and 18 presented to an ambulatory setting. The length of hospitalization ranged from 3 to 16 days with a median of 6 days, and the PICU stay ranged from 1 to 10 days with a median of 2 days.
Overall, the length of hospital stay and rate of PICU admission were not significantly different between the emergency presentation and outpatient presentation groups. Twenty-four patients entered the PICU, 13 at admission and 11 as transfers. However, the median number of days of symptoms prior to admission was significantly higher for outpatient cases (6 days vs. 4 days, P = .03).
One patient was readmitted to the hospital within 30 days for aseptic meningitis, and none of the patients died.
Initial symptoms were not significantly different for outpatient vs. emergency department patients. The most common initial manifestations of MIS-C included fever (100%), gastrointestinal symptoms (85%), and mucocutaneous symptoms (88%). Mucocutaneous symptoms included rash, oral mucosal changes, conjunctivitis, and hand/foot edema. In addition, 65% of the patients met at least 3 criteria for Kawasaki disease, the researchers noted.
The most common elevated labs at presentation regardless of setting were D-dimer (100%), C-reactive protein (97%), ferritin (97%), procalcitonin (97%), and serum IL-6 (94%).
The study findings were limited by the small sample size and focus on data from a single center. However, the results emphasize the varied presentations of MIS-C and the importance that both primary care and urgent care providers know the signs, as they are often the first point of health care for children with MIS-C, the researchers noted.
Keep looking for factors that put children at risk
“MIS-C is probably the most serious complication of COVID in children, so we as pediatricians on the front line need to know what it looks like,” Karalyn Kinsella, MD, a pediatrician in Cheshire, Conn., said in an interview.
Dr. Kinsella said she was surprised by the study finding that children’s length of hospital stay was not affected by presentation setting.
“I would have thought the kids presenting in an outpatient setting would take longer to diagnose, and therefore have a longer hospital stay,” she noted. Instead, the take-home message is that whether the MIS-C diagnosis occurs in the outpatient or emergency setting, the length of stay is the same, and that the most common symptoms are fever, gastrointestinal, mucocutaneous, and cardiac symptoms regardless of initial presentation setting, she said.
More research is needed, and future studies should examine “any potential underlying factors making these particular kids susceptible to MIS-C,” Dr. Kinsella added.
The researchers had no financial conflicts to disclose. Dr. Kinsella had no financial conflicts, but serves on the Pediatric News Editorial Advisory Board.
Length of hospital stay and the need for intensive care for pediatric COVID-19 patients with multisystem inflammatory syndrome in children was not significantly different for those who presented first as outpatients or emergency patients, based on data from 34 children.
Multisystem inflammatory syndrome in children (MIS-C) can be challenging to diagnose, as the key characteristics of fever, elevated inflammatory markers, and involvement of at least two organ systems often overlap with other illnesses, said Erin B. Treemarcki, DO, of the University of Utah, Salt Lake City, and colleagues.
“Primary care and urgent care providers are often the first point of health care for children with symptoms of MIS-C,” the researchers wrote. In a study (Poster 142) presented at the annual meeting of the Pediatric Academic Societies, held virtually, the researchers conducted a retrospective review of 34 patients younger than 21 years who were hospitalized with MIS-C at a single center between April 2020 and December 2020. The average age of the patients was 7.9 years, 68% were male, 82% were White, and 53% first presented to an outpatient clinic.
Sixteen patients presented to an emergency department and 18 presented to an ambulatory setting. The length of hospitalization ranged from 3 to 16 days with a median of 6 days, and the PICU stay ranged from 1 to 10 days with a median of 2 days.
Overall, the length of hospital stay and rate of PICU admission were not significantly different between the emergency presentation and outpatient presentation groups. Twenty-four patients entered the PICU, 13 at admission and 11 as transfers. However, the median number of days of symptoms prior to admission was significantly higher for outpatient cases (6 days vs. 4 days, P = .03).
One patient was readmitted to the hospital within 30 days for aseptic meningitis, and none of the patients died.
Initial symptoms were not significantly different for outpatient vs. emergency department patients. The most common initial manifestations of MIS-C included fever (100%), gastrointestinal symptoms (85%), and mucocutaneous symptoms (88%). Mucocutaneous symptoms included rash, oral mucosal changes, conjunctivitis, and hand/foot edema. In addition, 65% of the patients met at least 3 criteria for Kawasaki disease, the researchers noted.
The most common elevated labs at presentation regardless of setting were D-dimer (100%), C-reactive protein (97%), ferritin (97%), procalcitonin (97%), and serum IL-6 (94%).
The study findings were limited by the small sample size and focus on data from a single center. However, the results emphasize the varied presentations of MIS-C and the importance that both primary care and urgent care providers know the signs, as they are often the first point of health care for children with MIS-C, the researchers noted.
Keep looking for factors that put children at risk
“MIS-C is probably the most serious complication of COVID in children, so we as pediatricians on the front line need to know what it looks like,” Karalyn Kinsella, MD, a pediatrician in Cheshire, Conn., said in an interview.
Dr. Kinsella said she was surprised by the study finding that children’s length of hospital stay was not affected by presentation setting.
“I would have thought the kids presenting in an outpatient setting would take longer to diagnose, and therefore have a longer hospital stay,” she noted. Instead, the take-home message is that whether the MIS-C diagnosis occurs in the outpatient or emergency setting, the length of stay is the same, and that the most common symptoms are fever, gastrointestinal, mucocutaneous, and cardiac symptoms regardless of initial presentation setting, she said.
More research is needed, and future studies should examine “any potential underlying factors making these particular kids susceptible to MIS-C,” Dr. Kinsella added.
The researchers had no financial conflicts to disclose. Dr. Kinsella had no financial conflicts, but serves on the Pediatric News Editorial Advisory Board.
Length of hospital stay and the need for intensive care for pediatric COVID-19 patients with multisystem inflammatory syndrome in children was not significantly different for those who presented first as outpatients or emergency patients, based on data from 34 children.
Multisystem inflammatory syndrome in children (MIS-C) can be challenging to diagnose, as the key characteristics of fever, elevated inflammatory markers, and involvement of at least two organ systems often overlap with other illnesses, said Erin B. Treemarcki, DO, of the University of Utah, Salt Lake City, and colleagues.
“Primary care and urgent care providers are often the first point of health care for children with symptoms of MIS-C,” the researchers wrote. In a study (Poster 142) presented at the annual meeting of the Pediatric Academic Societies, held virtually, the researchers conducted a retrospective review of 34 patients younger than 21 years who were hospitalized with MIS-C at a single center between April 2020 and December 2020. The average age of the patients was 7.9 years, 68% were male, 82% were White, and 53% first presented to an outpatient clinic.
Sixteen patients presented to an emergency department and 18 presented to an ambulatory setting. The length of hospitalization ranged from 3 to 16 days with a median of 6 days, and the PICU stay ranged from 1 to 10 days with a median of 2 days.
Overall, the length of hospital stay and rate of PICU admission were not significantly different between the emergency presentation and outpatient presentation groups. Twenty-four patients entered the PICU, 13 at admission and 11 as transfers. However, the median number of days of symptoms prior to admission was significantly higher for outpatient cases (6 days vs. 4 days, P = .03).
One patient was readmitted to the hospital within 30 days for aseptic meningitis, and none of the patients died.
Initial symptoms were not significantly different for outpatient vs. emergency department patients. The most common initial manifestations of MIS-C included fever (100%), gastrointestinal symptoms (85%), and mucocutaneous symptoms (88%). Mucocutaneous symptoms included rash, oral mucosal changes, conjunctivitis, and hand/foot edema. In addition, 65% of the patients met at least 3 criteria for Kawasaki disease, the researchers noted.
The most common elevated labs at presentation regardless of setting were D-dimer (100%), C-reactive protein (97%), ferritin (97%), procalcitonin (97%), and serum IL-6 (94%).
The study findings were limited by the small sample size and focus on data from a single center. However, the results emphasize the varied presentations of MIS-C and the importance that both primary care and urgent care providers know the signs, as they are often the first point of health care for children with MIS-C, the researchers noted.
Keep looking for factors that put children at risk
“MIS-C is probably the most serious complication of COVID in children, so we as pediatricians on the front line need to know what it looks like,” Karalyn Kinsella, MD, a pediatrician in Cheshire, Conn., said in an interview.
Dr. Kinsella said she was surprised by the study finding that children’s length of hospital stay was not affected by presentation setting.
“I would have thought the kids presenting in an outpatient setting would take longer to diagnose, and therefore have a longer hospital stay,” she noted. Instead, the take-home message is that whether the MIS-C diagnosis occurs in the outpatient or emergency setting, the length of stay is the same, and that the most common symptoms are fever, gastrointestinal, mucocutaneous, and cardiac symptoms regardless of initial presentation setting, she said.
More research is needed, and future studies should examine “any potential underlying factors making these particular kids susceptible to MIS-C,” Dr. Kinsella added.
The researchers had no financial conflicts to disclose. Dr. Kinsella had no financial conflicts, but serves on the Pediatric News Editorial Advisory Board.
FROM PAS 2021

Risk factors identified for late seizure relapse after epilepsy surgery
according to a new study on the factors most associated with seizure recurrence in drug-resistant epilepsy.
“As our study analyzed late seizure relapse, our results are not applicable for short‐term seizure control. Vice versa, results for short‐term outcomes should not be transferred to long‐term outcomes,” Stephan Petrik of the Epilepsy Center at the University of Freiburg (Germany) and colleagues wrote. The study was published in the May 2021 issue of Epilepsia.
To assess the variables that increase risk of late seizure recurrence following surgery, the researchers retrospectively studied the medical records of patients who underwent resective epilepsy surgery at the University Hospital Freiburg (Germany) between 1999 and 2015. Of the 1,278 initial patients, a group of 99 participants (7.7%) with seizure relapses after at least 2 years of complete seizure freedom were matched with controls experiencing long-term seizure freedom. The two groups had similar mean durations of epilepsy from onset to surgery: 13.9 years in the relapse group and 13.0 years in the control group.
The mean follow-up was 9.7 years (standard deviation, 4.0; range, 2.9-18.5) in the relapse group and 8.2 years (SD, 3.5; range, 2.2-18.3) in the control group. The mean time to late seizure recurrence was 56.6 months, and two-thirds of patients relapsed in the 5 years after surgery. Twenty of the relapse patients only experienced a single seizure, and 41% of the patients who reported more than one seizure had a frequency of less than one per month.
The type of resection had no discernible impact on outcomes, although anterior temporal lobe resection did trend toward being associated with recurrence (odds ratio, 2.7; 95% confidence interval, 0.93-8.89; P = .06). Incomplete resection was significantly associated with late relapse but did not seem to affect timing: the mean duration of seizure freedom was 56.5 months with complete resection and 58.5 months with incomplete resection (P = .62). Additional preoperative PET scans were performed on 45% of patients in the relapse group, compared with 29% in the control group.
After multivariate analysis, predictors for late relapse included incomplete resection (OR, 3.81; 95% CI, 1.79-8.53; P < .001); the existence of additional, potentially epileptogenic lesions in the contralateral hemisphere on presurgical MRI (OR, 3.36; 95% CI, 1.18-10.62; P = .03); epilepsy onset during the first year of life (OR, 4.24; 95% CI, 1.4-15.89; P = .02); and preoperative PET scans being performed (OR, 2.47; 95% CI, 1.25-4.97; P = .01). Though use of preoperative and postoperative antiepileptic drugs (AEDs) was higher in the relapse group, along with complete withdrawal being more common in the control group (68%, compared with 51%), neither was deemed significant in multivariate analysis.
What to do about seizure relapse risk factors
“This is one of the best analyses of the factors that contribute to late seizure relapse,” Gregory K. Bergey, MD, director of the Johns Hopkins Epilepsy Center in Baltimore, said in an interview. “Am I surprised by their results? Not necessarily.”
What did jump out, he said, was AED use not being a predictor of recurrence, as well as all the patients with late relapse having lesional epilepsy. “As they point out, you can have relapse with nonlesional epilepsy, but very often it happens in the first year or 2. If someone is 2 years out and doesn’t have a lesion, they’re probably more likely to remain seizure free.”
Despite the researchers’ comprehensive review of risk factors, the question remains: What to do with this information?
“They’ve done a very good job of identifying that 7.7% of 1,200 who are at risk of a late relapse,” he said. “Now, take those patients with high-risk factors and launch a trial where you keep medicines the same or do something that would alter that outcome.”
“The problem is,” he added, “that’s a 10-year study. It’s easy for me to sit here and call for one of those. But still, as valuable as this was, it’s a retrospective study. Now you have to say, what are the implications of this? What can we do in the prospective fashion?”
The authors acknowledged their study’s other limitations, including a lack of information on the reasons for an incomplete resection, a notable decrease in follow-up visits more than 5 years after surgery, and potential selection bias. They added, however, that “matching by age at surgery, gender, and time to relapse/last follow‐up” should have helped reduce any significant bias.
No potential conflicts of interest were disclosed.
according to a new study on the factors most associated with seizure recurrence in drug-resistant epilepsy.
“As our study analyzed late seizure relapse, our results are not applicable for short‐term seizure control. Vice versa, results for short‐term outcomes should not be transferred to long‐term outcomes,” Stephan Petrik of the Epilepsy Center at the University of Freiburg (Germany) and colleagues wrote. The study was published in the May 2021 issue of Epilepsia.
To assess the variables that increase risk of late seizure recurrence following surgery, the researchers retrospectively studied the medical records of patients who underwent resective epilepsy surgery at the University Hospital Freiburg (Germany) between 1999 and 2015. Of the 1,278 initial patients, a group of 99 participants (7.7%) with seizure relapses after at least 2 years of complete seizure freedom were matched with controls experiencing long-term seizure freedom. The two groups had similar mean durations of epilepsy from onset to surgery: 13.9 years in the relapse group and 13.0 years in the control group.
