User login
Breaking the Diagnostic Bottleneck in RA
As head of the clinical laboratory at the San Juan University Hospital in Alicante, Spain, Maria Salinas, PhD, is passionate about the role she and her colleagues can play in clinical decision-making.
Her mission is the identification of “hidden diseases,” as she calls them, chronic conditions for which early identification and intervention can change the course of the illness. Her lab has been a leader over the past decade in using technology to partner with clinicians to promote the appropriate use of testing and clinical decision-making.
An example of a disease ripe for this type of intervention is rheumatoid arthritis (RA), the most common form of autoimmune arthritis, affecting around 1.3 million people in the United States. The prognosis for patients is better the earlier treatment begins.
But the
Amy S. Kehl, MD, an attending rheumatologist at Cedars-Sinai Medical Center in Los Angeles, who also sees patients at Saint John’s Physician Partners in Santa Monica, California, recommends a workup for inflammatory arthritis for patients presenting with the new onset of joint pain and swelling, primarily of small joints, although larger joints can be involved. The workup includes markers of inflammation such as an erythrocyte sedimentation rate and C-reactive protein, which are typically elevated and can be used to monitor the progression of the disease. Similarly, the presence of anemia is consistent with RA and helpful in tracking response to treatment.
But pinning down the diagnosis requires the presence of autoimmune antibodies. Guidelines from the American College of Rheumatology require a positive result for either rheumatoid factor (RF) or anti-cyclic citrullinated peptide (anti-CCP) antibody to definitively determine whether a patient has RA (Table).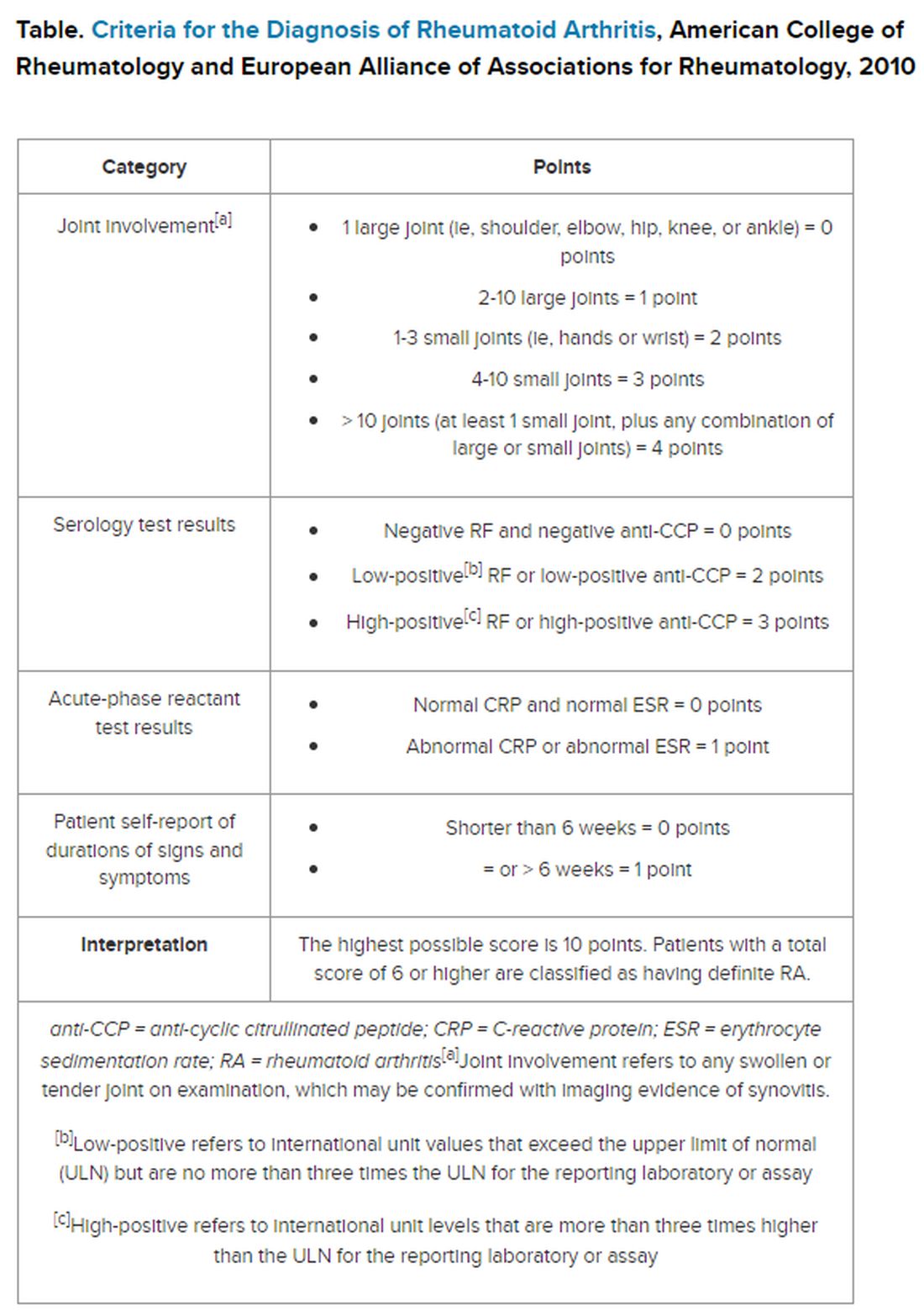
“Classically, I find that the primary care physicians include a rheumatoid factor, not always a CCP, and may include other antibodies, including an ANA [antinuclear antibody] test, as part of that workup,” Dr. Kehl said. The problem with that strategy is that although the RF test does detect 60%-80% of patients with RA, it is positive in many other autoimmune conditions. Although the ANA might be positive in patients with RA, it is nonspecific and does not confirm the diagnosis of RA.
Up to 50% of autoimmune antibody tests are inappropriately ordered. And for rheumatologists, that leads to unnecessary referrals of patients with musculoskeletal complaints who do not meet objective clinical criteria for joint disease.
“These tests get ordered almost reflexively, and sometimes they’re ordered as part of a panel that includes a rheumatoid factor and an ANA, and it’s not necessarily going to be a high-yield test,” Dr. Kehl said. Superfluous tests and referrals often cause unnecessary anxiety in patients, as well as drive up costs, she added.
Dr. Salinas made the same observation in her hospital lab, which also serves nine primary care centers. She documented an upward trend in orders for RF testing in her hospital lab between 2011 and 2019. Dr. Salinas also noted that the anti-CCP antibody test was not commonly requested, although it has more utility in the diagnosis. Like the RF, it detects 50%-70% of patients with RA but has 95% specificity, resulting in far fewer false-positive results.
The appearance of both RF and anti-CCP antibodies often predicts a rapid progression to clinical disease. Dr. Kehl wants symptomatic patients with positive results for both markers to be seen right away by a rheumatologist. “We do know from studies that bony erosions can develop as early as a month or months after the onset of an inflammatory arthritis,” she said.
To address this need, Dr. Salinas worked with rheumatologists and primary care clinicians to develop an algorithm that called for reflex testing of samples from patients with positive RF results (> 30 IU/mL) for anti-CCP antibodies. If the anti-CCP antibody result was > 40 IU/mL, a comment in the lab report suggested rheumatology referral. The lab turned down requests to test sample for RF if the patient had a negative result in the previous 12 months — but it would perform the test if the clinician repeated the request.
The results were encouraging, Dr. Salinas said. “The main result in this study was that we really identified more hidden cases of patients with rheumatoid arthritis,” she told this news organization.
Compared with baseline trends, during the study period from April 2019 to January 2021 her lab demonstrated:
- Reduced RF tests conducted by canceling 16% of tests ordered for patients with negative RF result in the previous 12 months
- Fewer unnecessary referrals, from 22% in the baseline period to 8% during the intervention period
- A smaller percentage of missed patients, from 21% to 16%
To be sure, pre- and post-implementation comparisons are difficult when the implementation period happens to coincide with the emergence of SARS-CoV-2.
Although fewer patients were seen and fewer lab tests were ordered overall in Alicante during the COVID-19 pandemic, the proportion of tests ordered for RF testing dropped, and all the patients identified with double positives for RF and anti-CCP antibodies were referred to rheumatology, suggesting evidence of benefit.
Dr. Kehl said the practice of using clinical decision support systems could be used in the United States. “I thought this was an important study,” she said. Electronic health records systems “have all these capabilities where we can include best practice alerts when you order a test to make sure that it’s clinically warranted and cost-effective.”
Dr. Salinas and Dr. Kehl reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
As head of the clinical laboratory at the San Juan University Hospital in Alicante, Spain, Maria Salinas, PhD, is passionate about the role she and her colleagues can play in clinical decision-making.
Her mission is the identification of “hidden diseases,” as she calls them, chronic conditions for which early identification and intervention can change the course of the illness. Her lab has been a leader over the past decade in using technology to partner with clinicians to promote the appropriate use of testing and clinical decision-making.
An example of a disease ripe for this type of intervention is rheumatoid arthritis (RA), the most common form of autoimmune arthritis, affecting around 1.3 million people in the United States. The prognosis for patients is better the earlier treatment begins.
But the
Amy S. Kehl, MD, an attending rheumatologist at Cedars-Sinai Medical Center in Los Angeles, who also sees patients at Saint John’s Physician Partners in Santa Monica, California, recommends a workup for inflammatory arthritis for patients presenting with the new onset of joint pain and swelling, primarily of small joints, although larger joints can be involved. The workup includes markers of inflammation such as an erythrocyte sedimentation rate and C-reactive protein, which are typically elevated and can be used to monitor the progression of the disease. Similarly, the presence of anemia is consistent with RA and helpful in tracking response to treatment.
But pinning down the diagnosis requires the presence of autoimmune antibodies. Guidelines from the American College of Rheumatology require a positive result for either rheumatoid factor (RF) or anti-cyclic citrullinated peptide (anti-CCP) antibody to definitively determine whether a patient has RA (Table).
“Classically, I find that the primary care physicians include a rheumatoid factor, not always a CCP, and may include other antibodies, including an ANA [antinuclear antibody] test, as part of that workup,” Dr. Kehl said. The problem with that strategy is that although the RF test does detect 60%-80% of patients with RA, it is positive in many other autoimmune conditions. Although the ANA might be positive in patients with RA, it is nonspecific and does not confirm the diagnosis of RA.
Up to 50% of autoimmune antibody tests are inappropriately ordered. And for rheumatologists, that leads to unnecessary referrals of patients with musculoskeletal complaints who do not meet objective clinical criteria for joint disease.
“These tests get ordered almost reflexively, and sometimes they’re ordered as part of a panel that includes a rheumatoid factor and an ANA, and it’s not necessarily going to be a high-yield test,” Dr. Kehl said. Superfluous tests and referrals often cause unnecessary anxiety in patients, as well as drive up costs, she added.
Dr. Salinas made the same observation in her hospital lab, which also serves nine primary care centers. She documented an upward trend in orders for RF testing in her hospital lab between 2011 and 2019. Dr. Salinas also noted that the anti-CCP antibody test was not commonly requested, although it has more utility in the diagnosis. Like the RF, it detects 50%-70% of patients with RA but has 95% specificity, resulting in far fewer false-positive results.
The appearance of both RF and anti-CCP antibodies often predicts a rapid progression to clinical disease. Dr. Kehl wants symptomatic patients with positive results for both markers to be seen right away by a rheumatologist. “We do know from studies that bony erosions can develop as early as a month or months after the onset of an inflammatory arthritis,” she said.
To address this need, Dr. Salinas worked with rheumatologists and primary care clinicians to develop an algorithm that called for reflex testing of samples from patients with positive RF results (> 30 IU/mL) for anti-CCP antibodies. If the anti-CCP antibody result was > 40 IU/mL, a comment in the lab report suggested rheumatology referral. The lab turned down requests to test sample for RF if the patient had a negative result in the previous 12 months — but it would perform the test if the clinician repeated the request.
The results were encouraging, Dr. Salinas said. “The main result in this study was that we really identified more hidden cases of patients with rheumatoid arthritis,” she told this news organization.
Compared with baseline trends, during the study period from April 2019 to January 2021 her lab demonstrated:
- Reduced RF tests conducted by canceling 16% of tests ordered for patients with negative RF result in the previous 12 months
- Fewer unnecessary referrals, from 22% in the baseline period to 8% during the intervention period
- A smaller percentage of missed patients, from 21% to 16%
To be sure, pre- and post-implementation comparisons are difficult when the implementation period happens to coincide with the emergence of SARS-CoV-2.
Although fewer patients were seen and fewer lab tests were ordered overall in Alicante during the COVID-19 pandemic, the proportion of tests ordered for RF testing dropped, and all the patients identified with double positives for RF and anti-CCP antibodies were referred to rheumatology, suggesting evidence of benefit.
Dr. Kehl said the practice of using clinical decision support systems could be used in the United States. “I thought this was an important study,” she said. Electronic health records systems “have all these capabilities where we can include best practice alerts when you order a test to make sure that it’s clinically warranted and cost-effective.”
Dr. Salinas and Dr. Kehl reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
As head of the clinical laboratory at the San Juan University Hospital in Alicante, Spain, Maria Salinas, PhD, is passionate about the role she and her colleagues can play in clinical decision-making.
Her mission is the identification of “hidden diseases,” as she calls them, chronic conditions for which early identification and intervention can change the course of the illness. Her lab has been a leader over the past decade in using technology to partner with clinicians to promote the appropriate use of testing and clinical decision-making.
An example of a disease ripe for this type of intervention is rheumatoid arthritis (RA), the most common form of autoimmune arthritis, affecting around 1.3 million people in the United States. The prognosis for patients is better the earlier treatment begins.
But the
Amy S. Kehl, MD, an attending rheumatologist at Cedars-Sinai Medical Center in Los Angeles, who also sees patients at Saint John’s Physician Partners in Santa Monica, California, recommends a workup for inflammatory arthritis for patients presenting with the new onset of joint pain and swelling, primarily of small joints, although larger joints can be involved. The workup includes markers of inflammation such as an erythrocyte sedimentation rate and C-reactive protein, which are typically elevated and can be used to monitor the progression of the disease. Similarly, the presence of anemia is consistent with RA and helpful in tracking response to treatment.
But pinning down the diagnosis requires the presence of autoimmune antibodies. Guidelines from the American College of Rheumatology require a positive result for either rheumatoid factor (RF) or anti-cyclic citrullinated peptide (anti-CCP) antibody to definitively determine whether a patient has RA (Table).
“Classically, I find that the primary care physicians include a rheumatoid factor, not always a CCP, and may include other antibodies, including an ANA [antinuclear antibody] test, as part of that workup,” Dr. Kehl said. The problem with that strategy is that although the RF test does detect 60%-80% of patients with RA, it is positive in many other autoimmune conditions. Although the ANA might be positive in patients with RA, it is nonspecific and does not confirm the diagnosis of RA.
Up to 50% of autoimmune antibody tests are inappropriately ordered. And for rheumatologists, that leads to unnecessary referrals of patients with musculoskeletal complaints who do not meet objective clinical criteria for joint disease.
“These tests get ordered almost reflexively, and sometimes they’re ordered as part of a panel that includes a rheumatoid factor and an ANA, and it’s not necessarily going to be a high-yield test,” Dr. Kehl said. Superfluous tests and referrals often cause unnecessary anxiety in patients, as well as drive up costs, she added.
Dr. Salinas made the same observation in her hospital lab, which also serves nine primary care centers. She documented an upward trend in orders for RF testing in her hospital lab between 2011 and 2019. Dr. Salinas also noted that the anti-CCP antibody test was not commonly requested, although it has more utility in the diagnosis. Like the RF, it detects 50%-70% of patients with RA but has 95% specificity, resulting in far fewer false-positive results.
The appearance of both RF and anti-CCP antibodies often predicts a rapid progression to clinical disease. Dr. Kehl wants symptomatic patients with positive results for both markers to be seen right away by a rheumatologist. “We do know from studies that bony erosions can develop as early as a month or months after the onset of an inflammatory arthritis,” she said.
To address this need, Dr. Salinas worked with rheumatologists and primary care clinicians to develop an algorithm that called for reflex testing of samples from patients with positive RF results (> 30 IU/mL) for anti-CCP antibodies. If the anti-CCP antibody result was > 40 IU/mL, a comment in the lab report suggested rheumatology referral. The lab turned down requests to test sample for RF if the patient had a negative result in the previous 12 months — but it would perform the test if the clinician repeated the request.
The results were encouraging, Dr. Salinas said. “The main result in this study was that we really identified more hidden cases of patients with rheumatoid arthritis,” she told this news organization.
Compared with baseline trends, during the study period from April 2019 to January 2021 her lab demonstrated:
- Reduced RF tests conducted by canceling 16% of tests ordered for patients with negative RF result in the previous 12 months
- Fewer unnecessary referrals, from 22% in the baseline period to 8% during the intervention period
- A smaller percentage of missed patients, from 21% to 16%
To be sure, pre- and post-implementation comparisons are difficult when the implementation period happens to coincide with the emergence of SARS-CoV-2.
Although fewer patients were seen and fewer lab tests were ordered overall in Alicante during the COVID-19 pandemic, the proportion of tests ordered for RF testing dropped, and all the patients identified with double positives for RF and anti-CCP antibodies were referred to rheumatology, suggesting evidence of benefit.
Dr. Kehl said the practice of using clinical decision support systems could be used in the United States. “I thought this was an important study,” she said. Electronic health records systems “have all these capabilities where we can include best practice alerts when you order a test to make sure that it’s clinically warranted and cost-effective.”
Dr. Salinas and Dr. Kehl reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
Noduloplaque on the Forehead
The Diagnosis: Giant Apocrine Hidrocystoma
Histopathology of the noduloplaque revealed an unremarkable epidermis with multilocular cystic spaces centered in the dermis. The cysts had a double-lined epithelium with inner columnar to cuboidal cells and outer myoepithelial cells (bottom quiz image). Columnar cells showing decapitation secretion could be appreciated at places indicating apocrine secretion (Figure). A final diagnosis of apocrine hidrocystoma was made.
.")
Hidrocystomas are rare, benign, cystic lesions derived either from apocrine or eccrine glands.1 Apocrine hidrocystoma usually manifests as asymptomatic, solitary, dome-shaped papules or nodules with a predilection for the head and neck region. Hidrocystomas can vary from flesh colored to blue, brown, or black. Pigmentation in hidrocystoma is seen in 5% to 80% of cases and is attributed to the Tyndall effect.1 The tumor usually is less than 20 mm in diameter; larger lesions are termed giant apocrine hidrocystoma.2 Apocrine hidrocystoma manifesting with multiple lesions and a size greater than 10 mm, as seen in our case, is uncommon.
Zaballos et al3 described dermoscopy of apocrine hidrocystoma in 22 patients. Hallmark dermoscopic findings were the presence of a homogeneous flesh-colored, yellowish, blue to pinkish-blue area involving the entire lesion with arborizing vessels and whitish structures.3 Similar dermoscopic findings were present in our patient. The homogeneous area histologically correlates to the multiloculated cysts located in the dermis. The exact reason for white structures is unknown; however, their visualization in apocrine hidrocystoma could be attributed to the alternation in collagen orientation secondary to the presence of large or multiple cysts in the dermis.
The presence of shiny white dots arranged in a square resembling a four-leaf clover (also known as white rosettes) was a unique dermoscopic finding in our patient. These rosettes can be appreciated only with polarized dermoscopy, and they have been described in actinic keratosis, seborrheic keratosis, squamous cell carcinoma, and basal cell carcinoma.4 The exact morphologic correlate of white rosettes is unknown but is postulated to be secondary to material inside adnexal openings in small rosettes and concentric perifollicular fibrosis in larger rosettes.4 In our patient, we believe the white rosettes can be attributed to the accumulated secretions in the dermal glands, which also were seen via histopathology. Dermoscopy also revealed increased peripheral, brown, networklike pigmentation, which was unique and could be secondary to the patient’s darker skin phenotype.
Differential diagnoses of apocrine hidrocystoma include both melanocytic and nonmelanocytic conditions such as epidermal cyst, nodular melanoma, nodular hidradenoma, syringoma, blue nevus, pilomatricoma, eccrine poroma, nodular Kaposi sarcoma, and venous lake.1 Histopathology showing large unilocular or multilocular dermal cysts with double lining comprising outer myoepithelial cells and inner columnar or cuboidal cell with decapitation secretion is paramount in confirming the diagnosis of apocrine hidrocystoma.
Dermoscopy can act as a valuable noninvasive modality in differentiating apocrine hidrocystoma from its melanocytic and nonmelanocytic differential diagnoses (Table).5-8 In our patient, the presence of a homogeneous pink to bluish area involving the entire lesion, linear branched vessels, and whitish structures on dermoscopy pointed to the diagnosis of apocrine hidrocystoma, which was further confirmed by characteristic histopathologic findings.

The treatment of apocrine hidrocystoma includes surgical excision for solitary lesions, with electrodesiccation and curettage, chemical cautery, and CO2 laser ablation employed for multiple lesions.1 Our patient was scheduled for CO2 laser ablation, considering the multiple lesions and size of the apocrine hidrocystoma but was subsequently lost to follow-up.
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp [published online August 15, 2020]. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703. doi:10.1111/j.1365-4632.2005.02512.x
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Haspeslagh M, Noë M, De Wispelaere I, et al. Rosettes and other white shiny structures in polarized dermoscopy: histological correlate and optical explanation. J Eur Acad Dermatol Venereol. 2016;30:311-313. doi:10.1111/jdv.13080
- Suh KS, Kang DY, Park JB, et al. Usefulness of dermoscopy in the differential diagnosis of ruptured and unruptured epidermal cysts. Ann Dermatol. 2017;29:33-38. doi:10.5021/ad.2017.29.1.33
- Serrano P, Lallas A, Del Pozo LJ, et al. Dermoscopy of nodular hidradenoma, a great masquerader: a morphological study of 28 cases. Dermatology. 2016;232:78-82. doi:10.1159/000441218
- Russo T, Piccolo V, Lallas A, et al. Dermoscopy of malignant skin tumours: what’s new? Dermatology. 2017;233:64-73. doi:10.1159/000472253
- Zaballos P, Llambrich A, Puig S, et al. Dermoscopic findings of pilomatricomas. Dermatology. 2008;217:225-230. doi:10.1159 /000148248
The Diagnosis: Giant Apocrine Hidrocystoma
Histopathology of the noduloplaque revealed an unremarkable epidermis with multilocular cystic spaces centered in the dermis. The cysts had a double-lined epithelium with inner columnar to cuboidal cells and outer myoepithelial cells (bottom quiz image). Columnar cells showing decapitation secretion could be appreciated at places indicating apocrine secretion (Figure). A final diagnosis of apocrine hidrocystoma was made.
Hidrocystomas are rare, benign, cystic lesions derived either from apocrine or eccrine glands.1 Apocrine hidrocystoma usually manifests as asymptomatic, solitary, dome-shaped papules or nodules with a predilection for the head and neck region. Hidrocystomas can vary from flesh colored to blue, brown, or black. Pigmentation in hidrocystoma is seen in 5% to 80% of cases and is attributed to the Tyndall effect.1 The tumor usually is less than 20 mm in diameter; larger lesions are termed giant apocrine hidrocystoma.2 Apocrine hidrocystoma manifesting with multiple lesions and a size greater than 10 mm, as seen in our case, is uncommon.
Zaballos et al3 described dermoscopy of apocrine hidrocystoma in 22 patients. Hallmark dermoscopic findings were the presence of a homogeneous flesh-colored, yellowish, blue to pinkish-blue area involving the entire lesion with arborizing vessels and whitish structures.3 Similar dermoscopic findings were present in our patient. The homogeneous area histologically correlates to the multiloculated cysts located in the dermis. The exact reason for white structures is unknown; however, their visualization in apocrine hidrocystoma could be attributed to the alternation in collagen orientation secondary to the presence of large or multiple cysts in the dermis.
The presence of shiny white dots arranged in a square resembling a four-leaf clover (also known as white rosettes) was a unique dermoscopic finding in our patient. These rosettes can be appreciated only with polarized dermoscopy, and they have been described in actinic keratosis, seborrheic keratosis, squamous cell carcinoma, and basal cell carcinoma.4 The exact morphologic correlate of white rosettes is unknown but is postulated to be secondary to material inside adnexal openings in small rosettes and concentric perifollicular fibrosis in larger rosettes.4 In our patient, we believe the white rosettes can be attributed to the accumulated secretions in the dermal glands, which also were seen via histopathology. Dermoscopy also revealed increased peripheral, brown, networklike pigmentation, which was unique and could be secondary to the patient’s darker skin phenotype.
Differential diagnoses of apocrine hidrocystoma include both melanocytic and nonmelanocytic conditions such as epidermal cyst, nodular melanoma, nodular hidradenoma, syringoma, blue nevus, pilomatricoma, eccrine poroma, nodular Kaposi sarcoma, and venous lake.1 Histopathology showing large unilocular or multilocular dermal cysts with double lining comprising outer myoepithelial cells and inner columnar or cuboidal cell with decapitation secretion is paramount in confirming the diagnosis of apocrine hidrocystoma.
Dermoscopy can act as a valuable noninvasive modality in differentiating apocrine hidrocystoma from its melanocytic and nonmelanocytic differential diagnoses (Table).5-8 In our patient, the presence of a homogeneous pink to bluish area involving the entire lesion, linear branched vessels, and whitish structures on dermoscopy pointed to the diagnosis of apocrine hidrocystoma, which was further confirmed by characteristic histopathologic findings.
The treatment of apocrine hidrocystoma includes surgical excision for solitary lesions, with electrodesiccation and curettage, chemical cautery, and CO2 laser ablation employed for multiple lesions.1 Our patient was scheduled for CO2 laser ablation, considering the multiple lesions and size of the apocrine hidrocystoma but was subsequently lost to follow-up.
The Diagnosis: Giant Apocrine Hidrocystoma
Histopathology of the noduloplaque revealed an unremarkable epidermis with multilocular cystic spaces centered in the dermis. The cysts had a double-lined epithelium with inner columnar to cuboidal cells and outer myoepithelial cells (bottom quiz image). Columnar cells showing decapitation secretion could be appreciated at places indicating apocrine secretion (Figure). A final diagnosis of apocrine hidrocystoma was made.
Hidrocystomas are rare, benign, cystic lesions derived either from apocrine or eccrine glands.1 Apocrine hidrocystoma usually manifests as asymptomatic, solitary, dome-shaped papules or nodules with a predilection for the head and neck region. Hidrocystomas can vary from flesh colored to blue, brown, or black. Pigmentation in hidrocystoma is seen in 5% to 80% of cases and is attributed to the Tyndall effect.1 The tumor usually is less than 20 mm in diameter; larger lesions are termed giant apocrine hidrocystoma.2 Apocrine hidrocystoma manifesting with multiple lesions and a size greater than 10 mm, as seen in our case, is uncommon.
Zaballos et al3 described dermoscopy of apocrine hidrocystoma in 22 patients. Hallmark dermoscopic findings were the presence of a homogeneous flesh-colored, yellowish, blue to pinkish-blue area involving the entire lesion with arborizing vessels and whitish structures.3 Similar dermoscopic findings were present in our patient. The homogeneous area histologically correlates to the multiloculated cysts located in the dermis. The exact reason for white structures is unknown; however, their visualization in apocrine hidrocystoma could be attributed to the alternation in collagen orientation secondary to the presence of large or multiple cysts in the dermis.
The presence of shiny white dots arranged in a square resembling a four-leaf clover (also known as white rosettes) was a unique dermoscopic finding in our patient. These rosettes can be appreciated only with polarized dermoscopy, and they have been described in actinic keratosis, seborrheic keratosis, squamous cell carcinoma, and basal cell carcinoma.4 The exact morphologic correlate of white rosettes is unknown but is postulated to be secondary to material inside adnexal openings in small rosettes and concentric perifollicular fibrosis in larger rosettes.4 In our patient, we believe the white rosettes can be attributed to the accumulated secretions in the dermal glands, which also were seen via histopathology. Dermoscopy also revealed increased peripheral, brown, networklike pigmentation, which was unique and could be secondary to the patient’s darker skin phenotype.
Differential diagnoses of apocrine hidrocystoma include both melanocytic and nonmelanocytic conditions such as epidermal cyst, nodular melanoma, nodular hidradenoma, syringoma, blue nevus, pilomatricoma, eccrine poroma, nodular Kaposi sarcoma, and venous lake.1 Histopathology showing large unilocular or multilocular dermal cysts with double lining comprising outer myoepithelial cells and inner columnar or cuboidal cell with decapitation secretion is paramount in confirming the diagnosis of apocrine hidrocystoma.