The mean follow-up was 9.7 years (standard deviation, 4.0; range, 2.9-18.5) in the relapse group and 8.2 years (SD, 3.5; range, 2.2-18.3) in the control group. The mean time to late seizure recurrence was 56.6 months, and two-thirds of patients relapsed in the 5 years after surgery. Twenty of the relapse patients only experienced a single seizure, and 41% of the patients who reported more than one seizure had a frequency of less than one per month.
The type of resection had no discernible impact on outcomes, although anterior temporal lobe resection did trend toward being associated with recurrence (odds ratio, 2.7; 95% confidence interval, 0.93-8.89; P = .06). Incomplete resection was significantly associated with late relapse but did not seem to affect timing: the mean duration of seizure freedom was 56.5 months with complete resection and 58.5 months with incomplete resection (P = .62). Additional preoperative PET scans were performed on 45% of patients in the relapse group, compared with 29% in the control group.
After multivariate analysis, predictors for late relapse included incomplete resection (OR, 3.81; 95% CI, 1.79-8.53; P < .001); the existence of additional, potentially epileptogenic lesions in the contralateral hemisphere on presurgical MRI (OR, 3.36; 95% CI, 1.18-10.62; P = .03); epilepsy onset during the first year of life (OR, 4.24; 95% CI, 1.4-15.89; P = .02); and preoperative PET scans being performed (OR, 2.47; 95% CI, 1.25-4.97; P = .01). Though use of preoperative and postoperative antiepileptic drugs (AEDs) was higher in the relapse group, along with complete withdrawal being more common in the control group (68%, compared with 51%), neither was deemed significant in multivariate analysis.
What to do about seizure relapse risk factors
“This is one of the best analyses of the factors that contribute to late seizure relapse,” Gregory K. Bergey, MD, director of the Johns Hopkins Epilepsy Center in Baltimore, said in an interview. “Am I surprised by their results? Not necessarily.”
What did jump out, he said, was AED use not being a predictor of recurrence, as well as all the patients with late relapse having lesional epilepsy. “As they point out, you can have relapse with nonlesional epilepsy, but very often it happens in the first year or 2. If someone is 2 years out and doesn’t have a lesion, they’re probably more likely to remain seizure free.”
Despite the researchers’ comprehensive review of risk factors, the question remains: What to do with this information?
“They’ve done a very good job of identifying that 7.7% of 1,200 who are at risk of a late relapse,” he said. “Now, take those patients with high-risk factors and launch a trial where you keep medicines the same or do something that would alter that outcome.”
“The problem is,” he added, “that’s a 10-year study. It’s easy for me to sit here and call for one of those. But still, as valuable as this was, it’s a retrospective study. Now you have to say, what are the implications of this? What can we do in the prospective fashion?”
The authors acknowledged their study’s other limitations, including a lack of information on the reasons for an incomplete resection, a notable decrease in follow-up visits more than 5 years after surgery, and potential selection bias. They added, however, that “matching by age at surgery, gender, and time to relapse/last follow‐up” should have helped reduce any significant bias.
No potential conflicts of interest were disclosed.
according to a new study on the factors most associated with seizure recurrence in drug-resistant epilepsy.
“As our study analyzed late seizure relapse, our results are not applicable for short‐term seizure control. Vice versa, results for short‐term outcomes should not be transferred to long‐term outcomes,” Stephan Petrik of the Epilepsy Center at the University of Freiburg (Germany) and colleagues wrote. The study was published in the May 2021 issue of Epilepsia.
To assess the variables that increase risk of late seizure recurrence following surgery, the researchers retrospectively studied the medical records of patients who underwent resective epilepsy surgery at the University Hospital Freiburg (Germany) between 1999 and 2015. Of the 1,278 initial patients, a group of 99 participants (7.7%) with seizure relapses after at least 2 years of complete seizure freedom were matched with controls experiencing long-term seizure freedom. The two groups had similar mean durations of epilepsy from onset to surgery: 13.9 years in the relapse group and 13.0 years in the control group.
The mean follow-up was 9.7 years (standard deviation, 4.0; range, 2.9-18.5) in the relapse group and 8.2 years (SD, 3.5; range, 2.2-18.3) in the control group. The mean time to late seizure recurrence was 56.6 months, and two-thirds of patients relapsed in the 5 years after surgery. Twenty of the relapse patients only experienced a single seizure, and 41% of the patients who reported more than one seizure had a frequency of less than one per month.
The type of resection had no discernible impact on outcomes, although anterior temporal lobe resection did trend toward being associated with recurrence (odds ratio, 2.7; 95% confidence interval, 0.93-8.89; P = .06). Incomplete resection was significantly associated with late relapse but did not seem to affect timing: the mean duration of seizure freedom was 56.5 months with complete resection and 58.5 months with incomplete resection (P = .62). Additional preoperative PET scans were performed on 45% of patients in the relapse group, compared with 29% in the control group.
After multivariate analysis, predictors for late relapse included incomplete resection (OR, 3.81; 95% CI, 1.79-8.53; P < .001); the existence of additional, potentially epileptogenic lesions in the contralateral hemisphere on presurgical MRI (OR, 3.36; 95% CI, 1.18-10.62; P = .03); epilepsy onset during the first year of life (OR, 4.24; 95% CI, 1.4-15.89; P = .02); and preoperative PET scans being performed (OR, 2.47; 95% CI, 1.25-4.97; P = .01). Though use of preoperative and postoperative antiepileptic drugs (AEDs) was higher in the relapse group, along with complete withdrawal being more common in the control group (68%, compared with 51%), neither was deemed significant in multivariate analysis.
What to do about seizure relapse risk factors
“This is one of the best analyses of the factors that contribute to late seizure relapse,” Gregory K. Bergey, MD, director of the Johns Hopkins Epilepsy Center in Baltimore, said in an interview. “Am I surprised by their results? Not necessarily.”
What did jump out, he said, was AED use not being a predictor of recurrence, as well as all the patients with late relapse having lesional epilepsy. “As they point out, you can have relapse with nonlesional epilepsy, but very often it happens in the first year or 2. If someone is 2 years out and doesn’t have a lesion, they’re probably more likely to remain seizure free.”
Despite the researchers’ comprehensive review of risk factors, the question remains: What to do with this information?
“They’ve done a very good job of identifying that 7.7% of 1,200 who are at risk of a late relapse,” he said. “Now, take those patients with high-risk factors and launch a trial where you keep medicines the same or do something that would alter that outcome.”
“The problem is,” he added, “that’s a 10-year study. It’s easy for me to sit here and call for one of those. But still, as valuable as this was, it’s a retrospective study. Now you have to say, what are the implications of this? What can we do in the prospective fashion?”
The authors acknowledged their study’s other limitations, including a lack of information on the reasons for an incomplete resection, a notable decrease in follow-up visits more than 5 years after surgery, and potential selection bias. They added, however, that “matching by age at surgery, gender, and time to relapse/last follow‐up” should have helped reduce any significant bias.
No potential conflicts of interest were disclosed.
FROM EPILEPSIA
SHM Converge Daily News -- Wrap-up
Click here for the wrap-up issue of the SHM Converge Daily News newsletter.
Click here for the wrap-up issue of the SHM Converge Daily News newsletter.
Click here for the wrap-up issue of the SHM Converge Daily News newsletter.
A new take on breathing and a performance-enhancing placebo
No ifs, ands, or butt ventilators
Breathing, on most days, is a pretty simple task. You inhale, the oxygen goes in, fills your lungs, becomes carbon dioxide, and is exhaled. But as certain recent events have made very clear, some diseases make this task difficult, which is where ventilators come in. The issue is, some patients can’t really use ventilators.
Enter a new study from Japan, which tested the ability of mice and pigs to absorb oxygen through the rectum. Yes, breathing through the butt. It’s not actually such a far-fetched idea; several aquatic animals such as sea cucumbers and catfish absorb oxygen through their intestines, and as any drunken frat boy can tell you after a good butt chug, other chemicals can absolutely be absorbed by human intestines.
After an initial successful experiment where a group of mice had their intestines scrubbed, had pure oxygen inserted enterally, and were exposed to a hypoxic environment, the researchers decided to step up their game and avoid the exhaustive act of digestive scrubbing by enlisting the aid of something out of science fiction: perfluorocarbon. If you haven’t seen “The Abyss,” this liquid can absorb massive amounts of oxygen, so you can actually breathe it in the same way you do with air.

In part two of the experiment, a group of hypoxic mice and pigs had perfluorocarbon inserted into their anuses, while another group got saline solution. The saline group did not fare well, but the animals that got perfluorocarbon had their hypoxic symptoms relieved within minutes.
The effectiveness of this procedure in humans clearly has yet to be tested, and while it may not be useful in all, or even most, situations, it is always beneficial to have more ways to combat a problem. Just don’t tell the frat boys: They’ll be hooking oxygen tanks up to their butts and chanting: “Breathe! Breathe! Breathe!”
Better, stronger, faster … pinker
Many people, most of whom aren’t even athletes, commit huge amounts of time, effort, and expense to improve their athletic performance. But what if there’s an easier way?
Research conducted at the University of Westminster (England) showed that participants could, with one fairly simple intervention, get on a treadmill and run 212 meters further in 30 minutes, increasing their speed by an average of 4.4%. Not only that, but “feelings of pleasure were also enhanced, meaning participants found running more enjoyable,” according to a statement from the university.
Is this amazing intervention a new wonder drug? No. Is it a super special nutritional supplement? Negatory. An energy drink that “gives you wiiings”? Nope. The latest designer steroid? Nyet.
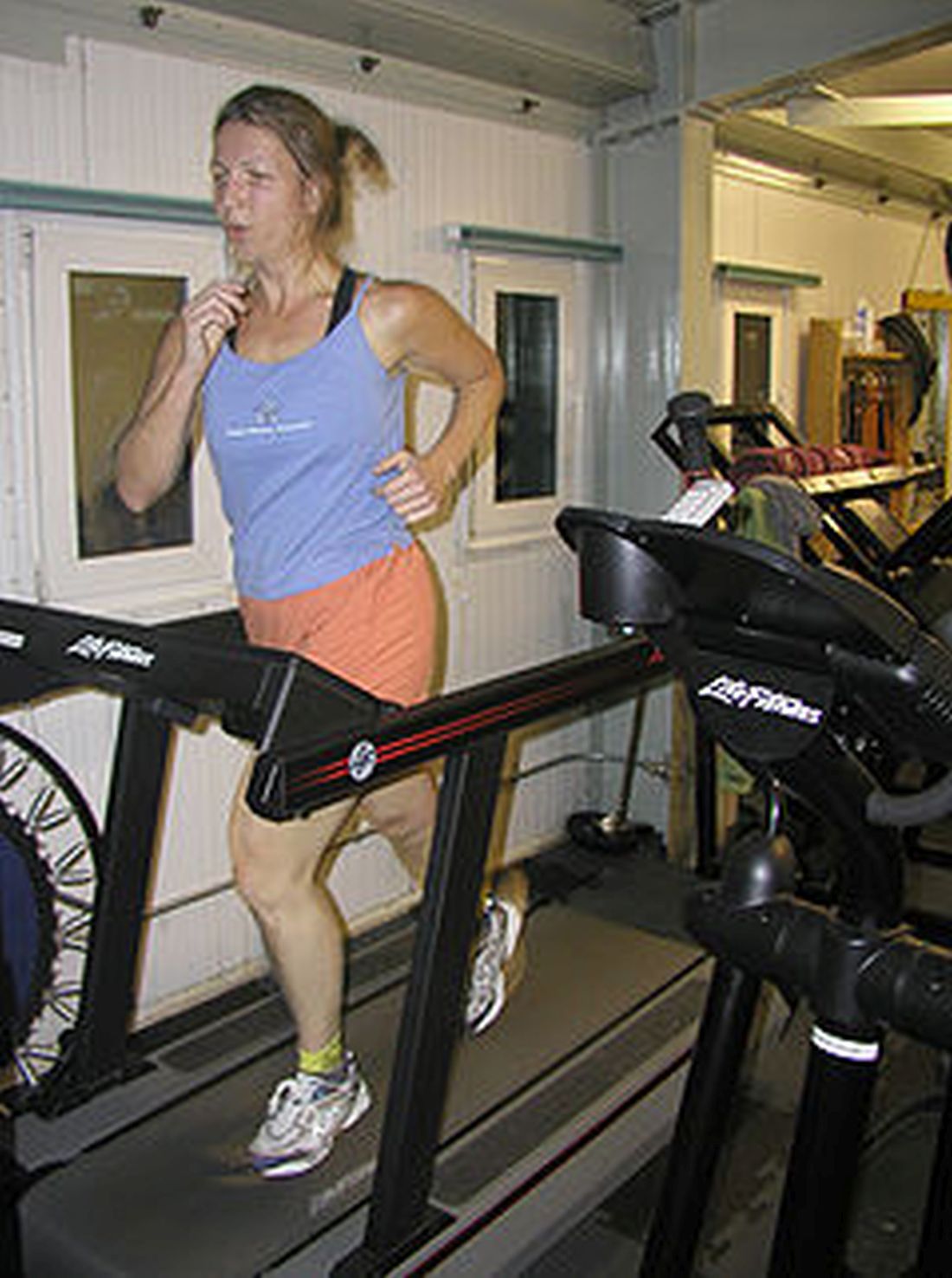
Like we said, it’s simple, and it’s pink. Literally, the color pink. We will explain.
Each of the 10 study subjects completed two 30-minute trials on the treadmill. For one, they were given a clear, artificially sweetened drink while they were running. For the other, they received the exact same drink colored pink with food dye. Pink did better. So to recap the last month in our column, faster looks pink, and skinny smells like lemons.
Once again, science demonstrates that you can’t go wrong by fooling a brain. Next week, LOTME tries to find out if purple makes you funnier.
Hey … I’m singing here!
Noise pollution has been linked to plenty of negative outcomes, but the latest target is the poor baby zebra finch.
Researchers at the Max Planck Institute of Ornithology in Germany say traffic noise disrupts the timing of vocal development and impairs learning in the flying finches. The noise was also shown to suppress their immune systems, because of lingering stress.

The good news is that the birds with noise-induced stress sang as much as their peers in a control group, so the delay in development “was not due to a lack of vocal practice,” according to researchers. However, one long-term effect could be that zebra finch birdsongs could change over time due to noise-induced copying errors. Imagine a really long game of birdsong telephone – the song at the beginning is unlikely to be the song years from now.
While not mentioned in the study, one could also imagine that due to all that exposure to traffic, young zebra finches could be developing a salty dialect and impatience with fellow finches taking up too much space on the same tree branch. Hopefully, they don’t give others “the bird.”
Slimy soap
Remember at the beginning of the pandemic when it was almost impossible to find sufficient hand-washing supplies? Just when you thought you’d tried everything, there is soap made from snail slime.
Snail slime, surprisingly, has many beneficial properties for humans. The slime has antiaging and skin healing properties and is actually used in some Korean beauty supplies. The snails even use the slime to help fix their shells if they become damaged.

Happily, no snails are harmed in the slime extraction and making of the soap. Snail farmer Damien Desrochers says, “I only touch it with my finger, you see it’s not violent, it’s simple.”
As you can probably imagine, a lot of slime is needed to have a steady supply of this soap, so Mr. Desrochers has systems in place to get enough slime. Approximately 40 snails are needed to make 15 bars of soap, and he hopes to produce about 3,000 bars in the first year.
Nothing really surprises us anymore in the beauty world: People put eggs in their hair and bee venom on their skin, so what’s wrong with a little snail slime?
No ifs, ands, or butt ventilators
Breathing, on most days, is a pretty simple task. You inhale, the oxygen goes in, fills your lungs, becomes carbon dioxide, and is exhaled. But as certain recent events have made very clear, some diseases make this task difficult, which is where ventilators come in. The issue is, some patients can’t really use ventilators.
Enter a new study from Japan, which tested the ability of mice and pigs to absorb oxygen through the rectum. Yes, breathing through the butt. It’s not actually such a far-fetched idea; several aquatic animals such as sea cucumbers and catfish absorb oxygen through their intestines, and as any drunken frat boy can tell you after a good butt chug, other chemicals can absolutely be absorbed by human intestines.
After an initial successful experiment where a group of mice had their intestines scrubbed, had pure oxygen inserted enterally, and were exposed to a hypoxic environment, the researchers decided to step up their game and avoid the exhaustive act of digestive scrubbing by enlisting the aid of something out of science fiction: perfluorocarbon. If you haven’t seen “The Abyss,” this liquid can absorb massive amounts of oxygen, so you can actually breathe it in the same way you do with air.
In part two of the experiment, a group of hypoxic mice and pigs had perfluorocarbon inserted into their anuses, while another group got saline solution. The saline group did not fare well, but the animals that got perfluorocarbon had their hypoxic symptoms relieved within minutes.
The effectiveness of this procedure in humans clearly has yet to be tested, and while it may not be useful in all, or even most, situations, it is always beneficial to have more ways to combat a problem. Just don’t tell the frat boys: They’ll be hooking oxygen tanks up to their butts and chanting: “Breathe! Breathe! Breathe!”
Better, stronger, faster … pinker
Many people, most of whom aren’t even athletes, commit huge amounts of time, effort, and expense to improve their athletic performance. But what if there’s an easier way?
Research conducted at the University of Westminster (England) showed that participants could, with one fairly simple intervention, get on a treadmill and run 212 meters further in 30 minutes, increasing their speed by an average of 4.4%. Not only that, but “feelings of pleasure were also enhanced, meaning participants found running more enjoyable,” according to a statement from the university.
Is this amazing intervention a new wonder drug? No. Is it a super special nutritional supplement? Negatory. An energy drink that “gives you wiiings”? Nope. The latest designer steroid? Nyet.
Like we said, it’s simple, and it’s pink. Literally, the color pink. We will explain.
Each of the 10 study subjects completed two 30-minute trials on the treadmill. For one, they were given a clear, artificially sweetened drink while they were running. For the other, they received the exact same drink colored pink with food dye. Pink did better. So to recap the last month in our column, faster looks pink, and skinny smells like lemons.
Once again, science demonstrates that you can’t go wrong by fooling a brain. Next week, LOTME tries to find out if purple makes you funnier.
Hey … I’m singing here!
Noise pollution has been linked to plenty of negative outcomes, but the latest target is the poor baby zebra finch.
Researchers at the Max Planck Institute of Ornithology in Germany say traffic noise disrupts the timing of vocal development and impairs learning in the flying finches. The noise was also shown to suppress their immune systems, because of lingering stress.
The good news is that the birds with noise-induced stress sang as much as their peers in a control group, so the delay in development “was not due to a lack of vocal practice,” according to researchers. However, one long-term effect could be that zebra finch birdsongs could change over time due to noise-induced copying errors. Imagine a really long game of birdsong telephone – the song at the beginning is unlikely to be the song years from now.
While not mentioned in the study, one could also imagine that due to all that exposure to traffic, young zebra finches could be developing a salty dialect and impatience with fellow finches taking up too much space on the same tree branch. Hopefully, they don’t give others “the bird.”
Slimy soap
Remember at the beginning of the pandemic when it was almost impossible to find sufficient hand-washing supplies? Just when you thought you’d tried everything, there is soap made from snail slime.
Snail slime, surprisingly, has many beneficial properties for humans. The slime has antiaging and skin healing properties and is actually used in some Korean beauty supplies. The snails even use the slime to help fix their shells if they become damaged.
Happily, no snails are harmed in the slime extraction and making of the soap. Snail farmer Damien Desrochers says, “I only touch it with my finger, you see it’s not violent, it’s simple.”
As you can probably imagine, a lot of slime is needed to have a steady supply of this soap, so Mr. Desrochers has systems in place to get enough slime. Approximately 40 snails are needed to make 15 bars of soap, and he hopes to produce about 3,000 bars in the first year.
Nothing really surprises us anymore in the beauty world: People put eggs in their hair and bee venom on their skin, so what’s wrong with a little snail slime?
No ifs, ands, or butt ventilators
Breathing, on most days, is a pretty simple task. You inhale, the oxygen goes in, fills your lungs, becomes carbon dioxide, and is exhaled. But as certain recent events have made very clear, some diseases make this task difficult, which is where ventilators come in. The issue is, some patients can’t really use ventilators.
Enter a new study from Japan, which tested the ability of mice and pigs to absorb oxygen through the rectum. Yes, breathing through the butt. It’s not actually such a far-fetched idea; several aquatic animals such as sea cucumbers and catfish absorb oxygen through their intestines, and as any drunken frat boy can tell you after a good butt chug, other chemicals can absolutely be absorbed by human intestines.
After an initial successful experiment where a group of mice had their intestines scrubbed, had pure oxygen inserted enterally, and were exposed to a hypoxic environment, the researchers decided to step up their game and avoid the exhaustive act of digestive scrubbing by enlisting the aid of something out of science fiction: perfluorocarbon. If you haven’t seen “The Abyss,” this liquid can absorb massive amounts of oxygen, so you can actually breathe it in the same way you do with air.
In part two of the experiment, a group of hypoxic mice and pigs had perfluorocarbon inserted into their anuses, while another group got saline solution. The saline group did not fare well, but the animals that got perfluorocarbon had their hypoxic symptoms relieved within minutes.
The effectiveness of this procedure in humans clearly has yet to be tested, and while it may not be useful in all, or even most, situations, it is always beneficial to have more ways to combat a problem. Just don’t tell the frat boys: They’ll be hooking oxygen tanks up to their butts and chanting: “Breathe! Breathe! Breathe!”
Better, stronger, faster … pinker
Many people, most of whom aren’t even athletes, commit huge amounts of time, effort, and expense to improve their athletic performance. But what if there’s an easier way?
Research conducted at the University of Westminster (England) showed that participants could, with one fairly simple intervention, get on a treadmill and run 212 meters further in 30 minutes, increasing their speed by an average of 4.4%. Not only that, but “feelings of pleasure were also enhanced, meaning participants found running more enjoyable,” according to a statement from the university.
Is this amazing intervention a new wonder drug? No. Is it a super special nutritional supplement? Negatory. An energy drink that “gives you wiiings”? Nope. The latest designer steroid? Nyet.
Like we said, it’s simple, and it’s pink. Literally, the color pink. We will explain.
Each of the 10 study subjects completed two 30-minute trials on the treadmill. For one, they were given a clear, artificially sweetened drink while they were running. For the other, they received the exact same drink colored pink with food dye. Pink did better. So to recap the last month in our column, faster looks pink, and skinny smells like lemons.
Once again, science demonstrates that you can’t go wrong by fooling a brain. Next week, LOTME tries to find out if purple makes you funnier.
Hey … I’m singing here!
Noise pollution has been linked to plenty of negative outcomes, but the latest target is the poor baby zebra finch.
Researchers at the Max Planck Institute of Ornithology in Germany say traffic noise disrupts the timing of vocal development and impairs learning in the flying finches. The noise was also shown to suppress their immune systems, because of lingering stress.
The good news is that the birds with noise-induced stress sang as much as their peers in a control group, so the delay in development “was not due to a lack of vocal practice,” according to researchers. However, one long-term effect could be that zebra finch birdsongs could change over time due to noise-induced copying errors. Imagine a really long game of birdsong telephone – the song at the beginning is unlikely to be the song years from now.
While not mentioned in the study, one could also imagine that due to all that exposure to traffic, young zebra finches could be developing a salty dialect and impatience with fellow finches taking up too much space on the same tree branch. Hopefully, they don’t give others “the bird.”
Slimy soap
Remember at the beginning of the pandemic when it was almost impossible to find sufficient hand-washing supplies? Just when you thought you’d tried everything, there is soap made from snail slime.
Snail slime, surprisingly, has many beneficial properties for humans. The slime has antiaging and skin healing properties and is actually used in some Korean beauty supplies. The snails even use the slime to help fix their shells if they become damaged.
Happily, no snails are harmed in the slime extraction and making of the soap. Snail farmer Damien Desrochers says, “I only touch it with my finger, you see it’s not violent, it’s simple.”
As you can probably imagine, a lot of slime is needed to have a steady supply of this soap, so Mr. Desrochers has systems in place to get enough slime. Approximately 40 snails are needed to make 15 bars of soap, and he hopes to produce about 3,000 bars in the first year.
Nothing really surprises us anymore in the beauty world: People put eggs in their hair and bee venom on their skin, so what’s wrong with a little snail slime?
Systematic review of radiofrequency microneedling studies unveiled
Of the according to results from a new systematic review.

“Most devices for aesthetic purposes induce denaturation and remodeling of collagen, elastin, and other dermal structures through tissue injury and stimulating the body’s wound-healing response,” lead study author Marcus G. Tan, MD, told this news organization during the annual conference of the American Society for Laser Medicine and Surgery. “Radiofrequency microneedling is no exception in this regard. RFMN creates perforations in the skin and delivers radiofrequency-generated thermal energy into the underlying tissue. However, RFMN is unique in that thermal energy is delivered in a fashion that produces a reverse temperature gradient to most ablative lasers.”
When using ablative lasers, which target water as its chromophore through selective photothermolysis, the temperature gradient is highest at the epidermis and papillary dermis, and decreases as it penetrates the deeper structures of the skin. In RFMN, radiofrequency energy is delivered directly to the target depth through the microneedle electrodes, thus creating a temperature gradient that is highest in the deep, target structures and cooler at the superficial structures. “This results in less unwanted epidermal heating and reduces the risk of postinflammatory hyperpigmentation,” explained Dr. Tan, a resident in the division of dermatology at the University of Ottawa.
“Because RFMN is unaffected by skin chromophores, it is essentially a ‘color-blind’ technology and safe for use in patients of all skin phototypes. In comparison to lasers, radiofrequency energy can also be delivered to deeper structures of the skin by increasing the length of microneedle electrodes. Despite these advantages of RFMN, this technology remains utilized less frequently compared to ablative lasers for its skin rejuvenating effects.”
To review high-quality medical literature related to RFMN, Dr. Tan and colleagues searched EMBASE and MEDLINE from inception to May 13, 2020, by using the terms “radiofrequency microneedling,” “fractional radiofrequency,” “radiofrequency needling,” or “radiofrequency percutaneous collagen induction.” They limited the analysis to dermatology-related randomized, split-body, or blinded studies with original data in humans. Of the 42 studies included in the final analysis, there were 14 studies of skin rejuvenation, 7 of acne scars, 6 of acne vulgaris, 5 each of striae and axillary hyperhidrosis, 2 of melasma, and 1 each of rosacea, cellulite, and androgenetic alopecia.
After reviewing the 42 studies, the study authors proposed that a strong recommendation for RFMN be made for skin rejuvenation, acne vulgaris, acne scars, and axillary hyperhidrosis, and a weak recommendation for the technology to be used for papulopustular rosacea, striae, and male-pattern androgenetic alopecia when used in conjunction with topical 5% minoxidil. There was insufficient evidence to make recommendations for its use in cellulite and melasma.
One finding that Dr. Tan described as “interesting” was the observation that RFMN was superior to Er:YAG fractional ablative lasers for treatment of rhytides on the lower face (i.e., the nasolabial, perioral, jawline and neck regions). “Secondly, we observed that one session of RFMN was able to achieve 37% efficacy of a surgical face-lift, but without any adverse effects,” Dr. Tan said. “Two-thirds of the patients who received surgical face-lift developed hypertrophic scarring requiring further scar management, compared to none of the patients receiving RFMN.”