Dermoscopy can act as a valuable noninvasive modality in differentiating apocrine hidrocystoma from its melanocytic and nonmelanocytic differential diagnoses (Table).5-8 In our patient, the presence of a homogeneous pink to bluish area involving the entire lesion, linear branched vessels, and whitish structures on dermoscopy pointed to the diagnosis of apocrine hidrocystoma, which was further confirmed by characteristic histopathologic findings.
The treatment of apocrine hidrocystoma includes surgical excision for solitary lesions, with electrodesiccation and curettage, chemical cautery, and CO2 laser ablation employed for multiple lesions.1 Our patient was scheduled for CO2 laser ablation, considering the multiple lesions and size of the apocrine hidrocystoma but was subsequently lost to follow-up.
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp [published online August 15, 2020]. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703. doi:10.1111/j.1365-4632.2005.02512.x
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Haspeslagh M, Noë M, De Wispelaere I, et al. Rosettes and other white shiny structures in polarized dermoscopy: histological correlate and optical explanation. J Eur Acad Dermatol Venereol. 2016;30:311-313. doi:10.1111/jdv.13080
- Suh KS, Kang DY, Park JB, et al. Usefulness of dermoscopy in the differential diagnosis of ruptured and unruptured epidermal cysts. Ann Dermatol. 2017;29:33-38. doi:10.5021/ad.2017.29.1.33
- Serrano P, Lallas A, Del Pozo LJ, et al. Dermoscopy of nodular hidradenoma, a great masquerader: a morphological study of 28 cases. Dermatology. 2016;232:78-82. doi:10.1159/000441218
- Russo T, Piccolo V, Lallas A, et al. Dermoscopy of malignant skin tumours: what’s new? Dermatology. 2017;233:64-73. doi:10.1159/000472253
- Zaballos P, Llambrich A, Puig S, et al. Dermoscopic findings of pilomatricomas. Dermatology. 2008;217:225-230. doi:10.1159 /000148248
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp [published online August 15, 2020]. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703. doi:10.1111/j.1365-4632.2005.02512.x
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Haspeslagh M, Noë M, De Wispelaere I, et al. Rosettes and other white shiny structures in polarized dermoscopy: histological correlate and optical explanation. J Eur Acad Dermatol Venereol. 2016;30:311-313. doi:10.1111/jdv.13080
- Suh KS, Kang DY, Park JB, et al. Usefulness of dermoscopy in the differential diagnosis of ruptured and unruptured epidermal cysts. Ann Dermatol. 2017;29:33-38. doi:10.5021/ad.2017.29.1.33
- Serrano P, Lallas A, Del Pozo LJ, et al. Dermoscopy of nodular hidradenoma, a great masquerader: a morphological study of 28 cases. Dermatology. 2016;232:78-82. doi:10.1159/000441218
- Russo T, Piccolo V, Lallas A, et al. Dermoscopy of malignant skin tumours: what’s new? Dermatology. 2017;233:64-73. doi:10.1159/000472253
- Zaballos P, Llambrich A, Puig S, et al. Dermoscopic findings of pilomatricomas. Dermatology. 2008;217:225-230. doi:10.1159 /000148248
A 21-year-old man presented with a raised lesion on the forehead that had started as a single papule 16 years prior and gradually increased in number and size. There were no associated symptoms and no history of seasonal variation in the size of the lesions. Physical examination revealed multiple erythematous to slightly bluish translucent papules that coalesced to form a 3×3-cm noduloplaque with cystic consistency on the right side of the forehead (top). Dermoscopic examination (middle) (polarized noncontact mode) revealed a homogeneous pink to bluish background, scattered linear vessels with branches (black arrows), multiple chrysalislike shiny white lines (blue arrows), and dots arranged in a 4-dot pattern (black circle) resembling a four-leaf clover. Increased peripheral, brown, networklike pigmentation (black stars) also was noted on dermoscopy. Histopathologic examination of the noduloplaque was performed (bottom).



Cell-Free DNA May Inform IBD Diagnosis
LAS VEGAS —
“We think for indeterminate colitis, our assay can be quite beneficial in helping a clinician resolve or provide some additional insight and information. We have a very robust ability to predict from just the plasma microbial cell free DNA alone [whether] individuals are in remission or mild or moderate or active disease,” Shiv Kale, PhD, said during a presentation of the results at the annual Crohn’s & Colitis Congress®, a partnership of the Crohn’s & Colitis Foundation and the American Gastroenterological Association. Dr. Kale is director of computational biomarker discovery at Karius Inc., a company whose Karius Test is also being developed as a rapid test for various infections and febrile neutropenia.
cfDNA has proved to be a useful biomarker for a range of conditions, including cancer screening, diagnosis and monitoring, prenatal testing, and organ monitoring following transplantation. The tests rely on the fact that both human and microbial cells release DNA after cell death, and it is readily detectable in plasma.
Dr. Kale described the company’s efforts to identify microbial species biomarkers and a classification scheme that he said leads to consistent performance.
The latest study included 196 patients with Crohn’s disease and 196 with ulcerative colitis, with each group including individuals in remission and with mild, moderate, and severe disease. All patients had an endoscopic assessment within 30 days of plasma measurements.
cfDNA distinguished between patients with Crohn’s disease, ulcerative colitis, and those who were asymptomatic. It distinguished between patients with IBD and those who were asymptomatic with a sensitivity of 99.5% and a specificity of 90%, which are equivalent to or better than other traditional measures to distinguish active IBD versus asymptomatic patients.
The researchers plan a follow-up study of 1,800 samples in partnership with the Crohn’s and Colitis Foundation to examine the ability of cfDNA to determine disease severity, as well as location of disease and Crohn’s disease subtypes.
An ‘Intriguing’ Method
During the Q&A session, one questioner pointed out that colorectal cancer and infections can produce microbial signatures that could be confounding the results. He asked: “Do you have a data set to compare [cfDNA findings] to serum from C. diff, norovirus, and colorectal cancer? And if you don’t, I strongly encourage you to do so.” Dr. Kale responded that the group is working on that.
Asked for comment, session moderator Dana Lukin, MD, AGAF, offered praise for the work.
“I think it’s an intriguing method that we haven’t seen used as a predictor in IBD. Using this as a biomarker of disease type is very intriguing for confirming a diagnosis. I think probably one of the most compelling points is the distinction between IBD and non-IBD. Our current serologic-based assays do not really do that very well. I think this is a big improvement on that,” said Dr. Lukin, associate professor of clinical medicine at Weill Cornell Medical College, New York.

He echoed the questioner’s comments, suggesting that a key question will be whether cfDNA can correlate with disease activity over time.
He also called for prospective studies to validate the approach.
If successful, the technology could have broad application. “Certainly in kids this might be useful or in folks that either aren’t as inclined to have invasive testing done, or for whatever logistic reasons it’s not easy,” said Dr. Lukin.
Dr. Kale is an employee of Karius. Dr. Lukin has no relevant financial disclosures..
LAS VEGAS —
“We think for indeterminate colitis, our assay can be quite beneficial in helping a clinician resolve or provide some additional insight and information. We have a very robust ability to predict from just the plasma microbial cell free DNA alone [whether] individuals are in remission or mild or moderate or active disease,” Shiv Kale, PhD, said during a presentation of the results at the annual Crohn’s & Colitis Congress®, a partnership of the Crohn’s & Colitis Foundation and the American Gastroenterological Association. Dr. Kale is director of computational biomarker discovery at Karius Inc., a company whose Karius Test is also being developed as a rapid test for various infections and febrile neutropenia.
cfDNA has proved to be a useful biomarker for a range of conditions, including cancer screening, diagnosis and monitoring, prenatal testing, and organ monitoring following transplantation. The tests rely on the fact that both human and microbial cells release DNA after cell death, and it is readily detectable in plasma.
Dr. Kale described the company’s efforts to identify microbial species biomarkers and a classification scheme that he said leads to consistent performance.
The latest study included 196 patients with Crohn’s disease and 196 with ulcerative colitis, with each group including individuals in remission and with mild, moderate, and severe disease. All patients had an endoscopic assessment within 30 days of plasma measurements.
cfDNA distinguished between patients with Crohn’s disease, ulcerative colitis, and those who were asymptomatic. It distinguished between patients with IBD and those who were asymptomatic with a sensitivity of 99.5% and a specificity of 90%, which are equivalent to or better than other traditional measures to distinguish active IBD versus asymptomatic patients.
The researchers plan a follow-up study of 1,800 samples in partnership with the Crohn’s and Colitis Foundation to examine the ability of cfDNA to determine disease severity, as well as location of disease and Crohn’s disease subtypes.
An ‘Intriguing’ Method
During the Q&A session, one questioner pointed out that colorectal cancer and infections can produce microbial signatures that could be confounding the results. He asked: “Do you have a data set to compare [cfDNA findings] to serum from C. diff, norovirus, and colorectal cancer? And if you don’t, I strongly encourage you to do so.” Dr. Kale responded that the group is working on that.
Asked for comment, session moderator Dana Lukin, MD, AGAF, offered praise for the work.
“I think it’s an intriguing method that we haven’t seen used as a predictor in IBD. Using this as a biomarker of disease type is very intriguing for confirming a diagnosis. I think probably one of the most compelling points is the distinction between IBD and non-IBD. Our current serologic-based assays do not really do that very well. I think this is a big improvement on that,” said Dr. Lukin, associate professor of clinical medicine at Weill Cornell Medical College, New York.
He echoed the questioner’s comments, suggesting that a key question will be whether cfDNA can correlate with disease activity over time.
He also called for prospective studies to validate the approach.
If successful, the technology could have broad application. “Certainly in kids this might be useful or in folks that either aren’t as inclined to have invasive testing done, or for whatever logistic reasons it’s not easy,” said Dr. Lukin.
Dr. Kale is an employee of Karius. Dr. Lukin has no relevant financial disclosures..
LAS VEGAS —
“We think for indeterminate colitis, our assay can be quite beneficial in helping a clinician resolve or provide some additional insight and information. We have a very robust ability to predict from just the plasma microbial cell free DNA alone [whether] individuals are in remission or mild or moderate or active disease,” Shiv Kale, PhD, said during a presentation of the results at the annual Crohn’s & Colitis Congress®, a partnership of the Crohn’s & Colitis Foundation and the American Gastroenterological Association. Dr. Kale is director of computational biomarker discovery at Karius Inc., a company whose Karius Test is also being developed as a rapid test for various infections and febrile neutropenia.
cfDNA has proved to be a useful biomarker for a range of conditions, including cancer screening, diagnosis and monitoring, prenatal testing, and organ monitoring following transplantation. The tests rely on the fact that both human and microbial cells release DNA after cell death, and it is readily detectable in plasma.
Dr. Kale described the company’s efforts to identify microbial species biomarkers and a classification scheme that he said leads to consistent performance.
The latest study included 196 patients with Crohn’s disease and 196 with ulcerative colitis, with each group including individuals in remission and with mild, moderate, and severe disease. All patients had an endoscopic assessment within 30 days of plasma measurements.
cfDNA distinguished between patients with Crohn’s disease, ulcerative colitis, and those who were asymptomatic. It distinguished between patients with IBD and those who were asymptomatic with a sensitivity of 99.5% and a specificity of 90%, which are equivalent to or better than other traditional measures to distinguish active IBD versus asymptomatic patients.
The researchers plan a follow-up study of 1,800 samples in partnership with the Crohn’s and Colitis Foundation to examine the ability of cfDNA to determine disease severity, as well as location of disease and Crohn’s disease subtypes.
An ‘Intriguing’ Method
During the Q&A session, one questioner pointed out that colorectal cancer and infections can produce microbial signatures that could be confounding the results. He asked: “Do you have a data set to compare [cfDNA findings] to serum from C. diff, norovirus, and colorectal cancer? And if you don’t, I strongly encourage you to do so.” Dr. Kale responded that the group is working on that.
Asked for comment, session moderator Dana Lukin, MD, AGAF, offered praise for the work.
“I think it’s an intriguing method that we haven’t seen used as a predictor in IBD. Using this as a biomarker of disease type is very intriguing for confirming a diagnosis. I think probably one of the most compelling points is the distinction between IBD and non-IBD. Our current serologic-based assays do not really do that very well. I think this is a big improvement on that,” said Dr. Lukin, associate professor of clinical medicine at Weill Cornell Medical College, New York.
He echoed the questioner’s comments, suggesting that a key question will be whether cfDNA can correlate with disease activity over time.
He also called for prospective studies to validate the approach.
If successful, the technology could have broad application. “Certainly in kids this might be useful or in folks that either aren’t as inclined to have invasive testing done, or for whatever logistic reasons it’s not easy,” said Dr. Lukin.
Dr. Kale is an employee of Karius. Dr. Lukin has no relevant financial disclosures..
FROM CROHN’S AND COLITIS CONGRESS
Targeting Fetus-derived Gdf15 May Curb Nausea and Vomiting of Pregnancy
, and targeting the hormone prophylactically may reduce this common gestational condition.
This protein acts on the brainstem to cause emesis, and, significantly, a mother’s prior exposure to it determines the degree of NVP severity she will experience, according to international researchers including Marlena Fejzo, PhD, a clinical assistant professor of population and public Health at Keck School of Medicine, University of Southern California, Los Angeles.
“GDF15 is at the mechanistic heart of NVP and HG [hyperemesis gravidarum],” Dr. Fejzo and colleagues wrote in Nature, pointing to the need for preventive and therapeutic strategies.

“My previous research showed an association between variation in the GDF15 gene and nausea and vomiting of pregnancy and HG, and this study takes it one step further by elucidating the mechanism. It confirms that the nausea and vomiting (N/V) hormone GDF15 is a major cause of NVP and HG,” Dr. Fejzo said.
The etiology of NVP remains poorly understood although it affects up to 80% of pregnancies. In the US, its severe form, HG, is the leading cause of hospitalization in early pregnancy and the second-leading reason for pregnancy hospitalization overall.
The immunoassay-based study showed that the majority of GDF15 in maternal blood during pregnancy comes from the fetal part of the placenta, and confirms previous studies reporting higher levels in pregnancies with more severe NVP, said Dr. Fejzo, who is who is a board member of the Hyperemesis Education and Research Foundation.