Based on their review, Dr. Tan and colleagues recommend that RFMN be offered as one of the therapeutic options for patients seeking treatment for skin rejuvenation, acne vulgaris, acne scars, and axillary hyperhidrosis. “It is usually tolerable with just topical anesthesia applied 30-60 minutes before treatment, and its side effects are transient and usually resolve after 5 days,” he said. “Patients should be counseled that the benefits of RFMN may have a slower onset, compared to other treatments, but it is progressive, durable, and can be used repeatedly and safely in all skin types including darker-skin phenotypes with minimal risk of adverse events.”

One of the abstract section chairs, Fernanda H. Sakamoto, MD, PhD, said that RFMN devices have become increasingly popular in recent years. “The paper presented by Tan et al. is very relevant, as it compares clinical indications, parameters, and results in search for evidence of efficacy and appropriate settings,” said Dr. Sakamoto, a dermatologist at the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, told this news organization. “The paper provides long-needed guidelines to clinicians and helps manage patients’ expectations.”
Dr. Tan acknowledged certain limitations of the study, including the lack of head-to-head studies comparing specific RFMN devices. “There are many RFMN devices available commercially, each with different capabilities and degrees of effectiveness,” he said. “With more research and technological advancements since the first radiofrequency device was approved in 2002, RFMN has made significant improvements. In general, the newer generation devices produce markedly better results.”
Dr. Tan reported having no financial disclosures. Dr. Sakamoto disclosed that she holds intellectual property rights with Accure Acne, Massachusetts General Hospital, and Lightwater Biosciences.
Of the according to results from a new systematic review.
“Most devices for aesthetic purposes induce denaturation and remodeling of collagen, elastin, and other dermal structures through tissue injury and stimulating the body’s wound-healing response,” lead study author Marcus G. Tan, MD, told this news organization during the annual conference of the American Society for Laser Medicine and Surgery. “Radiofrequency microneedling is no exception in this regard. RFMN creates perforations in the skin and delivers radiofrequency-generated thermal energy into the underlying tissue. However, RFMN is unique in that thermal energy is delivered in a fashion that produces a reverse temperature gradient to most ablative lasers.”
When using ablative lasers, which target water as its chromophore through selective photothermolysis, the temperature gradient is highest at the epidermis and papillary dermis, and decreases as it penetrates the deeper structures of the skin. In RFMN, radiofrequency energy is delivered directly to the target depth through the microneedle electrodes, thus creating a temperature gradient that is highest in the deep, target structures and cooler at the superficial structures. “This results in less unwanted epidermal heating and reduces the risk of postinflammatory hyperpigmentation,” explained Dr. Tan, a resident in the division of dermatology at the University of Ottawa.
“Because RFMN is unaffected by skin chromophores, it is essentially a ‘color-blind’ technology and safe for use in patients of all skin phototypes. In comparison to lasers, radiofrequency energy can also be delivered to deeper structures of the skin by increasing the length of microneedle electrodes. Despite these advantages of RFMN, this technology remains utilized less frequently compared to ablative lasers for its skin rejuvenating effects.”
To review high-quality medical literature related to RFMN, Dr. Tan and colleagues searched EMBASE and MEDLINE from inception to May 13, 2020, by using the terms “radiofrequency microneedling,” “fractional radiofrequency,” “radiofrequency needling,” or “radiofrequency percutaneous collagen induction.” They limited the analysis to dermatology-related randomized, split-body, or blinded studies with original data in humans. Of the 42 studies included in the final analysis, there were 14 studies of skin rejuvenation, 7 of acne scars, 6 of acne vulgaris, 5 each of striae and axillary hyperhidrosis, 2 of melasma, and 1 each of rosacea, cellulite, and androgenetic alopecia.
After reviewing the 42 studies, the study authors proposed that a strong recommendation for RFMN be made for skin rejuvenation, acne vulgaris, acne scars, and axillary hyperhidrosis, and a weak recommendation for the technology to be used for papulopustular rosacea, striae, and male-pattern androgenetic alopecia when used in conjunction with topical 5% minoxidil. There was insufficient evidence to make recommendations for its use in cellulite and melasma.
One finding that Dr. Tan described as “interesting” was the observation that RFMN was superior to Er:YAG fractional ablative lasers for treatment of rhytides on the lower face (i.e., the nasolabial, perioral, jawline and neck regions). “Secondly, we observed that one session of RFMN was able to achieve 37% efficacy of a surgical face-lift, but without any adverse effects,” Dr. Tan said. “Two-thirds of the patients who received surgical face-lift developed hypertrophic scarring requiring further scar management, compared to none of the patients receiving RFMN.”
Based on their review, Dr. Tan and colleagues recommend that RFMN be offered as one of the therapeutic options for patients seeking treatment for skin rejuvenation, acne vulgaris, acne scars, and axillary hyperhidrosis. “It is usually tolerable with just topical anesthesia applied 30-60 minutes before treatment, and its side effects are transient and usually resolve after 5 days,” he said. “Patients should be counseled that the benefits of RFMN may have a slower onset, compared to other treatments, but it is progressive, durable, and can be used repeatedly and safely in all skin types including darker-skin phenotypes with minimal risk of adverse events.”
One of the abstract section chairs, Fernanda H. Sakamoto, MD, PhD, said that RFMN devices have become increasingly popular in recent years. “The paper presented by Tan et al. is very relevant, as it compares clinical indications, parameters, and results in search for evidence of efficacy and appropriate settings,” said Dr. Sakamoto, a dermatologist at the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, told this news organization. “The paper provides long-needed guidelines to clinicians and helps manage patients’ expectations.”
Dr. Tan acknowledged certain limitations of the study, including the lack of head-to-head studies comparing specific RFMN devices. “There are many RFMN devices available commercially, each with different capabilities and degrees of effectiveness,” he said. “With more research and technological advancements since the first radiofrequency device was approved in 2002, RFMN has made significant improvements. In general, the newer generation devices produce markedly better results.”
Dr. Tan reported having no financial disclosures. Dr. Sakamoto disclosed that she holds intellectual property rights with Accure Acne, Massachusetts General Hospital, and Lightwater Biosciences.
Of the according to results from a new systematic review.
“Most devices for aesthetic purposes induce denaturation and remodeling of collagen, elastin, and other dermal structures through tissue injury and stimulating the body’s wound-healing response,” lead study author Marcus G. Tan, MD, told this news organization during the annual conference of the American Society for Laser Medicine and Surgery. “Radiofrequency microneedling is no exception in this regard. RFMN creates perforations in the skin and delivers radiofrequency-generated thermal energy into the underlying tissue. However, RFMN is unique in that thermal energy is delivered in a fashion that produces a reverse temperature gradient to most ablative lasers.”
When using ablative lasers, which target water as its chromophore through selective photothermolysis, the temperature gradient is highest at the epidermis and papillary dermis, and decreases as it penetrates the deeper structures of the skin. In RFMN, radiofrequency energy is delivered directly to the target depth through the microneedle electrodes, thus creating a temperature gradient that is highest in the deep, target structures and cooler at the superficial structures. “This results in less unwanted epidermal heating and reduces the risk of postinflammatory hyperpigmentation,” explained Dr. Tan, a resident in the division of dermatology at the University of Ottawa.
“Because RFMN is unaffected by skin chromophores, it is essentially a ‘color-blind’ technology and safe for use in patients of all skin phototypes. In comparison to lasers, radiofrequency energy can also be delivered to deeper structures of the skin by increasing the length of microneedle electrodes. Despite these advantages of RFMN, this technology remains utilized less frequently compared to ablative lasers for its skin rejuvenating effects.”
To review high-quality medical literature related to RFMN, Dr. Tan and colleagues searched EMBASE and MEDLINE from inception to May 13, 2020, by using the terms “radiofrequency microneedling,” “fractional radiofrequency,” “radiofrequency needling,” or “radiofrequency percutaneous collagen induction.” They limited the analysis to dermatology-related randomized, split-body, or blinded studies with original data in humans. Of the 42 studies included in the final analysis, there were 14 studies of skin rejuvenation, 7 of acne scars, 6 of acne vulgaris, 5 each of striae and axillary hyperhidrosis, 2 of melasma, and 1 each of rosacea, cellulite, and androgenetic alopecia.
After reviewing the 42 studies, the study authors proposed that a strong recommendation for RFMN be made for skin rejuvenation, acne vulgaris, acne scars, and axillary hyperhidrosis, and a weak recommendation for the technology to be used for papulopustular rosacea, striae, and male-pattern androgenetic alopecia when used in conjunction with topical 5% minoxidil. There was insufficient evidence to make recommendations for its use in cellulite and melasma.
One finding that Dr. Tan described as “interesting” was the observation that RFMN was superior to Er:YAG fractional ablative lasers for treatment of rhytides on the lower face (i.e., the nasolabial, perioral, jawline and neck regions). “Secondly, we observed that one session of RFMN was able to achieve 37% efficacy of a surgical face-lift, but without any adverse effects,” Dr. Tan said. “Two-thirds of the patients who received surgical face-lift developed hypertrophic scarring requiring further scar management, compared to none of the patients receiving RFMN.”
Based on their review, Dr. Tan and colleagues recommend that RFMN be offered as one of the therapeutic options for patients seeking treatment for skin rejuvenation, acne vulgaris, acne scars, and axillary hyperhidrosis. “It is usually tolerable with just topical anesthesia applied 30-60 minutes before treatment, and its side effects are transient and usually resolve after 5 days,” he said. “Patients should be counseled that the benefits of RFMN may have a slower onset, compared to other treatments, but it is progressive, durable, and can be used repeatedly and safely in all skin types including darker-skin phenotypes with minimal risk of adverse events.”
One of the abstract section chairs, Fernanda H. Sakamoto, MD, PhD, said that RFMN devices have become increasingly popular in recent years. “The paper presented by Tan et al. is very relevant, as it compares clinical indications, parameters, and results in search for evidence of efficacy and appropriate settings,” said Dr. Sakamoto, a dermatologist at the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, told this news organization. “The paper provides long-needed guidelines to clinicians and helps manage patients’ expectations.”
Dr. Tan acknowledged certain limitations of the study, including the lack of head-to-head studies comparing specific RFMN devices. “There are many RFMN devices available commercially, each with different capabilities and degrees of effectiveness,” he said. “With more research and technological advancements since the first radiofrequency device was approved in 2002, RFMN has made significant improvements. In general, the newer generation devices produce markedly better results.”
Dr. Tan reported having no financial disclosures. Dr. Sakamoto disclosed that she holds intellectual property rights with Accure Acne, Massachusetts General Hospital, and Lightwater Biosciences.
FROM ASLMS 2021
Photobiomodulation reduced acute radiodermatitis severity in head and neck cancer patients
The delivery of , according to results from the first randomized study of its kind.

“The use of light therapy-based applications for cancer therapy-related adverse events has steadily increased in the past 40 years,” lead study author Jolien Robijns, MSc, PhD, told this news organization during the annual conference of the American Society for Laser Medicine and Surgery. “The most well-known and studied indication of photobiomodulation therapy in supportive cancer care is oral mucositis,” she said, referring to a recent systematic review, which found that based on the available evidence, PBMT is an effective therapy for the prevention of oral mucositis, using well-defined PBM parameters in specific patient populations. “Various internationally well-recognized health organizations in oncology recommend PBMT to prevent and manage oral mucositis,” she added.
Based on the wound-healing and anti-inflammatory properties of PBMT, several studies have investigated its use for the prevention and management of acute radiodermatitis (ARD) since the 1990s, said Dr. Robijns, a postdoctoral researcher at Limburg Clinical Research Center in Hasselt, Belgium. Under the supervision of Jeroen Mebis, MD, PhD, at the Limburg Oncologic Laser Institute, she and her colleagues have been conducting clinical research on PBMT and ARD since 2014, with successful results. In 2020 they published a narrative review, which showed that based on nine clinical trials, PBMT could effectively reduce the incidence of severe ARD, decrease accompanying pain, and improve patients’ quality of life.
For the current study, known as the DERMISHEAD trial and published online March 9, 2021, in Radiotherapy and Oncology, investigators at Limburg Oncology Center at Jessa Hospital in Hasselt, and Hasselt University, recruited head and neck cancer patients who underwent bilateral radiotherapy with or without chemotherapy, for a total dose of 30-35 x 2 Gy . All patients received standard skin care combined with two PBMT or sham sessions twice per week during the complete course of RT, which resulted in 14 total sessions.
As described in the Radiotherapy and Oncology study, the commercially available device used for PBMT “consists of two laser diodes with different wavelengths (808-905 nm), peak powers (1.1-25 W), and emission modes (continuous and pulsed). Both diodes work simultaneously and synchronously with coincident propagation axes (average radiant power 3.3 W). The energy density (fluence) was set at 4 J/cm2 based on earlier recommendations and on our clinical experience.” A blinded study nurse used Radiation Therapy Oncology Group criteria to evaluate the skin reactions.
After 303 patients were initially assessed for eligibility, 46 patients were enrolled in DERMISHEAD (18 in the placebo group and 28 in the PBMT group). At the end of radiotherapy, 77.8% of patients in the placebo group had a grade 2 or 3 skin reaction, compared with 28.6% of patients in the PBMT group (P = .001).
“The DERMISHEAD trial proved that PBMT significantly reduces the severity of ARD,” Dr. Robijns said. “Thereby, it improves the patients’ quality of life during their radiotherapy course. The trial supports the further implementation of PBM in the supportive care of cancer patients undergoing radiotherapy.”
The results are similar to those in the TRANSDERMIS trial, in which Dr. Robijns and her colleagues used PMBT to treat breast cancer patients.
“However, an interesting difference is that the percentage decrease in severe ARD was higher in the DERMISHEAD trial than in the TRANSDERMIS trial: 49% vs. 23%, respectively,” she noted. “This difference can be rationalized because in total, more control head and neck cancer patients developed grade 3 ARD than did control breast cancer patients (17% vs. 5%). A possible explanation of this finding can be related to the difference in treatment regimens and radiotherapy parameters between the two trials.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Conn., who was asked to comment on the study, said that acute radiation dermatitis “can be very painful and distressing to patients, and over time, the skin changes can create long-term problems. Prevention of acute and chronic radiation dermatitis is worthwhile, particularly for patients at risk.”
This study, she added, “shows a benefit of photobiomodulation therapy as a potential preventative treatment. Notably, patients did not always follow up appropriately for the therapy, and the authors said that it is yet another thing that patients need to keep track of, in addition to their cancer therapy visits. Thus, optimally, it would be useful to have a biomarker of which patients would most benefit from treatments that prevent/potentiate radiation dermatitis.”
Dr. Robijns acknowledged certain limitations of the trial, including its small sample size and the scarcity of clinical trials on PBM and acute radiation dermatitis. “More studies are needed,” she said. “Future studies should focus on randomized controlled study designs with well-described and complete PBMT parameters in a larger and more diverse patient population. This would enable the implementation of PBM in the field of ARD and supportive cancer care, which would enhance wound care management and improve the patient’s quality of life.”
This work won a “best of clinical applications” abstract award from the ASLMS.
The research is part of the Limburg Clinical Research Center UHasselt-ZOL-Jessa, financially supported by the foundation Limburg Sterk Merk, province of Limburg, Flemish Government, Hasselt University, Ziekenhuis Oost-Limburg, and Jessa Hospital. The research is also funded by Kom op tegen Kanker (Stand up to Cancer), the Flemish Cancer Society, Limburgs Kankerfonds, and ASA Srl. Dr. Robijns reported having no financial disclosures.
The delivery of , according to results from the first randomized study of its kind.
“The use of light therapy-based applications for cancer therapy-related adverse events has steadily increased in the past 40 years,” lead study author Jolien Robijns, MSc, PhD, told this news organization during the annual conference of the American Society for Laser Medicine and Surgery. “The most well-known and studied indication of photobiomodulation therapy in supportive cancer care is oral mucositis,” she said, referring to a recent systematic review, which found that based on the available evidence, PBMT is an effective therapy for the prevention of oral mucositis, using well-defined PBM parameters in specific patient populations. “Various internationally well-recognized health organizations in oncology recommend PBMT to prevent and manage oral mucositis,” she added.
Based on the wound-healing and anti-inflammatory properties of PBMT, several studies have investigated its use for the prevention and management of acute radiodermatitis (ARD) since the 1990s, said Dr. Robijns, a postdoctoral researcher at Limburg Clinical Research Center in Hasselt, Belgium. Under the supervision of Jeroen Mebis, MD, PhD, at the Limburg Oncologic Laser Institute, she and her colleagues have been conducting clinical research on PBMT and ARD since 2014, with successful results. In 2020 they published a narrative review, which showed that based on nine clinical trials, PBMT could effectively reduce the incidence of severe ARD, decrease accompanying pain, and improve patients’ quality of life.
For the current study, known as the DERMISHEAD trial and published online March 9, 2021, in Radiotherapy and Oncology, investigators at Limburg Oncology Center at Jessa Hospital in Hasselt, and Hasselt University, recruited head and neck cancer patients who underwent bilateral radiotherapy with or without chemotherapy, for a total dose of 30-35 x 2 Gy . All patients received standard skin care combined with two PBMT or sham sessions twice per week during the complete course of RT, which resulted in 14 total sessions.
As described in the Radiotherapy and Oncology study, the commercially available device used for PBMT “consists of two laser diodes with different wavelengths (808-905 nm), peak powers (1.1-25 W), and emission modes (continuous and pulsed). Both diodes work simultaneously and synchronously with coincident propagation axes (average radiant power 3.3 W). The energy density (fluence) was set at 4 J/cm2 based on earlier recommendations and on our clinical experience.” A blinded study nurse used Radiation Therapy Oncology Group criteria to evaluate the skin reactions.
After 303 patients were initially assessed for eligibility, 46 patients were enrolled in DERMISHEAD (18 in the placebo group and 28 in the PBMT group). At the end of radiotherapy, 77.8% of patients in the placebo group had a grade 2 or 3 skin reaction, compared with 28.6% of patients in the PBMT group (P = .001).
“The DERMISHEAD trial proved that PBMT significantly reduces the severity of ARD,” Dr. Robijns said. “Thereby, it improves the patients’ quality of life during their radiotherapy course. The trial supports the further implementation of PBM in the supportive care of cancer patients undergoing radiotherapy.”
The results are similar to those in the TRANSDERMIS trial, in which Dr. Robijns and her colleagues used PMBT to treat breast cancer patients.
“However, an interesting difference is that the percentage decrease in severe ARD was higher in the DERMISHEAD trial than in the TRANSDERMIS trial: 49% vs. 23%, respectively,” she noted. “This difference can be rationalized because in total, more control head and neck cancer patients developed grade 3 ARD than did control breast cancer patients (17% vs. 5%). A possible explanation of this finding can be related to the difference in treatment regimens and radiotherapy parameters between the two trials.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Conn., who was asked to comment on the study, said that acute radiation dermatitis “can be very painful and distressing to patients, and over time, the skin changes can create long-term problems. Prevention of acute and chronic radiation dermatitis is worthwhile, particularly for patients at risk.”
This study, she added, “shows a benefit of photobiomodulation therapy as a potential preventative treatment. Notably, patients did not always follow up appropriately for the therapy, and the authors said that it is yet another thing that patients need to keep track of, in addition to their cancer therapy visits. Thus, optimally, it would be useful to have a biomarker of which patients would most benefit from treatments that prevent/potentiate radiation dermatitis.”
Dr. Robijns acknowledged certain limitations of the trial, including its small sample size and the scarcity of clinical trials on PBM and acute radiation dermatitis. “More studies are needed,” she said. “Future studies should focus on randomized controlled study designs with well-described and complete PBMT parameters in a larger and more diverse patient population. This would enable the implementation of PBM in the field of ARD and supportive cancer care, which would enhance wound care management and improve the patient’s quality of life.”
This work won a “best of clinical applications” abstract award from the ASLMS.
The research is part of the Limburg Clinical Research Center UHasselt-ZOL-Jessa, financially supported by the foundation Limburg Sterk Merk, province of Limburg, Flemish Government, Hasselt University, Ziekenhuis Oost-Limburg, and Jessa Hospital. The research is also funded by Kom op tegen Kanker (Stand up to Cancer), the Flemish Cancer Society, Limburgs Kankerfonds, and ASA Srl. Dr. Robijns reported having no financial disclosures.
The delivery of , according to results from the first randomized study of its kind.
“The use of light therapy-based applications for cancer therapy-related adverse events has steadily increased in the past 40 years,” lead study author Jolien Robijns, MSc, PhD, told this news organization during the annual conference of the American Society for Laser Medicine and Surgery. “The most well-known and studied indication of photobiomodulation therapy in supportive cancer care is oral mucositis,” she said, referring to a recent systematic review, which found that based on the available evidence, PBMT is an effective therapy for the prevention of oral mucositis, using well-defined PBM parameters in specific patient populations. “Various internationally well-recognized health organizations in oncology recommend PBMT to prevent and manage oral mucositis,” she added.
Based on the wound-healing and anti-inflammatory properties of PBMT, several studies have investigated its use for the prevention and management of acute radiodermatitis (ARD) since the 1990s, said Dr. Robijns, a postdoctoral researcher at Limburg Clinical Research Center in Hasselt, Belgium. Under the supervision of Jeroen Mebis, MD, PhD, at the Limburg Oncologic Laser Institute, she and her colleagues have been conducting clinical research on PBMT and ARD since 2014, with successful results. In 2020 they published a narrative review, which showed that based on nine clinical trials, PBMT could effectively reduce the incidence of severe ARD, decrease accompanying pain, and improve patients’ quality of life.
For the current study, known as the DERMISHEAD trial and published online March 9, 2021, in Radiotherapy and Oncology, investigators at Limburg Oncology Center at Jessa Hospital in Hasselt, and Hasselt University, recruited head and neck cancer patients who underwent bilateral radiotherapy with or without chemotherapy, for a total dose of 30-35 x 2 Gy . All patients received standard skin care combined with two PBMT or sham sessions twice per week during the complete course of RT, which resulted in 14 total sessions.
As described in the Radiotherapy and Oncology study, the commercially available device used for PBMT “consists of two laser diodes with different wavelengths (808-905 nm), peak powers (1.1-25 W), and emission modes (continuous and pulsed). Both diodes work simultaneously and synchronously with coincident propagation axes (average radiant power 3.3 W). The energy density (fluence) was set at 4 J/cm2 based on earlier recommendations and on our clinical experience.” A blinded study nurse used Radiation Therapy Oncology Group criteria to evaluate the skin reactions.
After 303 patients were initially assessed for eligibility, 46 patients were enrolled in DERMISHEAD (18 in the placebo group and 28 in the PBMT group). At the end of radiotherapy, 77.8% of patients in the placebo group had a grade 2 or 3 skin reaction, compared with 28.6% of patients in the PBMT group (P = .001).
“The DERMISHEAD trial proved that PBMT significantly reduces the severity of ARD,” Dr. Robijns said. “Thereby, it improves the patients’ quality of life during their radiotherapy course. The trial supports the further implementation of PBM in the supportive care of cancer patients undergoing radiotherapy.”
The results are similar to those in the TRANSDERMIS trial, in which Dr. Robijns and her colleagues used PMBT to treat breast cancer patients.
“However, an interesting difference is that the percentage decrease in severe ARD was higher in the DERMISHEAD trial than in the TRANSDERMIS trial: 49% vs. 23%, respectively,” she noted. “This difference can be rationalized because in total, more control head and neck cancer patients developed grade 3 ARD than did control breast cancer patients (17% vs. 5%). A possible explanation of this finding can be related to the difference in treatment regimens and radiotherapy parameters between the two trials.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Conn., who was asked to comment on the study, said that acute radiation dermatitis “can be very painful and distressing to patients, and over time, the skin changes can create long-term problems. Prevention of acute and chronic radiation dermatitis is worthwhile, particularly for patients at risk.”
This study, she added, “shows a benefit of photobiomodulation therapy as a potential preventative treatment. Notably, patients did not always follow up appropriately for the therapy, and the authors said that it is yet another thing that patients need to keep track of, in addition to their cancer therapy visits. Thus, optimally, it would be useful to have a biomarker of which patients would most benefit from treatments that prevent/potentiate radiation dermatitis.”
Dr. Robijns acknowledged certain limitations of the trial, including its small sample size and the scarcity of clinical trials on PBM and acute radiation dermatitis. “More studies are needed,” she said. “Future studies should focus on randomized controlled study designs with well-described and complete PBMT parameters in a larger and more diverse patient population. This would enable the implementation of PBM in the field of ARD and supportive cancer care, which would enhance wound care management and improve the patient’s quality of life.”
This work won a “best of clinical applications” abstract award from the ASLMS.
The research is part of the Limburg Clinical Research Center UHasselt-ZOL-Jessa, financially supported by the foundation Limburg Sterk Merk, province of Limburg, Flemish Government, Hasselt University, Ziekenhuis Oost-Limburg, and Jessa Hospital. The research is also funded by Kom op tegen Kanker (Stand up to Cancer), the Flemish Cancer Society, Limburgs Kankerfonds, and ASA Srl. Dr. Robijns reported having no financial disclosures.
FROM ASLMS 2021
Painful ingrown nail

Paronychia, or an ingrown nail, is caused by the introduction of bacteria into the nail fold, which can cause cellulitis and/or abscess formation. Typically, a single nail is involved, although multiple nails may be affected in rare, chemotherapy-induced cases. Generally, nail grooming behaviors, abnormal or scarred nail matrix architecture, nail biting, and wet work are the causes of acute or chronic disease.1
Conservative options are a reasonable first choice when there is no abscess. These include soaks 2 to 3 times daily for 15 to 20 minutes in hot water or water treated with an antiseptic, such as chlorhexidine. Topical or oral antibiotics aimed at suspected organisms may be helpful. Staphylococcus aureus, including methicillin-resistant Staphylococcus aureus, is a common isolate. Psuedomonas is characteristic in cases involving trauma to the hand and frequent workplace water exposure, as can happen in foodservice, farming, and marine industries.
When an abscess has formed (as in this case), or when conservative treatments fail, incision and drainage or partial nail avulsion can be curative and successful with, or without, antibiotics. Patients with recurrent paronychia in the same location may benefit from partial nail avulsion with partial matrix removal. This can be done surgically or with phenol.
In this case, the patient underwent lateral partial nail avulsion, which also served as an incision and drainage. She was not given antibiotics and her nail regrew within 6 months; there was no recurrence. She was counseled not to cut the nail plate too aggressively.
Text and photos courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. (Photo copyright retained.)
Rigopoulos D, Larios G, Gregoriou S, et al. Acute and chronic paronychia. Am Fam Physician. 2008;77:339-346.
Paronychia, or an ingrown nail, is caused by the introduction of bacteria into the nail fold, which can cause cellulitis and/or abscess formation. Typically, a single nail is involved, although multiple nails may be affected in rare, chemotherapy-induced cases. Generally, nail grooming behaviors, abnormal or scarred nail matrix architecture, nail biting, and wet work are the causes of acute or chronic disease.1
Conservative options are a reasonable first choice when there is no abscess. These include soaks 2 to 3 times daily for 15 to 20 minutes in hot water or water treated with an antiseptic, such as chlorhexidine. Topical or oral antibiotics aimed at suspected organisms may be helpful. Staphylococcus aureus, including methicillin-resistant Staphylococcus aureus, is a common isolate. Psuedomonas is characteristic in cases involving trauma to the hand and frequent workplace water exposure, as can happen in foodservice, farming, and marine industries.