“However, what was really fascinating and surprising is that prior to pregnancy the women who have more severe NVP symptoms actually have lower levels of the hormone.”
Although the gene variant linked to HG was previously associated with higher circulating levels in maternal blood, counterintuitively, this new research showed that women with abnormally high levels prior to pregnancy have either no or very little NVP, said Dr. Fejzo. “That suggests that in humans higher levels may lead to a desensitization to the high levels of the hormone in pregnancy. Then we also proved that desensitization can occur in a mouse model.”
According to Erin Higgins, MD, a clinical assistant professor of obgyn and reproductive biology at the Cleveland Clinic, Cleveland, Ohio, who was not involved in the study, “This is an exciting finding that may help us to better target treatment of N/V in pregnancy. Factors for NVP have been identified, but to my knowledge there has not been a clear etiology.”

Dr. Higgins cautioned, however, that the GDF15 gene seems important in normal placentation, “so it’s not as simple as blocking the gene or its receptor.” But since preconception exposure to GDF15 might decrease nausea and vomiting once a woman is pregnant, prophylactic treatment may be possible, and metformin has been suggested as a possibility, she said.
The study findings emerged from immunoassays on maternal blood samples collected at about 15 weeks (first trimester and early second trimester), from women with NVP (n = 168) or seen at a hospital for HG (n = 57). Results were compared with those from controls having similar characteristics but no significant symptoms.
Interestingly, GDF15 is also associated with cachexia, a condition similar to HG and characterized by loss of appetite and weight loss, Dr. Fejzo noted. “The hormone can be produced by malignant tumors at levels similar to those seen in pregnancy, and symptoms can be reduced by blocking GDF15 or its receptor, GFRAL. Clinical trials are already underway in cancer patients to test this.”
She is seeking funding to test the impact of increasing GDF15 levels prior to pregnancy in patients who previously experienced HG. “I am confident that desensitizing patients by increasing GDF15 prior to pregnancy and by lowering GDF15 levels during pregnancy will work. But we need to make sure we do safety studies and get the dosing and duration right, and that will take some time.”
Desensitization will need testing first in HG, where the risk for adverse maternal and fetal outcomes is high, so the benefit will outweigh any possible risk of testing medication in pregnancy, she continued. “It will take some time before we get to patients with normal NVP, but I do believe eventually the new findings will result in game-changing therapeutics for the condition.”
Dr. Higgins added, “Even if this isn’t the golden ticket, researchers and clinicians are working toward improvements in the treatment of NVP. We’ve already come a long way in recent years with the development of treatment algorithms and the advent of doxylamine/pyridoxine.”
This study was supported primarily by the Medical Research Council UK and National Institute for Health and Care Research UK, with additional support from various research funding organizations, including Novo Nordisk Foundation.
Dr. Fejzo is a paid consultant for Materna Biosciences and NGM Biopharmaceuticals, and a board member and science adviser for the Hyperemesis Education and Research Foundation.
Numerous study co-authors disclosed financial relationships with private-sector companies, including employment and patent ownership.
Dr. Higgins disclosed no competing interests relevant to her comments but is an instructor for Organon.
, and targeting the hormone prophylactically may reduce this common gestational condition.
This protein acts on the brainstem to cause emesis, and, significantly, a mother’s prior exposure to it determines the degree of NVP severity she will experience, according to international researchers including Marlena Fejzo, PhD, a clinical assistant professor of population and public Health at Keck School of Medicine, University of Southern California, Los Angeles.
“GDF15 is at the mechanistic heart of NVP and HG [hyperemesis gravidarum],” Dr. Fejzo and colleagues wrote in Nature, pointing to the need for preventive and therapeutic strategies.
“My previous research showed an association between variation in the GDF15 gene and nausea and vomiting of pregnancy and HG, and this study takes it one step further by elucidating the mechanism. It confirms that the nausea and vomiting (N/V) hormone GDF15 is a major cause of NVP and HG,” Dr. Fejzo said.
The etiology of NVP remains poorly understood although it affects up to 80% of pregnancies. In the US, its severe form, HG, is the leading cause of hospitalization in early pregnancy and the second-leading reason for pregnancy hospitalization overall.
The immunoassay-based study showed that the majority of GDF15 in maternal blood during pregnancy comes from the fetal part of the placenta, and confirms previous studies reporting higher levels in pregnancies with more severe NVP, said Dr. Fejzo, who is who is a board member of the Hyperemesis Education and Research Foundation.
“However, what was really fascinating and surprising is that prior to pregnancy the women who have more severe NVP symptoms actually have lower levels of the hormone.”
Although the gene variant linked to HG was previously associated with higher circulating levels in maternal blood, counterintuitively, this new research showed that women with abnormally high levels prior to pregnancy have either no or very little NVP, said Dr. Fejzo. “That suggests that in humans higher levels may lead to a desensitization to the high levels of the hormone in pregnancy. Then we also proved that desensitization can occur in a mouse model.”
According to Erin Higgins, MD, a clinical assistant professor of obgyn and reproductive biology at the Cleveland Clinic, Cleveland, Ohio, who was not involved in the study, “This is an exciting finding that may help us to better target treatment of N/V in pregnancy. Factors for NVP have been identified, but to my knowledge there has not been a clear etiology.”
Dr. Higgins cautioned, however, that the GDF15 gene seems important in normal placentation, “so it’s not as simple as blocking the gene or its receptor.” But since preconception exposure to GDF15 might decrease nausea and vomiting once a woman is pregnant, prophylactic treatment may be possible, and metformin has been suggested as a possibility, she said.
The study findings emerged from immunoassays on maternal blood samples collected at about 15 weeks (first trimester and early second trimester), from women with NVP (n = 168) or seen at a hospital for HG (n = 57). Results were compared with those from controls having similar characteristics but no significant symptoms.
Interestingly, GDF15 is also associated with cachexia, a condition similar to HG and characterized by loss of appetite and weight loss, Dr. Fejzo noted. “The hormone can be produced by malignant tumors at levels similar to those seen in pregnancy, and symptoms can be reduced by blocking GDF15 or its receptor, GFRAL. Clinical trials are already underway in cancer patients to test this.”
She is seeking funding to test the impact of increasing GDF15 levels prior to pregnancy in patients who previously experienced HG. “I am confident that desensitizing patients by increasing GDF15 prior to pregnancy and by lowering GDF15 levels during pregnancy will work. But we need to make sure we do safety studies and get the dosing and duration right, and that will take some time.”
Desensitization will need testing first in HG, where the risk for adverse maternal and fetal outcomes is high, so the benefit will outweigh any possible risk of testing medication in pregnancy, she continued. “It will take some time before we get to patients with normal NVP, but I do believe eventually the new findings will result in game-changing therapeutics for the condition.”
Dr. Higgins added, “Even if this isn’t the golden ticket, researchers and clinicians are working toward improvements in the treatment of NVP. We’ve already come a long way in recent years with the development of treatment algorithms and the advent of doxylamine/pyridoxine.”
This study was supported primarily by the Medical Research Council UK and National Institute for Health and Care Research UK, with additional support from various research funding organizations, including Novo Nordisk Foundation.
Dr. Fejzo is a paid consultant for Materna Biosciences and NGM Biopharmaceuticals, and a board member and science adviser for the Hyperemesis Education and Research Foundation.
Numerous study co-authors disclosed financial relationships with private-sector companies, including employment and patent ownership.
Dr. Higgins disclosed no competing interests relevant to her comments but is an instructor for Organon.
, and targeting the hormone prophylactically may reduce this common gestational condition.
This protein acts on the brainstem to cause emesis, and, significantly, a mother’s prior exposure to it determines the degree of NVP severity she will experience, according to international researchers including Marlena Fejzo, PhD, a clinical assistant professor of population and public Health at Keck School of Medicine, University of Southern California, Los Angeles.
“GDF15 is at the mechanistic heart of NVP and HG [hyperemesis gravidarum],” Dr. Fejzo and colleagues wrote in Nature, pointing to the need for preventive and therapeutic strategies.
“My previous research showed an association between variation in the GDF15 gene and nausea and vomiting of pregnancy and HG, and this study takes it one step further by elucidating the mechanism. It confirms that the nausea and vomiting (N/V) hormone GDF15 is a major cause of NVP and HG,” Dr. Fejzo said.
The etiology of NVP remains poorly understood although it affects up to 80% of pregnancies. In the US, its severe form, HG, is the leading cause of hospitalization in early pregnancy and the second-leading reason for pregnancy hospitalization overall.
The immunoassay-based study showed that the majority of GDF15 in maternal blood during pregnancy comes from the fetal part of the placenta, and confirms previous studies reporting higher levels in pregnancies with more severe NVP, said Dr. Fejzo, who is who is a board member of the Hyperemesis Education and Research Foundation.
“However, what was really fascinating and surprising is that prior to pregnancy the women who have more severe NVP symptoms actually have lower levels of the hormone.”
Although the gene variant linked to HG was previously associated with higher circulating levels in maternal blood, counterintuitively, this new research showed that women with abnormally high levels prior to pregnancy have either no or very little NVP, said Dr. Fejzo. “That suggests that in humans higher levels may lead to a desensitization to the high levels of the hormone in pregnancy. Then we also proved that desensitization can occur in a mouse model.”
According to Erin Higgins, MD, a clinical assistant professor of obgyn and reproductive biology at the Cleveland Clinic, Cleveland, Ohio, who was not involved in the study, “This is an exciting finding that may help us to better target treatment of N/V in pregnancy. Factors for NVP have been identified, but to my knowledge there has not been a clear etiology.”
Dr. Higgins cautioned, however, that the GDF15 gene seems important in normal placentation, “so it’s not as simple as blocking the gene or its receptor.” But since preconception exposure to GDF15 might decrease nausea and vomiting once a woman is pregnant, prophylactic treatment may be possible, and metformin has been suggested as a possibility, she said.
The study findings emerged from immunoassays on maternal blood samples collected at about 15 weeks (first trimester and early second trimester), from women with NVP (n = 168) or seen at a hospital for HG (n = 57). Results were compared with those from controls having similar characteristics but no significant symptoms.
Interestingly, GDF15 is also associated with cachexia, a condition similar to HG and characterized by loss of appetite and weight loss, Dr. Fejzo noted. “The hormone can be produced by malignant tumors at levels similar to those seen in pregnancy, and symptoms can be reduced by blocking GDF15 or its receptor, GFRAL. Clinical trials are already underway in cancer patients to test this.”
She is seeking funding to test the impact of increasing GDF15 levels prior to pregnancy in patients who previously experienced HG. “I am confident that desensitizing patients by increasing GDF15 prior to pregnancy and by lowering GDF15 levels during pregnancy will work. But we need to make sure we do safety studies and get the dosing and duration right, and that will take some time.”
Desensitization will need testing first in HG, where the risk for adverse maternal and fetal outcomes is high, so the benefit will outweigh any possible risk of testing medication in pregnancy, she continued. “It will take some time before we get to patients with normal NVP, but I do believe eventually the new findings will result in game-changing therapeutics for the condition.”
Dr. Higgins added, “Even if this isn’t the golden ticket, researchers and clinicians are working toward improvements in the treatment of NVP. We’ve already come a long way in recent years with the development of treatment algorithms and the advent of doxylamine/pyridoxine.”
This study was supported primarily by the Medical Research Council UK and National Institute for Health and Care Research UK, with additional support from various research funding organizations, including Novo Nordisk Foundation.
Dr. Fejzo is a paid consultant for Materna Biosciences and NGM Biopharmaceuticals, and a board member and science adviser for the Hyperemesis Education and Research Foundation.
Numerous study co-authors disclosed financial relationships with private-sector companies, including employment and patent ownership.
Dr. Higgins disclosed no competing interests relevant to her comments but is an instructor for Organon.
FROM NATURE
Nonblanching, Erythematous, Cerebriform Plaques on the Foot
The Diagnosis: Coral Dermatitis
At 3-week follow-up, the patient demonstrated remarkable improvement in the intensity and size of the erythematous cerebriform plaques following daily application of triamcinolone acetonide cream 0.1% (Figure). The lesion disappeared after several months and did not recur. The delayed presentation of symptoms with a history of incidental coral contact during snorkeling most likely represents the type IV hypersensitivity reaction seen in the diagnosis of coral dermatitis, an extraordinarily rare form of contact dermatitis.1 Not all coral trigger skin reactions. Species of coral that contain nematocysts in their tentacles (aptly named stinging capsules) are responsible for the sting preceding coral dermatitis, as the nematocysts eject a coiled filament in response to human tactile stimulation that injects toxins into the epidermis.2

Acute, delayed, or chronic cutaneous changes follow envenomation. Acute responses arise immediately to a few hours after initial contact and are considered an irritant contact dermatitis.3 Local tissue histamine release and cascades of cytotoxic reactions often result in the characteristic urticarial or vesiculobullous plaques in addition to necrosis, piloerection, and localized lymphadenopathy.2-4 Although relatively uncommon, there may be rapid onset of systemic symptoms such as fever, malaise, hives, nausea, or emesis. Cardiopulmonary events, hepatotoxicity, renal failure, or anaphylaxis are rare.2 Histopathology of biopsy specimens reveals epidermal spongiosis with microvesicles and papillary dermal edema.1,5 In comparison, delayed reactions occur within days to weeks and exhibit epidermal parakeratosis, spongiosis, basal layer vacuolization, focal necrosis, lymphocyte exocytosis, and papillary dermal edema with extravasated erythrocytes.1,6 Clinically, it may present as linear rows of erythematous papules with burning and pruritus.6 Chronic reactions manifest after months as difficult-to-treat, persistent lichenoid dermatitis occasionally accompanied by granulomatous changes.1,2,4 Primary prevention measures after initial contact include an acetic acid rinse and cold compression to wash away residual nematocysts in the affected area.4,7,8 If a rash develops, topical steroids are the mainstay of treatment.3,8
In tandem with toxic nematocysts, the rigid calcified bodies of coral provide an additional self-defense mechanism against human contact.2,4 The irregular haphazard nature of coral may catch novice divers off guard and lead to laceration of a mispositioned limb, thereby increasing the risk for secondary infections due to the introduction of calcium carbonate and toxic mucinous deposits at the wound site, warranting antibiotic treatment.2,4,7 Because tropical locales are home to other natural dangers that inflict disease and mimic early signs of coral dermatitis, reaching an accurate diagnosis can be difficult, particularly for lower limb lesions. In summary, the diagnosis of coral dermatitis can be rendered based on morphology of the lesion and clinical context (exposure to corals and delayed symptoms) as well as response to topical steroids.