When an abscess has formed (as in this case), or when conservative treatments fail, incision and drainage or partial nail avulsion can be curative and successful with, or without, antibiotics. Patients with recurrent paronychia in the same location may benefit from partial nail avulsion with partial matrix removal. This can be done surgically or with phenol.
In this case, the patient underwent lateral partial nail avulsion, which also served as an incision and drainage. She was not given antibiotics and her nail regrew within 6 months; there was no recurrence. She was counseled not to cut the nail plate too aggressively.
Text and photos courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. (Photo copyright retained.)
Paronychia, or an ingrown nail, is caused by the introduction of bacteria into the nail fold, which can cause cellulitis and/or abscess formation. Typically, a single nail is involved, although multiple nails may be affected in rare, chemotherapy-induced cases. Generally, nail grooming behaviors, abnormal or scarred nail matrix architecture, nail biting, and wet work are the causes of acute or chronic disease.1
Conservative options are a reasonable first choice when there is no abscess. These include soaks 2 to 3 times daily for 15 to 20 minutes in hot water or water treated with an antiseptic, such as chlorhexidine. Topical or oral antibiotics aimed at suspected organisms may be helpful. Staphylococcus aureus, including methicillin-resistant Staphylococcus aureus, is a common isolate. Psuedomonas is characteristic in cases involving trauma to the hand and frequent workplace water exposure, as can happen in foodservice, farming, and marine industries.
When an abscess has formed (as in this case), or when conservative treatments fail, incision and drainage or partial nail avulsion can be curative and successful with, or without, antibiotics. Patients with recurrent paronychia in the same location may benefit from partial nail avulsion with partial matrix removal. This can be done surgically or with phenol.
In this case, the patient underwent lateral partial nail avulsion, which also served as an incision and drainage. She was not given antibiotics and her nail regrew within 6 months; there was no recurrence. She was counseled not to cut the nail plate too aggressively.
Text and photos courtesy of Jonathan Karnes, MD, medical director, MDFMR Dermatology Services, Augusta, ME. (Photo copyright retained.)
Rigopoulos D, Larios G, Gregoriou S, et al. Acute and chronic paronychia. Am Fam Physician. 2008;77:339-346.
Rigopoulos D, Larios G, Gregoriou S, et al. Acute and chronic paronychia. Am Fam Physician. 2008;77:339-346.

Medical homes a boon to patients with bleeding disorders
As bleeding disorders are increasingly recognized as a national health priority, hematologists are focusing on how the patient-centered medical home – a widely accepted concept in primary care and in some specialties – can improve outcomes and quality life for their patients.

The patient-centered medical home is a model of health care delivery in which patients receive comprehensive, accessible care that is fully integrated across all providers and elements of a healthcare system.1 The concept emerged in the 1960s among pediatricians seeking to better coordinate care for children with complex medical needs. Since then, the patient-centered medical home has become a globally recognized standard – not only in primary care, but also in specialties such as endocrinology, oncology, and geriatric medicine. The movement to establish medical homes for patients with bleeding disorders is more recent and is receiving national attention.
Why a medical home?
The advent of prophylactic therapies for bleeding disorders has vastly improved the outlook for many patients compared to just a few decades ago. However, treatment options remain limited, and patients who have severe disease or complications – such as an inadequate treatment response or the development of inhibitory antibodies to replacement clotting factors – are at risk for recurrent breakthrough bleeding that can lead to synovitis and ultimately culminate in progressive, irreversible joint damage. The resulting pain and limitation of motion greatly compromises patients’ quality of life across physical, psychological, and social domains, undermines their ability to live and work independently, and greatly increases treatment costs.2-4 Family members, too, face high stress and lower quality of life when they struggle to obtain and manage treatment while caring for loved ones with bleeding disorders.5
For patients with bleeding disorders, a patient-centered medical home can help address or surmount these challenges, said Amy Shapiro, MD, medical director of the Indiana Hemophilia and Thrombosis Center in Indianapolis, Ind., which was the first hemophilia treatment center in the country to be formally certified as a medical home.
Dr. Shapiro explained that a patient-centered medical home leverages the care of an integrated multidisciplinary team to help optimize therapies and patient outcomes across all domains of life. She sees the medical home concept as a natural fit for patients with bleeding disorders, given the complexity of their needs and the number of specialties involved. “When you have hemophilia, you don’t just need a hematologist to manage your care. You need nurses, physical therapists, and social workers. You need coordinated care for genetic counseling. You also need to coordinate dental hygiene and surgical interventions, if these are required. Patients need nutrition counseling, and they may need assistance with education or career options if too many days are missed from work or school. Patients or their families may need counseling on choosing the right insurance program so they don’t choose a plan that may create more hardships for them because of their chronic disorder.”
Meeting these needs requires the help of an integrated care team, which many individuals with bleeding disorders lack. “If you are just out there in the community and you have medical issues that need to be dealt with, often the individuals themselves have to coordinate their own care, including their medications and their appointments with different specialists,” said Dr. Shapiro. “For example, a care provider may tell a patient that they need a physical therapist and give them some names, and then the patient has to take it from there and not only find the provider, but also determine if their insurance provides coverage.”
A medical home takes a completely different approach, she explained. “At my center, when we say you need a physical therapist, we have a physical therapist on staff. Our therapist provides an assessment and determines the need for ongoing PT and whether that can be done at home with a plan and intermittent oversight, or whether the patient needs a referral, and whether the person the patient is referred to needs education on how to provide PT for someone with hemophilia. A medical home provides all this in one place. It is a place where patients know they will receive either direct services, or support to shepherd their care and outcomes, and oversight of that support as well.”
Few studies have directly assessed the medical home model in the setting of bleeding disorders, but a number have evaluated the impact of integrated care, a more general term for the practice of coordinating multidisciplinary care to improve access and outcomes while eliminating redundancies and unnecessary costs. In a recent systematic review and meta-analysis of 27 nonrandomized studies of patients with hemophilia, integrated care was linked to lower mortality, fewer emergency room visits and hospitalizations, shorter lengths of stay in the hospital, and fewer missed days of school and work.6 Such findings, combined with promising outcomes data from studies of patient-centered medical homes in other disease settings, suggest that the patient-centered medical home can significantly benefit patients with bleeding disorders and their families and caregivers.
Creating a medical home
Establishing a patient-centered medical home can be challenging, involving a plethora of stakeholders and a considerable investment of time, energy, and resources. Organizations such as the National Committee for Quality Assurance and the Accreditation Association for Ambulatory Health Care have formal certification programs to help ensure that an inpatient or outpatient center that calls itself a medical home truly is one.7-8
The certification process requires centers to document activities in areas such as patients’ rights and responsibilities, administration and governance, patient and care team relationships, clinical records and other health information, and quality, comprehensiveness, continuity, and accessibility.7 Achieving certification is rigorous, often requiring centers to document compliance with more than 100 policies, procedures, and standards.
For the Indiana Hemophilia and Thrombosis Center, becoming certified as a medical home “was a multiyear process and an ongoing process,” said Dr. Shapiro. “It involves documentation of quality improvement initiatives, obtaining input from patients to document their satisfaction, and looking at all types of systems within our center and how we integrate care so that all those systems function together. It’s a difficult process, but treatment centers are a medical home for patients with bleeding disorders, and this is an effort to provide some documentation on a national level of how we’re doing everything that we are doing.”
She noted that the process of obtaining medical home certification may require an even higher level of commitment if a bleeding disorder (hemophilia) treatment center is embedded in a university or academic medical center. In this case, more stakeholders are involved, and more hoops may need to be jumped through to implement processes that meet medical home standards while still adhering to any requirements at the organizational level.
Certification programs for patient-centered medical homes are not designed around specific disorders or diseases, but a closer look at their compliance metrics underscores how medical homes can benefit patients with bleeding disorders. For example, to receive medical home certification from the Accreditation Association for Ambulatory Health Care, a center needs to be able to document that patients’ care is not transferred without first making arrangements with a receiving health care provider, that the quality improvement programs are peer-led, and that these programs assess and address diverse measures of clinical performance, cost-effectiveness, and administrative functioning.7-9
Medical homes, the NHPCC, and Healthy People 2030
Creating patient-centered medical homes for patients with bleeding disorders is now a quality improvement objective of the National Hemophilia Program Coordinating Center, or NHPCC. Established in 2012 and funded by the federal Health Resources and Services Administration, the NHPCC partners with the eight regional hemophilia networks and more than 140 federally funded hemophilia treatment centers across the United States to identify gaps, standardize and improve access to care, and share and promote best practices for the treatment and management of blood disorders.10
In the United States, receiving care in a hemophilia treatment center (which, despite its name, typically offers care for other disorders such as von Willebrand disease) has been linked to lower mortality and fewer hospitalizations related to bleeding complications.11 To continue to improve on these outcomes, the NHPCC, regional networks, and hemophilia treatment centers are prioritizing medical homes and ranking their establishments alongside core objectives such as bettering patient and family engagement and improving the transition from pediatric to adult care.12
As part of this quality improvement work, the NHPCC, regional leadership, and hemophilia treatment centers meet regularly to identify needs and priorities, plan programs, and ensure that each center is meeting the goals and objectives set out by its federal grant.13 Such partnerships help improve and integrate care within a coordinated national framework, Dr. Shapiro said. “We all are charged with this same mission,” she added. “That doesn’t mean that every treatment center looks exactly the same, has the same number of staff, or does everything the same way, but we all have the same mission, and we know what that is. That is the work of the NHPCC, to determine and document that and help level and improve care throughout the country.”
The NHPCC also engages other stakeholders, including consumer agencies and professional organizations. Recent achievements have included a first-ever national patient needs assessment, a tandem technical needs assessment of hemophilia treatment centers, an educational outreach program for genetic counselors, a webinar on transitioning care for adolescents, a national survey of the federal 340B Drug Pricing Program, and a survey of minority patients to identify and characterize problems such as language and insurance barriers, the lack of culturally appropriate educational materials on blood disorders, and difficulties getting transportation to treatment centers or educational programs.14
In part because of this advocacy work, the U.S. Department of Health and Human Services recently included hemophilia for the first time in Healthy People, its evidence-based set of decade-long objectives aimed at improving the health of all Americans. In Healthy People 2030, the specific objective for hemophilia is to reduce the proportion of patients with severe disease who experience more than four joint bleeds per year to 13.3% (the current estimate is 16.9%).15
For Healthy People to prioritize hemophilia for the first time alongside much more common conditions such as diabetes and heart disease reflects the challenges of managing bleeding disorders and the efforts by the NHPCC and other stakeholders to raise awareness about current needs. To track progress in meeting the Healthy People 2030 objective, the NHPCC will work with federal partners to analyze patient-level data gathered through the Centers for Disease Control’s Community Counts Registry for Bleeding Disorders Surveillance program, which collects data from hemophilia treatment centers across the United States and includes patients with all levels of disease severity.
“The inclusion of bleeding disorders in Healthy People 2030 is really very significant,” said Dr. Shapiro. “These are disorders that affect less than 200,000 Americans, which is the definition of a rare disease in this context. To have hemophilia considered as a national priority is very important, not only for hemophilia, but also for other rare diseases that may in the future also be considered as being as of national importance in this way.”
References
1. Rodriguez-Saldana J. 2019. The Patient-Centered Medical Home, Primary Care, and Diabetes. In: Rodriguez-Saldana J. (eds) The Diabetes Textbook. Springer, Cham.
2. J Comorb. 2011;1:51-59.
3. Eur J Haematol. 2018 Apr;100 Suppl 1:5-13.
4. Blood. 2003;102(7):2358-63.
5. Haemophilia. 2014 Jul;20(4):541-9.
6. Haemophilia. 2016;22(Suppl 3):31-40.
7. AAAHC. Medical Home.
8. NCQA. Patient-centered medical home (PCMH).
9. AAAHC, 2013. Medical Home On-Site Certification Handbook.
10. Centers for Disease Control and Prevention. HTC Population Profile.
11. Blood Transfus. 2014;12 Suppl 3(Suppl 3):e542-e548.
12. American Thrombosis and Hemostasis Network.
13. The Great Lakes Regional Hemophilia Network.
14. American Thrombosis and Hemostasis Network. What the NHPCC does.
15. U.S. Department of Health and Human Services. Healthy People 2030: Blood Disorders.
As bleeding disorders are increasingly recognized as a national health priority, hematologists are focusing on how the patient-centered medical home – a widely accepted concept in primary care and in some specialties – can improve outcomes and quality life for their patients.
The patient-centered medical home is a model of health care delivery in which patients receive comprehensive, accessible care that is fully integrated across all providers and elements of a healthcare system.1 The concept emerged in the 1960s among pediatricians seeking to better coordinate care for children with complex medical needs. Since then, the patient-centered medical home has become a globally recognized standard – not only in primary care, but also in specialties such as endocrinology, oncology, and geriatric medicine. The movement to establish medical homes for patients with bleeding disorders is more recent and is receiving national attention.
Why a medical home?
The advent of prophylactic therapies for bleeding disorders has vastly improved the outlook for many patients compared to just a few decades ago. However, treatment options remain limited, and patients who have severe disease or complications – such as an inadequate treatment response or the development of inhibitory antibodies to replacement clotting factors – are at risk for recurrent breakthrough bleeding that can lead to synovitis and ultimately culminate in progressive, irreversible joint damage. The resulting pain and limitation of motion greatly compromises patients’ quality of life across physical, psychological, and social domains, undermines their ability to live and work independently, and greatly increases treatment costs.2-4 Family members, too, face high stress and lower quality of life when they struggle to obtain and manage treatment while caring for loved ones with bleeding disorders.5
For patients with bleeding disorders, a patient-centered medical home can help address or surmount these challenges, said Amy Shapiro, MD, medical director of the Indiana Hemophilia and Thrombosis Center in Indianapolis, Ind., which was the first hemophilia treatment center in the country to be formally certified as a medical home.