The differential diagnosis includes accidental trauma. Variations in impact force and patient skin integrity lead to a number of possible cutaneous manifestations seen in accidental trauma,9 which includes contusions resulting from burst capillaries underneath intact skin, abrasions due to the superficial epidermis scuffing away, and lacerations caused by enough force to rip and split the skin, leaving subcutaneous tissue between the intact tissue.9,10 Typically, the pattern of injury can provide hints to match what object or organism caused the wound.9 However, delayed response and worsening symptoms, as seen in coral dermatitis, would be unusual in accidental trauma unless it is complicated by secondary infection (infectious dermatitis), which does not respond to topical steroids and requires antibiotic treatment.
Another differential diagnosis includes cutaneous larva migrans, which infests domesticated and stray animals. For example, hookworm larvae propagate their eggs inside the intestines of their host before fecal-soil transmission in sandy locales.11 Unexpecting beachgoers travel barefoot on this contaminated soil, offering ample opportunity for the parasite to burrow into the upper dermis.11,12 The clinical presentation includes signs and symptoms of creeping eruption such as pruritic, linear, serpiginous tracks. Topical treatment with thiabendazole requires application 3 times daily for 15 days, which increases the risk for nonadherence, yet this therapy proves advantageous if a patient does not tolerate oral agents due to systemic adverse effects.11,12 Oral agents (eg, ivermectin, albendazole) offer improved adherence with a single dose11,13; the cure rate was higher with a single dose of ivermectin 12 mg vs a single dose of albendazole 400 mg.13 The current suggested treatment is ivermectin 200 μg/kg by mouth daily for 1 or 2 days.14
The incidence of seabather’s eruption (also known as chinkui dermatitis) is highest during the summer season and fluctuates between epidemic and nonepidemic years.15,16 It occurs sporadically worldwide mostly in tropical climates due to trapping of larvae spawn of sea animals such as crustaceans in swimwear. Initially, it presents as a pruritic and burning sensation after exiting the water, manifesting as a macular, papular, or maculopapular rash on areas covered by the swimsuit.15,16 The sensation is worse in areas that are tightly banded on the swimsuit, including the waistband and elastic straps.15 Commonly, the affected individual will seek relief via a shower, which intensifies the burning, especially if the swimsuit has not been removed. The contaminated swimwear should be immediately discarded, as the trapped sea larvae’s nematocysts activate with the pressure and friction of movement.15 Seabather’s eruption typically resolves spontaneously within a week, but symptom management can be achieved with topical steroids (triamcinolone 0.1% or clobetasol 0.05%).15,16 Unlike coral dermatitis, in seabather’s eruption the symptoms are immediate and the location of the eruption coincides with areas covered by the swimsuit.
- Ahn HS, Yoon SY, Park HJ, et al. A patient with delayed contact dermatitis to coral and she displayed superficial granuloma. Ann Dermatol. 2009;21:95-97. doi:10.5021/ad.2009.21.1.95
- Haddad V Jr, Lupi O, Lonza JP, et al. Tropical dermatology: marine and aquatic dermatology. J Am Acad Dermatol. 2009;61:733-752. doi:10.1016/j.jaad.2009.01.046
- Salik J, Tang R. Images in clinical medicine. Coral dermatitis. N Engl J Med. 2015;373:E2. doi:10.1056/NEJMicm1412907
- Reese E, Depenbrock P. Water envenomations and stings. Curr Sports Med Rep. 2014;13:126-131. doi:10.1249/JSR.0000000000000042
- Addy JH. Red sea coral contact dermatitis. Int J Dermatol. 1991; 30:271-273. doi:10.1111/j.1365-4362.1991.tb04636.x
- Miracco C, Lalinga AV, Sbano P, et al. Delayed skin reaction to Red Sea coral injury showing superficial granulomas and atypical CD30+ lymphocytes: report of a case. Br J Dermatol. 2001;145:849-851. doi:10.1046/j.1365-2133.2001.04454.x
- Ceponis PJ, Cable R, Weaver LK. Don’t kick the coral! Wilderness Environ Med. 2017;28:153-155. doi:10.1016/j.wem.2017.01.025
- Tlougan BE, Podjasek JO, Adams BB. Aquatic sports dematoses. part 2-in the water: saltwater dermatoses. Int J Dermatol. 2010;49:994-1002. doi:10.1111/j.1365-4632.2010.04476.x
- Simon LV, Lopez RA, King KC. Blunt force trauma. StatPearls [Internet]. StatPearls Publishing; 2023. Accessed January 12, 2034. https://www.ncbi.nlm.nih.gov/books/NBK470338/
- Gentile S, Kneubuehl BP, Barrera V, et al. Fracture energy threshold in parry injuries due to sharp and blunt force. Int J Legal Med. 2019;133:1429-1435.
- Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814. doi:10.1086/313787
- Davies HD, Sakuls P, Keystone JS. Creeping eruption. A review of clinical presentation and management of 60 cases presenting to a tropical disease unit. Arch Dermatol. 1993;129:588-591. doi:10.1001 /archderm.129.5.588
- Caumes E, Carriere J, Datry A, et al. A randomized trial of ivermectin versus albendazole for the treatment of cutaneous larva migrans. Am J Trop Med Hyg. 1993;49:641-644. doi:10.4269 /ajtmh.1993.49.641
- Schuster A, Lesshafft H, Reichert F, et al. Hookworm-related cutaneous larva migrans in northern Brazil: resolution of clinical pathology after a single dose of ivermectin. Clin Infect Dis. 2013;57:1155-1157. doi:10.1093/cid/cit440
- Freudenthal AR, Joseph PR. Seabather’s eruption. N Engl J Med. 1993;329:542-544. doi:10.1056/NEJM199308193290805
- Odagawa S, Watari T, Yoshida M. Chinkui dermatitis: the sea bather’s eruption. QJM. 2022;115:100-101. doi:10.1093/qjmed/hcab277
The Diagnosis: Coral Dermatitis
At 3-week follow-up, the patient demonstrated remarkable improvement in the intensity and size of the erythematous cerebriform plaques following daily application of triamcinolone acetonide cream 0.1% (Figure). The lesion disappeared after several months and did not recur. The delayed presentation of symptoms with a history of incidental coral contact during snorkeling most likely represents the type IV hypersensitivity reaction seen in the diagnosis of coral dermatitis, an extraordinarily rare form of contact dermatitis.1 Not all coral trigger skin reactions. Species of coral that contain nematocysts in their tentacles (aptly named stinging capsules) are responsible for the sting preceding coral dermatitis, as the nematocysts eject a coiled filament in response to human tactile stimulation that injects toxins into the epidermis.2
Acute, delayed, or chronic cutaneous changes follow envenomation. Acute responses arise immediately to a few hours after initial contact and are considered an irritant contact dermatitis.3 Local tissue histamine release and cascades of cytotoxic reactions often result in the characteristic urticarial or vesiculobullous plaques in addition to necrosis, piloerection, and localized lymphadenopathy.2-4 Although relatively uncommon, there may be rapid onset of systemic symptoms such as fever, malaise, hives, nausea, or emesis. Cardiopulmonary events, hepatotoxicity, renal failure, or anaphylaxis are rare.2 Histopathology of biopsy specimens reveals epidermal spongiosis with microvesicles and papillary dermal edema.1,5 In comparison, delayed reactions occur within days to weeks and exhibit epidermal parakeratosis, spongiosis, basal layer vacuolization, focal necrosis, lymphocyte exocytosis, and papillary dermal edema with extravasated erythrocytes.1,6 Clinically, it may present as linear rows of erythematous papules with burning and pruritus.6 Chronic reactions manifest after months as difficult-to-treat, persistent lichenoid dermatitis occasionally accompanied by granulomatous changes.1,2,4 Primary prevention measures after initial contact include an acetic acid rinse and cold compression to wash away residual nematocysts in the affected area.4,7,8 If a rash develops, topical steroids are the mainstay of treatment.3,8
In tandem with toxic nematocysts, the rigid calcified bodies of coral provide an additional self-defense mechanism against human contact.2,4 The irregular haphazard nature of coral may catch novice divers off guard and lead to laceration of a mispositioned limb, thereby increasing the risk for secondary infections due to the introduction of calcium carbonate and toxic mucinous deposits at the wound site, warranting antibiotic treatment.2,4,7 Because tropical locales are home to other natural dangers that inflict disease and mimic early signs of coral dermatitis, reaching an accurate diagnosis can be difficult, particularly for lower limb lesions. In summary, the diagnosis of coral dermatitis can be rendered based on morphology of the lesion and clinical context (exposure to corals and delayed symptoms) as well as response to topical steroids.
The differential diagnosis includes accidental trauma. Variations in impact force and patient skin integrity lead to a number of possible cutaneous manifestations seen in accidental trauma,9 which includes contusions resulting from burst capillaries underneath intact skin, abrasions due to the superficial epidermis scuffing away, and lacerations caused by enough force to rip and split the skin, leaving subcutaneous tissue between the intact tissue.9,10 Typically, the pattern of injury can provide hints to match what object or organism caused the wound.9 However, delayed response and worsening symptoms, as seen in coral dermatitis, would be unusual in accidental trauma unless it is complicated by secondary infection (infectious dermatitis), which does not respond to topical steroids and requires antibiotic treatment.
Another differential diagnosis includes cutaneous larva migrans, which infests domesticated and stray animals. For example, hookworm larvae propagate their eggs inside the intestines of their host before fecal-soil transmission in sandy locales.11 Unexpecting beachgoers travel barefoot on this contaminated soil, offering ample opportunity for the parasite to burrow into the upper dermis.11,12 The clinical presentation includes signs and symptoms of creeping eruption such as pruritic, linear, serpiginous tracks. Topical treatment with thiabendazole requires application 3 times daily for 15 days, which increases the risk for nonadherence, yet this therapy proves advantageous if a patient does not tolerate oral agents due to systemic adverse effects.11,12 Oral agents (eg, ivermectin, albendazole) offer improved adherence with a single dose11,13; the cure rate was higher with a single dose of ivermectin 12 mg vs a single dose of albendazole 400 mg.13 The current suggested treatment is ivermectin 200 μg/kg by mouth daily for 1 or 2 days.14
The incidence of seabather’s eruption (also known as chinkui dermatitis) is highest during the summer season and fluctuates between epidemic and nonepidemic years.15,16 It occurs sporadically worldwide mostly in tropical climates due to trapping of larvae spawn of sea animals such as crustaceans in swimwear. Initially, it presents as a pruritic and burning sensation after exiting the water, manifesting as a macular, papular, or maculopapular rash on areas covered by the swimsuit.15,16 The sensation is worse in areas that are tightly banded on the swimsuit, including the waistband and elastic straps.15 Commonly, the affected individual will seek relief via a shower, which intensifies the burning, especially if the swimsuit has not been removed. The contaminated swimwear should be immediately discarded, as the trapped sea larvae’s nematocysts activate with the pressure and friction of movement.15 Seabather’s eruption typically resolves spontaneously within a week, but symptom management can be achieved with topical steroids (triamcinolone 0.1% or clobetasol 0.05%).15,16 Unlike coral dermatitis, in seabather’s eruption the symptoms are immediate and the location of the eruption coincides with areas covered by the swimsuit.
The Diagnosis: Coral Dermatitis
At 3-week follow-up, the patient demonstrated remarkable improvement in the intensity and size of the erythematous cerebriform plaques following daily application of triamcinolone acetonide cream 0.1% (Figure). The lesion disappeared after several months and did not recur. The delayed presentation of symptoms with a history of incidental coral contact during snorkeling most likely represents the type IV hypersensitivity reaction seen in the diagnosis of coral dermatitis, an extraordinarily rare form of contact dermatitis.1 Not all coral trigger skin reactions. Species of coral that contain nematocysts in their tentacles (aptly named stinging capsules) are responsible for the sting preceding coral dermatitis, as the nematocysts eject a coiled filament in response to human tactile stimulation that injects toxins into the epidermis.2
Acute, delayed, or chronic cutaneous changes follow envenomation. Acute responses arise immediately to a few hours after initial contact and are considered an irritant contact dermatitis.3 Local tissue histamine release and cascades of cytotoxic reactions often result in the characteristic urticarial or vesiculobullous plaques in addition to necrosis, piloerection, and localized lymphadenopathy.2-4 Although relatively uncommon, there may be rapid onset of systemic symptoms such as fever, malaise, hives, nausea, or emesis. Cardiopulmonary events, hepatotoxicity, renal failure, or anaphylaxis are rare.2 Histopathology of biopsy specimens reveals epidermal spongiosis with microvesicles and papillary dermal edema.1,5 In comparison, delayed reactions occur within days to weeks and exhibit epidermal parakeratosis, spongiosis, basal layer vacuolization, focal necrosis, lymphocyte exocytosis, and papillary dermal edema with extravasated erythrocytes.1,6 Clinically, it may present as linear rows of erythematous papules with burning and pruritus.6 Chronic reactions manifest after months as difficult-to-treat, persistent lichenoid dermatitis occasionally accompanied by granulomatous changes.1,2,4 Primary prevention measures after initial contact include an acetic acid rinse and cold compression to wash away residual nematocysts in the affected area.4,7,8 If a rash develops, topical steroids are the mainstay of treatment.3,8
In tandem with toxic nematocysts, the rigid calcified bodies of coral provide an additional self-defense mechanism against human contact.2,4 The irregular haphazard nature of coral may catch novice divers off guard and lead to laceration of a mispositioned limb, thereby increasing the risk for secondary infections due to the introduction of calcium carbonate and toxic mucinous deposits at the wound site, warranting antibiotic treatment.2,4,7 Because tropical locales are home to other natural dangers that inflict disease and mimic early signs of coral dermatitis, reaching an accurate diagnosis can be difficult, particularly for lower limb lesions. In summary, the diagnosis of coral dermatitis can be rendered based on morphology of the lesion and clinical context (exposure to corals and delayed symptoms) as well as response to topical steroids.
The differential diagnosis includes accidental trauma. Variations in impact force and patient skin integrity lead to a number of possible cutaneous manifestations seen in accidental trauma,9 which includes contusions resulting from burst capillaries underneath intact skin, abrasions due to the superficial epidermis scuffing away, and lacerations caused by enough force to rip and split the skin, leaving subcutaneous tissue between the intact tissue.9,10 Typically, the pattern of injury can provide hints to match what object or organism caused the wound.9 However, delayed response and worsening symptoms, as seen in coral dermatitis, would be unusual in accidental trauma unless it is complicated by secondary infection (infectious dermatitis), which does not respond to topical steroids and requires antibiotic treatment.
Another differential diagnosis includes cutaneous larva migrans, which infests domesticated and stray animals. For example, hookworm larvae propagate their eggs inside the intestines of their host before fecal-soil transmission in sandy locales.11 Unexpecting beachgoers travel barefoot on this contaminated soil, offering ample opportunity for the parasite to burrow into the upper dermis.11,12 The clinical presentation includes signs and symptoms of creeping eruption such as pruritic, linear, serpiginous tracks. Topical treatment with thiabendazole requires application 3 times daily for 15 days, which increases the risk for nonadherence, yet this therapy proves advantageous if a patient does not tolerate oral agents due to systemic adverse effects.11,12 Oral agents (eg, ivermectin, albendazole) offer improved adherence with a single dose11,13; the cure rate was higher with a single dose of ivermectin 12 mg vs a single dose of albendazole 400 mg.13 The current suggested treatment is ivermectin 200 μg/kg by mouth daily for 1 or 2 days.14
The incidence of seabather’s eruption (also known as chinkui dermatitis) is highest during the summer season and fluctuates between epidemic and nonepidemic years.15,16 It occurs sporadically worldwide mostly in tropical climates due to trapping of larvae spawn of sea animals such as crustaceans in swimwear. Initially, it presents as a pruritic and burning sensation after exiting the water, manifesting as a macular, papular, or maculopapular rash on areas covered by the swimsuit.15,16 The sensation is worse in areas that are tightly banded on the swimsuit, including the waistband and elastic straps.15 Commonly, the affected individual will seek relief via a shower, which intensifies the burning, especially if the swimsuit has not been removed. The contaminated swimwear should be immediately discarded, as the trapped sea larvae’s nematocysts activate with the pressure and friction of movement.15 Seabather’s eruption typically resolves spontaneously within a week, but symptom management can be achieved with topical steroids (triamcinolone 0.1% or clobetasol 0.05%).15,16 Unlike coral dermatitis, in seabather’s eruption the symptoms are immediate and the location of the eruption coincides with areas covered by the swimsuit.
- Ahn HS, Yoon SY, Park HJ, et al. A patient with delayed contact dermatitis to coral and she displayed superficial granuloma. Ann Dermatol. 2009;21:95-97. doi:10.5021/ad.2009.21.1.95
- Haddad V Jr, Lupi O, Lonza JP, et al. Tropical dermatology: marine and aquatic dermatology. J Am Acad Dermatol. 2009;61:733-752. doi:10.1016/j.jaad.2009.01.046
- Salik J, Tang R. Images in clinical medicine. Coral dermatitis. N Engl J Med. 2015;373:E2. doi:10.1056/NEJMicm1412907
- Reese E, Depenbrock P. Water envenomations and stings. Curr Sports Med Rep. 2014;13:126-131. doi:10.1249/JSR.0000000000000042
- Addy JH. Red sea coral contact dermatitis. Int J Dermatol. 1991; 30:271-273. doi:10.1111/j.1365-4362.1991.tb04636.x
- Miracco C, Lalinga AV, Sbano P, et al. Delayed skin reaction to Red Sea coral injury showing superficial granulomas and atypical CD30+ lymphocytes: report of a case. Br J Dermatol. 2001;145:849-851. doi:10.1046/j.1365-2133.2001.04454.x
- Ceponis PJ, Cable R, Weaver LK. Don’t kick the coral! Wilderness Environ Med. 2017;28:153-155. doi:10.1016/j.wem.2017.01.025
- Tlougan BE, Podjasek JO, Adams BB. Aquatic sports dematoses. part 2-in the water: saltwater dermatoses. Int J Dermatol. 2010;49:994-1002. doi:10.1111/j.1365-4632.2010.04476.x
- Simon LV, Lopez RA, King KC. Blunt force trauma. StatPearls [Internet]. StatPearls Publishing; 2023. Accessed January 12, 2034. https://www.ncbi.nlm.nih.gov/books/NBK470338/
- Gentile S, Kneubuehl BP, Barrera V, et al. Fracture energy threshold in parry injuries due to sharp and blunt force. Int J Legal Med. 2019;133:1429-1435.
- Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814. doi:10.1086/313787
- Davies HD, Sakuls P, Keystone JS. Creeping eruption. A review of clinical presentation and management of 60 cases presenting to a tropical disease unit. Arch Dermatol. 1993;129:588-591. doi:10.1001 /archderm.129.5.588
- Caumes E, Carriere J, Datry A, et al. A randomized trial of ivermectin versus albendazole for the treatment of cutaneous larva migrans. Am J Trop Med Hyg. 1993;49:641-644. doi:10.4269 /ajtmh.1993.49.641
- Schuster A, Lesshafft H, Reichert F, et al. Hookworm-related cutaneous larva migrans in northern Brazil: resolution of clinical pathology after a single dose of ivermectin. Clin Infect Dis. 2013;57:1155-1157. doi:10.1093/cid/cit440
- Freudenthal AR, Joseph PR. Seabather’s eruption. N Engl J Med. 1993;329:542-544. doi:10.1056/NEJM199308193290805
- Odagawa S, Watari T, Yoshida M. Chinkui dermatitis: the sea bather’s eruption. QJM. 2022;115:100-101. doi:10.1093/qjmed/hcab277
- Ahn HS, Yoon SY, Park HJ, et al. A patient with delayed contact dermatitis to coral and she displayed superficial granuloma. Ann Dermatol. 2009;21:95-97. doi:10.5021/ad.2009.21.1.95
- Haddad V Jr, Lupi O, Lonza JP, et al. Tropical dermatology: marine and aquatic dermatology. J Am Acad Dermatol. 2009;61:733-752. doi:10.1016/j.jaad.2009.01.046
- Salik J, Tang R. Images in clinical medicine. Coral dermatitis. N Engl J Med. 2015;373:E2. doi:10.1056/NEJMicm1412907
- Reese E, Depenbrock P. Water envenomations and stings. Curr Sports Med Rep. 2014;13:126-131. doi:10.1249/JSR.0000000000000042
- Addy JH. Red sea coral contact dermatitis. Int J Dermatol. 1991; 30:271-273. doi:10.1111/j.1365-4362.1991.tb04636.x
- Miracco C, Lalinga AV, Sbano P, et al. Delayed skin reaction to Red Sea coral injury showing superficial granulomas and atypical CD30+ lymphocytes: report of a case. Br J Dermatol. 2001;145:849-851. doi:10.1046/j.1365-2133.2001.04454.x
- Ceponis PJ, Cable R, Weaver LK. Don’t kick the coral! Wilderness Environ Med. 2017;28:153-155. doi:10.1016/j.wem.2017.01.025
- Tlougan BE, Podjasek JO, Adams BB. Aquatic sports dematoses. part 2-in the water: saltwater dermatoses. Int J Dermatol. 2010;49:994-1002. doi:10.1111/j.1365-4632.2010.04476.x
- Simon LV, Lopez RA, King KC. Blunt force trauma. StatPearls [Internet]. StatPearls Publishing; 2023. Accessed January 12, 2034. https://www.ncbi.nlm.nih.gov/books/NBK470338/
- Gentile S, Kneubuehl BP, Barrera V, et al. Fracture energy threshold in parry injuries due to sharp and blunt force. Int J Legal Med. 2019;133:1429-1435.
- Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814. doi:10.1086/313787
- Davies HD, Sakuls P, Keystone JS. Creeping eruption. A review of clinical presentation and management of 60 cases presenting to a tropical disease unit. Arch Dermatol. 1993;129:588-591. doi:10.1001 /archderm.129.5.588
- Caumes E, Carriere J, Datry A, et al. A randomized trial of ivermectin versus albendazole for the treatment of cutaneous larva migrans. Am J Trop Med Hyg. 1993;49:641-644. doi:10.4269 /ajtmh.1993.49.641
- Schuster A, Lesshafft H, Reichert F, et al. Hookworm-related cutaneous larva migrans in northern Brazil: resolution of clinical pathology after a single dose of ivermectin. Clin Infect Dis. 2013;57:1155-1157. doi:10.1093/cid/cit440
- Freudenthal AR, Joseph PR. Seabather’s eruption. N Engl J Med. 1993;329:542-544. doi:10.1056/NEJM199308193290805
- Odagawa S, Watari T, Yoshida M. Chinkui dermatitis: the sea bather’s eruption. QJM. 2022;115:100-101. doi:10.1093/qjmed/hcab277
A 48-year-old otherwise healthy man presented with a tender lesion on the dorsal aspect of the right foot with dysesthesia and progressive pruritus that he originally noticed 9 days prior after snorkeling in the Caribbean. He recalled kicking what he assumed was a rock while swimming. Initially there was negligible discomfort; however, on day 7 the symptoms started to worsen and the lesion started to swell. Application of a gauze pad soaked in hydrogen peroxide 3% failed to alleviate symptoms. Physical examination revealed a 4-cm region of well-demarcated, nonblanching, erythematous plaques in a lattice pattern accompanied by edematous and bullous changes. Triamcinolone acetonide cream 0.1% was prescribed.


More Side Effects With Local Therapies for Prostate Cancer
These were the findings of a retrospective cohort study in JAMA Network Open.
The standard treatment of advanced prostate cancer is androgen deprivation therapy (ADT). “The role of local therapy has been debated for several years. Studies have shown that radiation therapy or radical prostatectomy can improve patient survival under certain conditions,” said Hubert Kübler, MD, director of the Clinic and Polyclinic for Urology and Pediatric Urology at the University Hospital Würzburg in Germany. “At academic centers, a local therapy is pursued for oligometastatic patients if they are fit enough.”
The hope is to spare patients the side effects of ADT over an extended period and thus improve their quality of life. “But what impact does local therapy itself have on the men’s quality of life, especially considering that the survival advantage gained may be relatively small?” wrote study author Saira Khan, PhD, MPH, assistant professor of surgery at Washington University School of Medicine in St. Louis, Missouri, and her colleagues.
Examining Side Effects
This question has not been thoroughly examined yet. “To our knowledge, this is one of the first studies investigating the side effects of local therapy in men with advanced prostate cancer for up to 5 years after treatment,” wrote the authors.
The cohort study included 5500 US veterans who were diagnosed with advanced prostate cancer between January 1999 and December 2013. The tumors were in stage T4 (tumor is fixed or has spread to adjacent structures), with regional lymph node metastases (N1), and partially detectable distant metastases (M1).
The average age was 68.7 years, and 31% received local therapy (eg, radiation therapy, radical prostatectomy, or both), and 69% received systemic therapy (eg, hormone therapy, chemotherapy, or both).
Types of Local Therapy
Combining radiation therapy and radical prostatectomy “diminishes the meaningfulness of the study results,” according to Dr. Kübler. “The issue should have been analyzed in much finer detail. Studies clearly show, for example, that radiation therapy consistently performs slightly worse than prostatectomy in terms of gastrointestinal complaints.”
In their paper, the researchers reported that the prevalence of side effects was high, regardless of the therapy. Overall, 916 men (75.2%) with initial local therapy and 897 men (67.1%) with initial systemic therapy reported experiencing at least one side effect lasting more than 2 years and up to 5 years.
In the first year after the initial therapy, men who underwent local therapy, compared with those who underwent systemic therapy, experienced more of the following symptoms:
- Gastrointestinal issues (odds ratio [OR], 4.08)
- Pain (OR, 1.57)
- Sexual dysfunction (OR, 2.96)
- Urinary problems, predominantly incontinence (OR, 2.25)
Lasting Side Effects
Even up to year 5 after the initial therapy, men with local therapy reported more gastrointestinal and sexual issues, as well as more frequent incontinence, than those with systemic therapy. Only the frequency of pain equalized between the two groups in the second year.
“Our results are consistent with the known side effect profile [of local therapy] in patients with clinically localized prostate cancer receiving surgery or radiation therapy instead of active surveillance,” wrote the authors.
The comparison in advanced prostate cancer, however, is not with active surveillance but with ADT. “As the study confirmed, ADT is associated with various side effects,” said Dr. Kübler. Nevertheless, it was associated with fewer side effects than local therapy in this study. The concept behind local therapy (improving prognosis while avoiding local problems) is challenging to reconcile with these results.
Contradictory Data
The results also contradict findings from other studies. Dr. Kübler pointed to the recently presented PEACE-1 study, where “local complications and issues were reduced through local therapy in high-volume and high-risk patients.”
The study did not consider subsequent interventions, such as how many patients needed transurethral manipulation in the later course of the disease to address local problems. “There are older data showing that a radical prostatectomy can reduce the need for further resections,” Dr. Kübler added.
“I find it difficult to reconcile these data with other data and with my personal experience,” said Dr. Kübler. However, he agreed with the study authors’ conclusion, emphasizing the importance of informing patients about expected side effects of local therapy in the context of potentially marginal improvements in survival.
Different Situation in Germany
“As practitioners, we sometimes underestimate the side effects we subject our patients to. We need to talk to our patients about the prognosis improvement that comes with side effects,” said Dr. Kübler. He added that a similar study in Germany might yield different results. “Dr. Khan and her colleagues examined a very specific patient population: Namely, veterans. This patient clientele often faces many social difficulties, and the treatment structure in US veterans’ care differs significantly from ours.”
This article was translated from the Medscape German edition. A version of this article appeared on Medscape.com.
These were the findings of a retrospective cohort study in JAMA Network Open.
The standard treatment of advanced prostate cancer is androgen deprivation therapy (ADT). “The role of local therapy has been debated for several years. Studies have shown that radiation therapy or radical prostatectomy can improve patient survival under certain conditions,” said Hubert Kübler, MD, director of the Clinic and Polyclinic for Urology and Pediatric Urology at the University Hospital Würzburg in Germany. “At academic centers, a local therapy is pursued for oligometastatic patients if they are fit enough.”
The hope is to spare patients the side effects of ADT over an extended period and thus improve their quality of life. “But what impact does local therapy itself have on the men’s quality of life, especially considering that the survival advantage gained may be relatively small?” wrote study author Saira Khan, PhD, MPH, assistant professor of surgery at Washington University School of Medicine in St. Louis, Missouri, and her colleagues.