Dr. Shapiro explained that a patient-centered medical home leverages the care of an integrated multidisciplinary team to help optimize therapies and patient outcomes across all domains of life. She sees the medical home concept as a natural fit for patients with bleeding disorders, given the complexity of their needs and the number of specialties involved. “When you have hemophilia, you don’t just need a hematologist to manage your care. You need nurses, physical therapists, and social workers. You need coordinated care for genetic counseling. You also need to coordinate dental hygiene and surgical interventions, if these are required. Patients need nutrition counseling, and they may need assistance with education or career options if too many days are missed from work or school. Patients or their families may need counseling on choosing the right insurance program so they don’t choose a plan that may create more hardships for them because of their chronic disorder.”
Meeting these needs requires the help of an integrated care team, which many individuals with bleeding disorders lack. “If you are just out there in the community and you have medical issues that need to be dealt with, often the individuals themselves have to coordinate their own care, including their medications and their appointments with different specialists,” said Dr. Shapiro. “For example, a care provider may tell a patient that they need a physical therapist and give them some names, and then the patient has to take it from there and not only find the provider, but also determine if their insurance provides coverage.”
A medical home takes a completely different approach, she explained. “At my center, when we say you need a physical therapist, we have a physical therapist on staff. Our therapist provides an assessment and determines the need for ongoing PT and whether that can be done at home with a plan and intermittent oversight, or whether the patient needs a referral, and whether the person the patient is referred to needs education on how to provide PT for someone with hemophilia. A medical home provides all this in one place. It is a place where patients know they will receive either direct services, or support to shepherd their care and outcomes, and oversight of that support as well.”
Few studies have directly assessed the medical home model in the setting of bleeding disorders, but a number have evaluated the impact of integrated care, a more general term for the practice of coordinating multidisciplinary care to improve access and outcomes while eliminating redundancies and unnecessary costs. In a recent systematic review and meta-analysis of 27 nonrandomized studies of patients with hemophilia, integrated care was linked to lower mortality, fewer emergency room visits and hospitalizations, shorter lengths of stay in the hospital, and fewer missed days of school and work.6 Such findings, combined with promising outcomes data from studies of patient-centered medical homes in other disease settings, suggest that the patient-centered medical home can significantly benefit patients with bleeding disorders and their families and caregivers.
Creating a medical home
Establishing a patient-centered medical home can be challenging, involving a plethora of stakeholders and a considerable investment of time, energy, and resources. Organizations such as the National Committee for Quality Assurance and the Accreditation Association for Ambulatory Health Care have formal certification programs to help ensure that an inpatient or outpatient center that calls itself a medical home truly is one.7-8
The certification process requires centers to document activities in areas such as patients’ rights and responsibilities, administration and governance, patient and care team relationships, clinical records and other health information, and quality, comprehensiveness, continuity, and accessibility.7 Achieving certification is rigorous, often requiring centers to document compliance with more than 100 policies, procedures, and standards.
For the Indiana Hemophilia and Thrombosis Center, becoming certified as a medical home “was a multiyear process and an ongoing process,” said Dr. Shapiro. “It involves documentation of quality improvement initiatives, obtaining input from patients to document their satisfaction, and looking at all types of systems within our center and how we integrate care so that all those systems function together. It’s a difficult process, but treatment centers are a medical home for patients with bleeding disorders, and this is an effort to provide some documentation on a national level of how we’re doing everything that we are doing.”
She noted that the process of obtaining medical home certification may require an even higher level of commitment if a bleeding disorder (hemophilia) treatment center is embedded in a university or academic medical center. In this case, more stakeholders are involved, and more hoops may need to be jumped through to implement processes that meet medical home standards while still adhering to any requirements at the organizational level.
Certification programs for patient-centered medical homes are not designed around specific disorders or diseases, but a closer look at their compliance metrics underscores how medical homes can benefit patients with bleeding disorders. For example, to receive medical home certification from the Accreditation Association for Ambulatory Health Care, a center needs to be able to document that patients’ care is not transferred without first making arrangements with a receiving health care provider, that the quality improvement programs are peer-led, and that these programs assess and address diverse measures of clinical performance, cost-effectiveness, and administrative functioning.7-9
Medical homes, the NHPCC, and Healthy People 2030
Creating patient-centered medical homes for patients with bleeding disorders is now a quality improvement objective of the National Hemophilia Program Coordinating Center, or NHPCC. Established in 2012 and funded by the federal Health Resources and Services Administration, the NHPCC partners with the eight regional hemophilia networks and more than 140 federally funded hemophilia treatment centers across the United States to identify gaps, standardize and improve access to care, and share and promote best practices for the treatment and management of blood disorders.10
In the United States, receiving care in a hemophilia treatment center (which, despite its name, typically offers care for other disorders such as von Willebrand disease) has been linked to lower mortality and fewer hospitalizations related to bleeding complications.11 To continue to improve on these outcomes, the NHPCC, regional networks, and hemophilia treatment centers are prioritizing medical homes and ranking their establishments alongside core objectives such as bettering patient and family engagement and improving the transition from pediatric to adult care.12
As part of this quality improvement work, the NHPCC, regional leadership, and hemophilia treatment centers meet regularly to identify needs and priorities, plan programs, and ensure that each center is meeting the goals and objectives set out by its federal grant.13 Such partnerships help improve and integrate care within a coordinated national framework, Dr. Shapiro said. “We all are charged with this same mission,” she added. “That doesn’t mean that every treatment center looks exactly the same, has the same number of staff, or does everything the same way, but we all have the same mission, and we know what that is. That is the work of the NHPCC, to determine and document that and help level and improve care throughout the country.”
The NHPCC also engages other stakeholders, including consumer agencies and professional organizations. Recent achievements have included a first-ever national patient needs assessment, a tandem technical needs assessment of hemophilia treatment centers, an educational outreach program for genetic counselors, a webinar on transitioning care for adolescents, a national survey of the federal 340B Drug Pricing Program, and a survey of minority patients to identify and characterize problems such as language and insurance barriers, the lack of culturally appropriate educational materials on blood disorders, and difficulties getting transportation to treatment centers or educational programs.14
In part because of this advocacy work, the U.S. Department of Health and Human Services recently included hemophilia for the first time in Healthy People, its evidence-based set of decade-long objectives aimed at improving the health of all Americans. In Healthy People 2030, the specific objective for hemophilia is to reduce the proportion of patients with severe disease who experience more than four joint bleeds per year to 13.3% (the current estimate is 16.9%).15
For Healthy People to prioritize hemophilia for the first time alongside much more common conditions such as diabetes and heart disease reflects the challenges of managing bleeding disorders and the efforts by the NHPCC and other stakeholders to raise awareness about current needs. To track progress in meeting the Healthy People 2030 objective, the NHPCC will work with federal partners to analyze patient-level data gathered through the Centers for Disease Control’s Community Counts Registry for Bleeding Disorders Surveillance program, which collects data from hemophilia treatment centers across the United States and includes patients with all levels of disease severity.
“The inclusion of bleeding disorders in Healthy People 2030 is really very significant,” said Dr. Shapiro. “These are disorders that affect less than 200,000 Americans, which is the definition of a rare disease in this context. To have hemophilia considered as a national priority is very important, not only for hemophilia, but also for other rare diseases that may in the future also be considered as being as of national importance in this way.”
References
1. Rodriguez-Saldana J. 2019. The Patient-Centered Medical Home, Primary Care, and Diabetes. In: Rodriguez-Saldana J. (eds) The Diabetes Textbook. Springer, Cham.
2. J Comorb. 2011;1:51-59.
3. Eur J Haematol. 2018 Apr;100 Suppl 1:5-13.
4. Blood. 2003;102(7):2358-63.
5. Haemophilia. 2014 Jul;20(4):541-9.
6. Haemophilia. 2016;22(Suppl 3):31-40.
7. AAAHC. Medical Home.
8. NCQA. Patient-centered medical home (PCMH).
9. AAAHC, 2013. Medical Home On-Site Certification Handbook.
10. Centers for Disease Control and Prevention. HTC Population Profile.
11. Blood Transfus. 2014;12 Suppl 3(Suppl 3):e542-e548.
12. American Thrombosis and Hemostasis Network.
13. The Great Lakes Regional Hemophilia Network.
14. American Thrombosis and Hemostasis Network. What the NHPCC does.
15. U.S. Department of Health and Human Services. Healthy People 2030: Blood Disorders.
As bleeding disorders are increasingly recognized as a national health priority, hematologists are focusing on how the patient-centered medical home – a widely accepted concept in primary care and in some specialties – can improve outcomes and quality life for their patients.
The patient-centered medical home is a model of health care delivery in which patients receive comprehensive, accessible care that is fully integrated across all providers and elements of a healthcare system.1 The concept emerged in the 1960s among pediatricians seeking to better coordinate care for children with complex medical needs. Since then, the patient-centered medical home has become a globally recognized standard – not only in primary care, but also in specialties such as endocrinology, oncology, and geriatric medicine. The movement to establish medical homes for patients with bleeding disorders is more recent and is receiving national attention.
Why a medical home?
The advent of prophylactic therapies for bleeding disorders has vastly improved the outlook for many patients compared to just a few decades ago. However, treatment options remain limited, and patients who have severe disease or complications – such as an inadequate treatment response or the development of inhibitory antibodies to replacement clotting factors – are at risk for recurrent breakthrough bleeding that can lead to synovitis and ultimately culminate in progressive, irreversible joint damage. The resulting pain and limitation of motion greatly compromises patients’ quality of life across physical, psychological, and social domains, undermines their ability to live and work independently, and greatly increases treatment costs.2-4 Family members, too, face high stress and lower quality of life when they struggle to obtain and manage treatment while caring for loved ones with bleeding disorders.5
For patients with bleeding disorders, a patient-centered medical home can help address or surmount these challenges, said Amy Shapiro, MD, medical director of the Indiana Hemophilia and Thrombosis Center in Indianapolis, Ind., which was the first hemophilia treatment center in the country to be formally certified as a medical home.
Dr. Shapiro explained that a patient-centered medical home leverages the care of an integrated multidisciplinary team to help optimize therapies and patient outcomes across all domains of life. She sees the medical home concept as a natural fit for patients with bleeding disorders, given the complexity of their needs and the number of specialties involved. “When you have hemophilia, you don’t just need a hematologist to manage your care. You need nurses, physical therapists, and social workers. You need coordinated care for genetic counseling. You also need to coordinate dental hygiene and surgical interventions, if these are required. Patients need nutrition counseling, and they may need assistance with education or career options if too many days are missed from work or school. Patients or their families may need counseling on choosing the right insurance program so they don’t choose a plan that may create more hardships for them because of their chronic disorder.”
Meeting these needs requires the help of an integrated care team, which many individuals with bleeding disorders lack. “If you are just out there in the community and you have medical issues that need to be dealt with, often the individuals themselves have to coordinate their own care, including their medications and their appointments with different specialists,” said Dr. Shapiro. “For example, a care provider may tell a patient that they need a physical therapist and give them some names, and then the patient has to take it from there and not only find the provider, but also determine if their insurance provides coverage.”
A medical home takes a completely different approach, she explained. “At my center, when we say you need a physical therapist, we have a physical therapist on staff. Our therapist provides an assessment and determines the need for ongoing PT and whether that can be done at home with a plan and intermittent oversight, or whether the patient needs a referral, and whether the person the patient is referred to needs education on how to provide PT for someone with hemophilia. A medical home provides all this in one place. It is a place where patients know they will receive either direct services, or support to shepherd their care and outcomes, and oversight of that support as well.”
Few studies have directly assessed the medical home model in the setting of bleeding disorders, but a number have evaluated the impact of integrated care, a more general term for the practice of coordinating multidisciplinary care to improve access and outcomes while eliminating redundancies and unnecessary costs. In a recent systematic review and meta-analysis of 27 nonrandomized studies of patients with hemophilia, integrated care was linked to lower mortality, fewer emergency room visits and hospitalizations, shorter lengths of stay in the hospital, and fewer missed days of school and work.6 Such findings, combined with promising outcomes data from studies of patient-centered medical homes in other disease settings, suggest that the patient-centered medical home can significantly benefit patients with bleeding disorders and their families and caregivers.
Creating a medical home
Establishing a patient-centered medical home can be challenging, involving a plethora of stakeholders and a considerable investment of time, energy, and resources. Organizations such as the National Committee for Quality Assurance and the Accreditation Association for Ambulatory Health Care have formal certification programs to help ensure that an inpatient or outpatient center that calls itself a medical home truly is one.7-8
The certification process requires centers to document activities in areas such as patients’ rights and responsibilities, administration and governance, patient and care team relationships, clinical records and other health information, and quality, comprehensiveness, continuity, and accessibility.7 Achieving certification is rigorous, often requiring centers to document compliance with more than 100 policies, procedures, and standards.
For the Indiana Hemophilia and Thrombosis Center, becoming certified as a medical home “was a multiyear process and an ongoing process,” said Dr. Shapiro. “It involves documentation of quality improvement initiatives, obtaining input from patients to document their satisfaction, and looking at all types of systems within our center and how we integrate care so that all those systems function together. It’s a difficult process, but treatment centers are a medical home for patients with bleeding disorders, and this is an effort to provide some documentation on a national level of how we’re doing everything that we are doing.”
She noted that the process of obtaining medical home certification may require an even higher level of commitment if a bleeding disorder (hemophilia) treatment center is embedded in a university or academic medical center. In this case, more stakeholders are involved, and more hoops may need to be jumped through to implement processes that meet medical home standards while still adhering to any requirements at the organizational level.
Certification programs for patient-centered medical homes are not designed around specific disorders or diseases, but a closer look at their compliance metrics underscores how medical homes can benefit patients with bleeding disorders. For example, to receive medical home certification from the Accreditation Association for Ambulatory Health Care, a center needs to be able to document that patients’ care is not transferred without first making arrangements with a receiving health care provider, that the quality improvement programs are peer-led, and that these programs assess and address diverse measures of clinical performance, cost-effectiveness, and administrative functioning.7-9
Medical homes, the NHPCC, and Healthy People 2030
Creating patient-centered medical homes for patients with bleeding disorders is now a quality improvement objective of the National Hemophilia Program Coordinating Center, or NHPCC. Established in 2012 and funded by the federal Health Resources and Services Administration, the NHPCC partners with the eight regional hemophilia networks and more than 140 federally funded hemophilia treatment centers across the United States to identify gaps, standardize and improve access to care, and share and promote best practices for the treatment and management of blood disorders.10
In the United States, receiving care in a hemophilia treatment center (which, despite its name, typically offers care for other disorders such as von Willebrand disease) has been linked to lower mortality and fewer hospitalizations related to bleeding complications.11 To continue to improve on these outcomes, the NHPCC, regional networks, and hemophilia treatment centers are prioritizing medical homes and ranking their establishments alongside core objectives such as bettering patient and family engagement and improving the transition from pediatric to adult care.12
As part of this quality improvement work, the NHPCC, regional leadership, and hemophilia treatment centers meet regularly to identify needs and priorities, plan programs, and ensure that each center is meeting the goals and objectives set out by its federal grant.13 Such partnerships help improve and integrate care within a coordinated national framework, Dr. Shapiro said. “We all are charged with this same mission,” she added. “That doesn’t mean that every treatment center looks exactly the same, has the same number of staff, or does everything the same way, but we all have the same mission, and we know what that is. That is the work of the NHPCC, to determine and document that and help level and improve care throughout the country.”
The NHPCC also engages other stakeholders, including consumer agencies and professional organizations. Recent achievements have included a first-ever national patient needs assessment, a tandem technical needs assessment of hemophilia treatment centers, an educational outreach program for genetic counselors, a webinar on transitioning care for adolescents, a national survey of the federal 340B Drug Pricing Program, and a survey of minority patients to identify and characterize problems such as language and insurance barriers, the lack of culturally appropriate educational materials on blood disorders, and difficulties getting transportation to treatment centers or educational programs.14
In part because of this advocacy work, the U.S. Department of Health and Human Services recently included hemophilia for the first time in Healthy People, its evidence-based set of decade-long objectives aimed at improving the health of all Americans. In Healthy People 2030, the specific objective for hemophilia is to reduce the proportion of patients with severe disease who experience more than four joint bleeds per year to 13.3% (the current estimate is 16.9%).15
For Healthy People to prioritize hemophilia for the first time alongside much more common conditions such as diabetes and heart disease reflects the challenges of managing bleeding disorders and the efforts by the NHPCC and other stakeholders to raise awareness about current needs. To track progress in meeting the Healthy People 2030 objective, the NHPCC will work with federal partners to analyze patient-level data gathered through the Centers for Disease Control’s Community Counts Registry for Bleeding Disorders Surveillance program, which collects data from hemophilia treatment centers across the United States and includes patients with all levels of disease severity.
“The inclusion of bleeding disorders in Healthy People 2030 is really very significant,” said Dr. Shapiro. “These are disorders that affect less than 200,000 Americans, which is the definition of a rare disease in this context. To have hemophilia considered as a national priority is very important, not only for hemophilia, but also for other rare diseases that may in the future also be considered as being as of national importance in this way.”
References
1. Rodriguez-Saldana J. 2019. The Patient-Centered Medical Home, Primary Care, and Diabetes. In: Rodriguez-Saldana J. (eds) The Diabetes Textbook. Springer, Cham.
2. J Comorb. 2011;1:51-59.
3. Eur J Haematol. 2018 Apr;100 Suppl 1:5-13.
4. Blood. 2003;102(7):2358-63.
5. Haemophilia. 2014 Jul;20(4):541-9.
6. Haemophilia. 2016;22(Suppl 3):31-40.
7. AAAHC. Medical Home.
8. NCQA. Patient-centered medical home (PCMH).
9. AAAHC, 2013. Medical Home On-Site Certification Handbook.
10. Centers for Disease Control and Prevention. HTC Population Profile.
11. Blood Transfus. 2014;12 Suppl 3(Suppl 3):e542-e548.
12. American Thrombosis and Hemostasis Network.
13. The Great Lakes Regional Hemophilia Network.
14. American Thrombosis and Hemostasis Network. What the NHPCC does.
15. U.S. Department of Health and Human Services. Healthy People 2030: Blood Disorders.
Sex differences in COPD symptoms predict cardiac comorbidity
Sex-specific differences in the severity of symptoms and prevalence of comorbidities in patients with chronic obstructive pulmonary disease (COPD) may point to different criteria for diagnosing cardiac comorbidities in women and men, a retrospective analysis suggests.
Among 2,046 patients in the German COSYCONET (COPD and Systemic Consequences–Comorbidities Net) cohort, most functional parameters and comorbidities and several items on the COPD Assessment Test (CAT) differed significantly between men and women.
In addition, there were sex-specific differences in the association between symptoms and cardiac disease, Franziska C. Trudzinski, MD, from the University of Heidelberg (Germany), and colleagues reported.
(Note: Although the authors used the term “gender” to distinguish male from female, this news organization has used the term “sex” in this article to refer to biological attributes of individual patients rather than personal identity.)
“[Sex]-specific differences in COPD comprised not only differences in the level of symptoms, comorbidities, and functional alterations but also differences in their mutual relationships. This was reflected in different sets of predictors for cardiac disease,” they wrote in a thematic poster presented at the American Thoracic Society’s virtual international conference.
GOLD standard
The investigators conducted an analysis of data on 795 women and 1,251 men with GOLD (Global Initiative for Chronic Obstructive Lung Disease) class 1-3 disease from the COSYCONET COPD cohort.
They looked at the patients’ clinical history, comorbidities, lung function, CAT scores, and modified Medical Research Council (mMRC) dyspnea score.
The authors used multivariate regression analysis to model potential sex-related differences in the relationship between symptoms in general and CAT items in particular, and the pattern of comorbidities and functional alterations.
They also performed logistic regression analyses to identify predictors for cardiac disease, defined as myocardial infarction, heart failure, or coronary artery disease. The analyses were controlled for age, body mass index (BMI), smoking status, mMRC, CAT items, and z scores of forced expiratory volume in 1 second/forced vital capacity ratio.
and for CAT items of cough (item 1), phlegm (item 2), and energy (item 8; P < .05 for all comparisons).
In logistic regression analysis, predictors for cardiac disease in men were energy (CAT item 8), mMRC score, smoking status, BMI, age, and spirometric lung function.
In women, however, only age was significantly predictive for cardiac disease.
“Our findings give hints how diagnostic information might be used differently in men and women,” Dr. Trudzinski and colleagues wrote.
Reassuring data
David Mannino, MD, medical director of the COPD Foundation, who was not involved in the study, said in an interview that sex differences in COPD presentation and severity are common.
“In general, men and women report symptoms differently. For example, women don’t report a whole lot of chronic bronchitis and phlegm, although they may have it,” he said, “whereas men may report less dyspnea. It varies, but in general we know that men and women, even with the same type of disease, report symptoms differently.”
Comorbidities also differ between the sexes, he noted. Women more frequently have osteoporosis, and men more frequently have heart disease, as borne out in the study. The prevalence of heart disease among patients in the study was approximately 2.5 times higher in men than women.
“It’s reassuring, because what we’re seeing is similar to what we’ve seen in other [studies] with regards to comorbidities,” he said.
The study was sponsored by Philipps University Marburg Medical Center, Germany. The authors and Dr. Mannino have reported no relevant financial relationships.
A version of the article first appeared on Medscape.com.
Sex-specific differences in the severity of symptoms and prevalence of comorbidities in patients with chronic obstructive pulmonary disease (COPD) may point to different criteria for diagnosing cardiac comorbidities in women and men, a retrospective analysis suggests.
Among 2,046 patients in the German COSYCONET (COPD and Systemic Consequences–Comorbidities Net) cohort, most functional parameters and comorbidities and several items on the COPD Assessment Test (CAT) differed significantly between men and women.
In addition, there were sex-specific differences in the association between symptoms and cardiac disease, Franziska C. Trudzinski, MD, from the University of Heidelberg (Germany), and colleagues reported.
(Note: Although the authors used the term “gender” to distinguish male from female, this news organization has used the term “sex” in this article to refer to biological attributes of individual patients rather than personal identity.)
“[Sex]-specific differences in COPD comprised not only differences in the level of symptoms, comorbidities, and functional alterations but also differences in their mutual relationships. This was reflected in different sets of predictors for cardiac disease,” they wrote in a thematic poster presented at the American Thoracic Society’s virtual international conference.
GOLD standard
The investigators conducted an analysis of data on 795 women and 1,251 men with GOLD (Global Initiative for Chronic Obstructive Lung Disease) class 1-3 disease from the COSYCONET COPD cohort.
They looked at the patients’ clinical history, comorbidities, lung function, CAT scores, and modified Medical Research Council (mMRC) dyspnea score.
The authors used multivariate regression analysis to model potential sex-related differences in the relationship between symptoms in general and CAT items in particular, and the pattern of comorbidities and functional alterations.
They also performed logistic regression analyses to identify predictors for cardiac disease, defined as myocardial infarction, heart failure, or coronary artery disease. The analyses were controlled for age, body mass index (BMI), smoking status, mMRC, CAT items, and z scores of forced expiratory volume in 1 second/forced vital capacity ratio.
and for CAT items of cough (item 1), phlegm (item 2), and energy (item 8; P < .05 for all comparisons).
In logistic regression analysis, predictors for cardiac disease in men were energy (CAT item 8), mMRC score, smoking status, BMI, age, and spirometric lung function.
In women, however, only age was significantly predictive for cardiac disease.
“Our findings give hints how diagnostic information might be used differently in men and women,” Dr. Trudzinski and colleagues wrote.
Reassuring data
David Mannino, MD, medical director of the COPD Foundation, who was not involved in the study, said in an interview that sex differences in COPD presentation and severity are common.
“In general, men and women report symptoms differently. For example, women don’t report a whole lot of chronic bronchitis and phlegm, although they may have it,” he said, “whereas men may report less dyspnea. It varies, but in general we know that men and women, even with the same type of disease, report symptoms differently.”
Comorbidities also differ between the sexes, he noted. Women more frequently have osteoporosis, and men more frequently have heart disease, as borne out in the study. The prevalence of heart disease among patients in the study was approximately 2.5 times higher in men than women.
“It’s reassuring, because what we’re seeing is similar to what we’ve seen in other [studies] with regards to comorbidities,” he said.
The study was sponsored by Philipps University Marburg Medical Center, Germany. The authors and Dr. Mannino have reported no relevant financial relationships.
A version of the article first appeared on Medscape.com.
Sex-specific differences in the severity of symptoms and prevalence of comorbidities in patients with chronic obstructive pulmonary disease (COPD) may point to different criteria for diagnosing cardiac comorbidities in women and men, a retrospective analysis suggests.
Among 2,046 patients in the German COSYCONET (COPD and Systemic Consequences–Comorbidities Net) cohort, most functional parameters and comorbidities and several items on the COPD Assessment Test (CAT) differed significantly between men and women.
In addition, there were sex-specific differences in the association between symptoms and cardiac disease, Franziska C. Trudzinski, MD, from the University of Heidelberg (Germany), and colleagues reported.
(Note: Although the authors used the term “gender” to distinguish male from female, this news organization has used the term “sex” in this article to refer to biological attributes of individual patients rather than personal identity.)
“[Sex]-specific differences in COPD comprised not only differences in the level of symptoms, comorbidities, and functional alterations but also differences in their mutual relationships. This was reflected in different sets of predictors for cardiac disease,” they wrote in a thematic poster presented at the American Thoracic Society’s virtual international conference.
GOLD standard
The investigators conducted an analysis of data on 795 women and 1,251 men with GOLD (Global Initiative for Chronic Obstructive Lung Disease) class 1-3 disease from the COSYCONET COPD cohort.
They looked at the patients’ clinical history, comorbidities, lung function, CAT scores, and modified Medical Research Council (mMRC) dyspnea score.
The authors used multivariate regression analysis to model potential sex-related differences in the relationship between symptoms in general and CAT items in particular, and the pattern of comorbidities and functional alterations.
They also performed logistic regression analyses to identify predictors for cardiac disease, defined as myocardial infarction, heart failure, or coronary artery disease. The analyses were controlled for age, body mass index (BMI), smoking status, mMRC, CAT items, and z scores of forced expiratory volume in 1 second/forced vital capacity ratio.
and for CAT items of cough (item 1), phlegm (item 2), and energy (item 8; P < .05 for all comparisons).
In logistic regression analysis, predictors for cardiac disease in men were energy (CAT item 8), mMRC score, smoking status, BMI, age, and spirometric lung function.
In women, however, only age was significantly predictive for cardiac disease.
“Our findings give hints how diagnostic information might be used differently in men and women,” Dr. Trudzinski and colleagues wrote.
Reassuring data
David Mannino, MD, medical director of the COPD Foundation, who was not involved in the study, said in an interview that sex differences in COPD presentation and severity are common.
“In general, men and women report symptoms differently. For example, women don’t report a whole lot of chronic bronchitis and phlegm, although they may have it,” he said, “whereas men may report less dyspnea. It varies, but in general we know that men and women, even with the same type of disease, report symptoms differently.”
Comorbidities also differ between the sexes, he noted. Women more frequently have osteoporosis, and men more frequently have heart disease, as borne out in the study. The prevalence of heart disease among patients in the study was approximately 2.5 times higher in men than women.
“It’s reassuring, because what we’re seeing is similar to what we’ve seen in other [studies] with regards to comorbidities,” he said.
The study was sponsored by Philipps University Marburg Medical Center, Germany. The authors and Dr. Mannino have reported no relevant financial relationships.
A version of the article first appeared on Medscape.com.