Examining Side Effects
This question has not been thoroughly examined yet. “To our knowledge, this is one of the first studies investigating the side effects of local therapy in men with advanced prostate cancer for up to 5 years after treatment,” wrote the authors.
The cohort study included 5500 US veterans who were diagnosed with advanced prostate cancer between January 1999 and December 2013. The tumors were in stage T4 (tumor is fixed or has spread to adjacent structures), with regional lymph node metastases (N1), and partially detectable distant metastases (M1).
The average age was 68.7 years, and 31% received local therapy (eg, radiation therapy, radical prostatectomy, or both), and 69% received systemic therapy (eg, hormone therapy, chemotherapy, or both).
Types of Local Therapy
Combining radiation therapy and radical prostatectomy “diminishes the meaningfulness of the study results,” according to Dr. Kübler. “The issue should have been analyzed in much finer detail. Studies clearly show, for example, that radiation therapy consistently performs slightly worse than prostatectomy in terms of gastrointestinal complaints.”
In their paper, the researchers reported that the prevalence of side effects was high, regardless of the therapy. Overall, 916 men (75.2%) with initial local therapy and 897 men (67.1%) with initial systemic therapy reported experiencing at least one side effect lasting more than 2 years and up to 5 years.
In the first year after the initial therapy, men who underwent local therapy, compared with those who underwent systemic therapy, experienced more of the following symptoms:
- Gastrointestinal issues (odds ratio [OR], 4.08)
- Pain (OR, 1.57)
- Sexual dysfunction (OR, 2.96)
- Urinary problems, predominantly incontinence (OR, 2.25)
Lasting Side Effects
Even up to year 5 after the initial therapy, men with local therapy reported more gastrointestinal and sexual issues, as well as more frequent incontinence, than those with systemic therapy. Only the frequency of pain equalized between the two groups in the second year.
“Our results are consistent with the known side effect profile [of local therapy] in patients with clinically localized prostate cancer receiving surgery or radiation therapy instead of active surveillance,” wrote the authors.
The comparison in advanced prostate cancer, however, is not with active surveillance but with ADT. “As the study confirmed, ADT is associated with various side effects,” said Dr. Kübler. Nevertheless, it was associated with fewer side effects than local therapy in this study. The concept behind local therapy (improving prognosis while avoiding local problems) is challenging to reconcile with these results.
Contradictory Data
The results also contradict findings from other studies. Dr. Kübler pointed to the recently presented PEACE-1 study, where “local complications and issues were reduced through local therapy in high-volume and high-risk patients.”
The study did not consider subsequent interventions, such as how many patients needed transurethral manipulation in the later course of the disease to address local problems. “There are older data showing that a radical prostatectomy can reduce the need for further resections,” Dr. Kübler added.
“I find it difficult to reconcile these data with other data and with my personal experience,” said Dr. Kübler. However, he agreed with the study authors’ conclusion, emphasizing the importance of informing patients about expected side effects of local therapy in the context of potentially marginal improvements in survival.
Different Situation in Germany
“As practitioners, we sometimes underestimate the side effects we subject our patients to. We need to talk to our patients about the prognosis improvement that comes with side effects,” said Dr. Kübler. He added that a similar study in Germany might yield different results. “Dr. Khan and her colleagues examined a very specific patient population: Namely, veterans. This patient clientele often faces many social difficulties, and the treatment structure in US veterans’ care differs significantly from ours.”
This article was translated from the Medscape German edition. A version of this article appeared on Medscape.com.
These were the findings of a retrospective cohort study in JAMA Network Open.
The standard treatment of advanced prostate cancer is androgen deprivation therapy (ADT). “The role of local therapy has been debated for several years. Studies have shown that radiation therapy or radical prostatectomy can improve patient survival under certain conditions,” said Hubert Kübler, MD, director of the Clinic and Polyclinic for Urology and Pediatric Urology at the University Hospital Würzburg in Germany. “At academic centers, a local therapy is pursued for oligometastatic patients if they are fit enough.”
The hope is to spare patients the side effects of ADT over an extended period and thus improve their quality of life. “But what impact does local therapy itself have on the men’s quality of life, especially considering that the survival advantage gained may be relatively small?” wrote study author Saira Khan, PhD, MPH, assistant professor of surgery at Washington University School of Medicine in St. Louis, Missouri, and her colleagues.
Examining Side Effects
This question has not been thoroughly examined yet. “To our knowledge, this is one of the first studies investigating the side effects of local therapy in men with advanced prostate cancer for up to 5 years after treatment,” wrote the authors.
The cohort study included 5500 US veterans who were diagnosed with advanced prostate cancer between January 1999 and December 2013. The tumors were in stage T4 (tumor is fixed or has spread to adjacent structures), with regional lymph node metastases (N1), and partially detectable distant metastases (M1).
The average age was 68.7 years, and 31% received local therapy (eg, radiation therapy, radical prostatectomy, or both), and 69% received systemic therapy (eg, hormone therapy, chemotherapy, or both).
Types of Local Therapy
Combining radiation therapy and radical prostatectomy “diminishes the meaningfulness of the study results,” according to Dr. Kübler. “The issue should have been analyzed in much finer detail. Studies clearly show, for example, that radiation therapy consistently performs slightly worse than prostatectomy in terms of gastrointestinal complaints.”
In their paper, the researchers reported that the prevalence of side effects was high, regardless of the therapy. Overall, 916 men (75.2%) with initial local therapy and 897 men (67.1%) with initial systemic therapy reported experiencing at least one side effect lasting more than 2 years and up to 5 years.
In the first year after the initial therapy, men who underwent local therapy, compared with those who underwent systemic therapy, experienced more of the following symptoms:
- Gastrointestinal issues (odds ratio [OR], 4.08)
- Pain (OR, 1.57)
- Sexual dysfunction (OR, 2.96)
- Urinary problems, predominantly incontinence (OR, 2.25)
Lasting Side Effects
Even up to year 5 after the initial therapy, men with local therapy reported more gastrointestinal and sexual issues, as well as more frequent incontinence, than those with systemic therapy. Only the frequency of pain equalized between the two groups in the second year.
“Our results are consistent with the known side effect profile [of local therapy] in patients with clinically localized prostate cancer receiving surgery or radiation therapy instead of active surveillance,” wrote the authors.
The comparison in advanced prostate cancer, however, is not with active surveillance but with ADT. “As the study confirmed, ADT is associated with various side effects,” said Dr. Kübler. Nevertheless, it was associated with fewer side effects than local therapy in this study. The concept behind local therapy (improving prognosis while avoiding local problems) is challenging to reconcile with these results.
Contradictory Data
The results also contradict findings from other studies. Dr. Kübler pointed to the recently presented PEACE-1 study, where “local complications and issues were reduced through local therapy in high-volume and high-risk patients.”
The study did not consider subsequent interventions, such as how many patients needed transurethral manipulation in the later course of the disease to address local problems. “There are older data showing that a radical prostatectomy can reduce the need for further resections,” Dr. Kübler added.
“I find it difficult to reconcile these data with other data and with my personal experience,” said Dr. Kübler. However, he agreed with the study authors’ conclusion, emphasizing the importance of informing patients about expected side effects of local therapy in the context of potentially marginal improvements in survival.
Different Situation in Germany
“As practitioners, we sometimes underestimate the side effects we subject our patients to. We need to talk to our patients about the prognosis improvement that comes with side effects,” said Dr. Kübler. He added that a similar study in Germany might yield different results. “Dr. Khan and her colleagues examined a very specific patient population: Namely, veterans. This patient clientele often faces many social difficulties, and the treatment structure in US veterans’ care differs significantly from ours.”
This article was translated from the Medscape German edition. A version of this article appeared on Medscape.com.
FROM JAMA NETWORK OPEN
Study Evaluates Aesthetic Concerns in Asian Women
CHICAGO — presented at the annual meeting of the American Society for Dermatologic Surgery (ASDS),
“Asian Americans represent the fastest-expanding racial group in the US, underscoring the need for a comprehensive understanding of their specific aesthetic goals and needs,” said Annie Chiu, MD, a cosmetic dermatologist in Redondo Beach, California, who presented the results at the meeting. In an interview, she noted that most of this type of research has been done in White or Eurocentric populations, “so we really wanted to identify the top aesthetic concerns for Asian women as they tend to be different.”
In the study, an online survey was administered to aesthetically-inclined adults — defined as those who care about improving their appearance and are willing to go to a professional to do so — across different demographic groups in the United States. Respondents were surveyed about 41 facial and 31 body characteristics, identifying those they have and find bothersome. Maximum difference scaling was used to generate their most and least bothersome characteristics in each respective category.
Of the 3,974 women surveyed, 652 self-identified as female and Asian. The majority of Asian female respondents self-reported a Fitzpatrick skin type IV (East/Southeast [SE]Asian: 58%; Indian/Central or Southwest [CSW] Asian: 24%) or type V (East/SE Asian: 59%; Indian/CSW Asian: 30%). The findings reported at the meeting are specific to aesthetic concerns in Asian women.
Among the Asian female participants, the top three facial concerns were uneven skin color (40%), dull/dry skin (35%), and hair loss/thinning (34%). Top facial concerns in younger patients (under 30 years) were related to skin quality, such as dull skin (54%), acne scarring (51%), and large pore size (51%), whereas the most common concerns among those older than 57 years were related to under-eye bags or sagginess (60%), uneven skin tone (55%), and hair loss/thinning (47%).
Of note, acne scarring was noted as a top concern by the Indian/CSW Asian cohort. While the lines between the brows and skin sagging were top concerns in White female participants in the overall study, these concerns were not nearly as high among Asian female participants.
For all Asian female participants, the top body concerns were related to stubborn body fat in the stomach, sides, bra or back area, and arms. Stubborn body fat in the stomach area was the most frequent concern across generations (41% to 64%). East/SE Asian participants were more interested in receiving cosmetic treatments (91%) than the Indian/CSW Asian group (47%).
“Given that injectable treatments of neuromodulators and fillers are often what we focus aesthetic treatments around and thus, what we often center cosmetic consults on, it is important to remember to customize patient consultations and address specific needs with cultural sensitivity,” Dr. Chiu said. “We may not be properly recognizing and prioritizing patient discussion around concerns of dyspigmentation, skin quality, or hair thinning,” she continued, adding: “Ultimately as experts, it’s important we use this data along with what we know about structural and cutaneous differences in patients of different cultural backgrounds to optimize and prioritize treatments.”
Allergan Aesthetics, an AbbVie company, funded the study and participated in the trial design, research, analysis, data collection, interpretation of data, and the review. Dr. Chiu is a consultant, advisory board member, and investigator for Allergan, AbbVie,and Merz; and is a consultant and advisory board member for Galderma, Evolus, and Sofwave. Other authors disclosed ties with Allergan, Merz Aesthetics, Prollenium, Revance, Galderma, Alastin, Glo Pharma, and Teoxane. Two authors are AbbVie employees.
CHICAGO — presented at the annual meeting of the American Society for Dermatologic Surgery (ASDS),
“Asian Americans represent the fastest-expanding racial group in the US, underscoring the need for a comprehensive understanding of their specific aesthetic goals and needs,” said Annie Chiu, MD, a cosmetic dermatologist in Redondo Beach, California, who presented the results at the meeting. In an interview, she noted that most of this type of research has been done in White or Eurocentric populations, “so we really wanted to identify the top aesthetic concerns for Asian women as they tend to be different.”
In the study, an online survey was administered to aesthetically-inclined adults — defined as those who care about improving their appearance and are willing to go to a professional to do so — across different demographic groups in the United States. Respondents were surveyed about 41 facial and 31 body characteristics, identifying those they have and find bothersome. Maximum difference scaling was used to generate their most and least bothersome characteristics in each respective category.
Of the 3,974 women surveyed, 652 self-identified as female and Asian. The majority of Asian female respondents self-reported a Fitzpatrick skin type IV (East/Southeast [SE]Asian: 58%; Indian/Central or Southwest [CSW] Asian: 24%) or type V (East/SE Asian: 59%; Indian/CSW Asian: 30%). The findings reported at the meeting are specific to aesthetic concerns in Asian women.
Among the Asian female participants, the top three facial concerns were uneven skin color (40%), dull/dry skin (35%), and hair loss/thinning (34%). Top facial concerns in younger patients (under 30 years) were related to skin quality, such as dull skin (54%), acne scarring (51%), and large pore size (51%), whereas the most common concerns among those older than 57 years were related to under-eye bags or sagginess (60%), uneven skin tone (55%), and hair loss/thinning (47%).
Of note, acne scarring was noted as a top concern by the Indian/CSW Asian cohort. While the lines between the brows and skin sagging were top concerns in White female participants in the overall study, these concerns were not nearly as high among Asian female participants.
For all Asian female participants, the top body concerns were related to stubborn body fat in the stomach, sides, bra or back area, and arms. Stubborn body fat in the stomach area was the most frequent concern across generations (41% to 64%). East/SE Asian participants were more interested in receiving cosmetic treatments (91%) than the Indian/CSW Asian group (47%).
“Given that injectable treatments of neuromodulators and fillers are often what we focus aesthetic treatments around and thus, what we often center cosmetic consults on, it is important to remember to customize patient consultations and address specific needs with cultural sensitivity,” Dr. Chiu said. “We may not be properly recognizing and prioritizing patient discussion around concerns of dyspigmentation, skin quality, or hair thinning,” she continued, adding: “Ultimately as experts, it’s important we use this data along with what we know about structural and cutaneous differences in patients of different cultural backgrounds to optimize and prioritize treatments.”
Allergan Aesthetics, an AbbVie company, funded the study and participated in the trial design, research, analysis, data collection, interpretation of data, and the review. Dr. Chiu is a consultant, advisory board member, and investigator for Allergan, AbbVie,and Merz; and is a consultant and advisory board member for Galderma, Evolus, and Sofwave. Other authors disclosed ties with Allergan, Merz Aesthetics, Prollenium, Revance, Galderma, Alastin, Glo Pharma, and Teoxane. Two authors are AbbVie employees.
CHICAGO — presented at the annual meeting of the American Society for Dermatologic Surgery (ASDS),
“Asian Americans represent the fastest-expanding racial group in the US, underscoring the need for a comprehensive understanding of their specific aesthetic goals and needs,” said Annie Chiu, MD, a cosmetic dermatologist in Redondo Beach, California, who presented the results at the meeting. In an interview, she noted that most of this type of research has been done in White or Eurocentric populations, “so we really wanted to identify the top aesthetic concerns for Asian women as they tend to be different.”
In the study, an online survey was administered to aesthetically-inclined adults — defined as those who care about improving their appearance and are willing to go to a professional to do so — across different demographic groups in the United States. Respondents were surveyed about 41 facial and 31 body characteristics, identifying those they have and find bothersome. Maximum difference scaling was used to generate their most and least bothersome characteristics in each respective category.
Of the 3,974 women surveyed, 652 self-identified as female and Asian. The majority of Asian female respondents self-reported a Fitzpatrick skin type IV (East/Southeast [SE]Asian: 58%; Indian/Central or Southwest [CSW] Asian: 24%) or type V (East/SE Asian: 59%; Indian/CSW Asian: 30%). The findings reported at the meeting are specific to aesthetic concerns in Asian women.
Among the Asian female participants, the top three facial concerns were uneven skin color (40%), dull/dry skin (35%), and hair loss/thinning (34%). Top facial concerns in younger patients (under 30 years) were related to skin quality, such as dull skin (54%), acne scarring (51%), and large pore size (51%), whereas the most common concerns among those older than 57 years were related to under-eye bags or sagginess (60%), uneven skin tone (55%), and hair loss/thinning (47%).
Of note, acne scarring was noted as a top concern by the Indian/CSW Asian cohort. While the lines between the brows and skin sagging were top concerns in White female participants in the overall study, these concerns were not nearly as high among Asian female participants.
For all Asian female participants, the top body concerns were related to stubborn body fat in the stomach, sides, bra or back area, and arms. Stubborn body fat in the stomach area was the most frequent concern across generations (41% to 64%). East/SE Asian participants were more interested in receiving cosmetic treatments (91%) than the Indian/CSW Asian group (47%).
“Given that injectable treatments of neuromodulators and fillers are often what we focus aesthetic treatments around and thus, what we often center cosmetic consults on, it is important to remember to customize patient consultations and address specific needs with cultural sensitivity,” Dr. Chiu said. “We may not be properly recognizing and prioritizing patient discussion around concerns of dyspigmentation, skin quality, or hair thinning,” she continued, adding: “Ultimately as experts, it’s important we use this data along with what we know about structural and cutaneous differences in patients of different cultural backgrounds to optimize and prioritize treatments.”
Allergan Aesthetics, an AbbVie company, funded the study and participated in the trial design, research, analysis, data collection, interpretation of data, and the review. Dr. Chiu is a consultant, advisory board member, and investigator for Allergan, AbbVie,and Merz; and is a consultant and advisory board member for Galderma, Evolus, and Sofwave. Other authors disclosed ties with Allergan, Merz Aesthetics, Prollenium, Revance, Galderma, Alastin, Glo Pharma, and Teoxane. Two authors are AbbVie employees.
AT ASDS 2023
Colorectal Cancer Risk Increasing Across Successive Birth Cohorts
Colorectal cancer (CRC) epidemiology is changing due to a birth cohort effect, also called birth cohort CRC — the observed phenomena of the rising risk for CRC across successive generations of people born in 1960 and later — according to a new narrative review.
Birth cohort CRC is associated with increasing rectal cancer (greater than colon cancer) diagnosis and distant-stage (greater than local-stage) CRC diagnosis, and a rising incidence of early-onset CRC (EOCRC), defined as occurring before age 50.
Recognizing this birth cohort effect could improve the understanding of CRC risk factors, etiology, mechanisms, as well as the public health consequences of rising rates.
“The changing epidemiology means that we need to redouble our efforts at optimizing early detection and prevention of colorectal cancer,” Samir Gupta, MD, the review’s lead author and professor of gastroenterology at the University of California, San Diego, California, told this news organization. Dr. Gupta serves as the co-lead for the cancer control program at Moores Cancer Center at UC San Diego Health.
This requires “being alert for potential red flag signs and symptoms of colorectal cancer, such as iron deficiency anemia and rectal bleeding, that are otherwise unexplained, including for those under age 45,” he said.
We also should make “sure that all people eligible for screening — at age 45 and older — have every opportunity to get screened for colorectal cancer,” Dr. Gupta added.
The review was published online in Clinical Gastroenterology and Hepatology.
Tracking Birth Cohort Trends
CRC rates have increased in the United States among people born since the early 1960s, the authors wrote.
Generation X (individuals born in 1965-1980) experienced an increase in EOCRC, and rates subsequently increased in this generation after age 50. Rates are 1.22-fold higher among people born in 1965-1969 and 1.58-fold higher among those born 1975-1979 than among people born in 1950-1954.
Now rates are also increasing across younger generations, particularly among Millennials (individuals born in 1981-1996) as they enter mid-adulthood. Incidence rates are 1.89-fold higher among people born in 1980-1984 and 2.98-fold higher among those born in 1990-1994 than among individuals born in 1950-1954.
These birth cohort effects are evident globally, despite differences in population age structures, screening programs, and diagnostic strategies around the world. Due to this ongoing trend, physicians anticipate that CRC rates will likely continue to increase as higher-risk birth cohorts become older, the authors wrote.
Notably, four important shifts in CRC incidence are apparent, they noted. First, rates are steadily increasing up to age 50 and plateauing after age 60. Rectal cancers are now predominant through ages 50-59. Rates of distant-stage disease have increased most rapidly among ages 30-49 and more slowly decreased among ages 60-79 compared with those of local-stage disease. In addition, the increasing rates of EOCRC have been observed across all racial and ethnic groups since the early 1990s.
These shifts led to major changes in the types of patients diagnosed with CRC now vs 30 years ago, with a higher proportion being patients younger than 60, as well as Black, Asian or Pacific Islander, American Indian/Alaska Native, and Hispanic patients.
The combination of age-related increases in CRC and birth cohort–related trends will likely lead to substantial increases in the number of people diagnosed with CRC in coming years, especially as Generation X patients move into their 50s and 60s, the authors wrote.
Research and Clinical Implications
Birth cohort CRC, including increasing EOCRC incidence, likely is driven by a range of influences, including demographic, lifestyle, early life, environmental, genetic, and somatic factors, as well as interactions among them, the authors noted. Examples within these broad categories include male sex, food insecurity, income inequality, diabetes, alcohol use, less healthy dietary patterns, in utero exposure to certain medications, and microbiome concerns such as early life antibiotic exposure or dysbiosis.
“From a research perspective, this means that we need to think about risk factors and mechanisms that are associated with birth cohorts, not just age at diagnosis,” Dr. Gupta said. “To date, most studies of changing epidemiology have not taken into account birth cohort, such as whether someone is Generation X or later versus pre-Baby Boomer.”
Although additional research is needed, the epidemiology changes have several immediate clinical implications, Dr. Gupta said. For those younger than 45, it is critical to raise awareness about the signs and symptoms of CRC, such as hematochezia, iron deficiency anemia, and unintentional weight loss, as well as family history.
For ages 45 and older, a major focus should be placed on increasing screening participation and follow-up after abnormal results, addressing disparities in screening participation, and optimizing screening quality.
In addition, as CRC incidence continues to increase, health systems and policymakers should ensure every patient has access to guideline-appropriate care and innovative clinical trials, the authors wrote. This access may be particularly important to address the increasing burden of rectal cancer, as treatment approaches rapidly evolve toward more effective therapies, such as neoadjuvant chemotherapy and radiation prior to surgery, and with less-morbid treatments on the horizon, they added.
‘An Interesting Concept’
“Birth cohort CRC is an interesting concept that allows people to think of their CRC risk according to their birth cohort in addition to age,” Shuji Ogino, MD, PhD, chief of the Molecular Pathological Epidemiology program at Brigham & Women’s Hospital, Boston, Massachusetts, told this news organization.
Dr. Ogino, who wasn’t involved with this study, serves as a member of the cancer immunology and cancer epidemiology programs at the Dana-Farber Harvard Cancer Center. In studies of EOCRC, he and colleagues have found various biogeographical and pathogenic trends across age groups.
“More research is needed to disentangle the complex etiologies of birth cohort CRC and early-onset CRC,” Dr. Ogino said. “Tumor cells and tissues have certain past and ongoing pathological marks, which we can detect to better understand birth cohort CRC and early-onset CRC.”
The study was funded by several National Institutes of Health/National Cancer Institute grants. Dr. Gupta disclosed consulting for Geneoscopy, Guardant Health, Universal Diagnostics, InterVenn Bio, and CellMax. Another author reported consulting for Freenome, Exact Sciences, Medtronic, and Geneoscopy. Dr. Ogino reported no relevant financial disclosures.
A version of this article appeared on Medscape.com .
Colorectal cancer (CRC) epidemiology is changing due to a birth cohort effect, also called birth cohort CRC — the observed phenomena of the rising risk for CRC across successive generations of people born in 1960 and later — according to a new narrative review.
Birth cohort CRC is associated with increasing rectal cancer (greater than colon cancer) diagnosis and distant-stage (greater than local-stage) CRC diagnosis, and a rising incidence of early-onset CRC (EOCRC), defined as occurring before age 50.
Recognizing this birth cohort effect could improve the understanding of CRC risk factors, etiology, mechanisms, as well as the public health consequences of rising rates.
“The changing epidemiology means that we need to redouble our efforts at optimizing early detection and prevention of colorectal cancer,” Samir Gupta, MD, the review’s lead author and professor of gastroenterology at the University of California, San Diego, California, told this news organization. Dr. Gupta serves as the co-lead for the cancer control program at Moores Cancer Center at UC San Diego Health.
This requires “being alert for potential red flag signs and symptoms of colorectal cancer, such as iron deficiency anemia and rectal bleeding, that are otherwise unexplained, including for those under age 45,” he said.
We also should make “sure that all people eligible for screening — at age 45 and older — have every opportunity to get screened for colorectal cancer,” Dr. Gupta added.
The review was published online in Clinical Gastroenterology and Hepatology.
Tracking Birth Cohort Trends
CRC rates have increased in the United States among people born since the early 1960s, the authors wrote.
Generation X (individuals born in 1965-1980) experienced an increase in EOCRC, and rates subsequently increased in this generation after age 50. Rates are 1.22-fold higher among people born in 1965-1969 and 1.58-fold higher among those born 1975-1979 than among people born in 1950-1954.
Now rates are also increasing across younger generations, particularly among Millennials (individuals born in 1981-1996) as they enter mid-adulthood. Incidence rates are 1.89-fold higher among people born in 1980-1984 and 2.98-fold higher among those born in 1990-1994 than among individuals born in 1950-1954.
These birth cohort effects are evident globally, despite differences in population age structures, screening programs, and diagnostic strategies around the world. Due to this ongoing trend, physicians anticipate that CRC rates will likely continue to increase as higher-risk birth cohorts become older, the authors wrote.
Notably, four important shifts in CRC incidence are apparent, they noted. First, rates are steadily increasing up to age 50 and plateauing after age 60. Rectal cancers are now predominant through ages 50-59. Rates of distant-stage disease have increased most rapidly among ages 30-49 and more slowly decreased among ages 60-79 compared with those of local-stage disease. In addition, the increasing rates of EOCRC have been observed across all racial and ethnic groups since the early 1990s.
These shifts led to major changes in the types of patients diagnosed with CRC now vs 30 years ago, with a higher proportion being patients younger than 60, as well as Black, Asian or Pacific Islander, American Indian/Alaska Native, and Hispanic patients.
The combination of age-related increases in CRC and birth cohort–related trends will likely lead to substantial increases in the number of people diagnosed with CRC in coming years, especially as Generation X patients move into their 50s and 60s, the authors wrote.
Research and Clinical Implications
Birth cohort CRC, including increasing EOCRC incidence, likely is driven by a range of influences, including demographic, lifestyle, early life, environmental, genetic, and somatic factors, as well as interactions among them, the authors noted. Examples within these broad categories include male sex, food insecurity, income inequality, diabetes, alcohol use, less healthy dietary patterns, in utero exposure to certain medications, and microbiome concerns such as early life antibiotic exposure or dysbiosis.
“From a research perspective, this means that we need to think about risk factors and mechanisms that are associated with birth cohorts, not just age at diagnosis,” Dr. Gupta said. “To date, most studies of changing epidemiology have not taken into account birth cohort, such as whether someone is Generation X or later versus pre-Baby Boomer.”
Although additional research is needed, the epidemiology changes have several immediate clinical implications, Dr. Gupta said. For those younger than 45, it is critical to raise awareness about the signs and symptoms of CRC, such as hematochezia, iron deficiency anemia, and unintentional weight loss, as well as family history.
For ages 45 and older, a major focus should be placed on increasing screening participation and follow-up after abnormal results, addressing disparities in screening participation, and optimizing screening quality.
In addition, as CRC incidence continues to increase, health systems and policymakers should ensure every patient has access to guideline-appropriate care and innovative clinical trials, the authors wrote. This access may be particularly important to address the increasing burden of rectal cancer, as treatment approaches rapidly evolve toward more effective therapies, such as neoadjuvant chemotherapy and radiation prior to surgery, and with less-morbid treatments on the horizon, they added.
‘An Interesting Concept’
“Birth cohort CRC is an interesting concept that allows people to think of their CRC risk according to their birth cohort in addition to age,” Shuji Ogino, MD, PhD, chief of the Molecular Pathological Epidemiology program at Brigham & Women’s Hospital, Boston, Massachusetts, told this news organization.
Dr. Ogino, who wasn’t involved with this study, serves as a member of the cancer immunology and cancer epidemiology programs at the Dana-Farber Harvard Cancer Center. In studies of EOCRC, he and colleagues have found various biogeographical and pathogenic trends across age groups.
“More research is needed to disentangle the complex etiologies of birth cohort CRC and early-onset CRC,” Dr. Ogino said. “Tumor cells and tissues have certain past and ongoing pathological marks, which we can detect to better understand birth cohort CRC and early-onset CRC.”
The study was funded by several National Institutes of Health/National Cancer Institute grants. Dr. Gupta disclosed consulting for Geneoscopy, Guardant Health, Universal Diagnostics, InterVenn Bio, and CellMax. Another author reported consulting for Freenome, Exact Sciences, Medtronic, and Geneoscopy. Dr. Ogino reported no relevant financial disclosures.
A version of this article appeared on Medscape.com .
Colorectal cancer (CRC) epidemiology is changing due to a birth cohort effect, also called birth cohort CRC — the observed phenomena of the rising risk for CRC across successive generations of people born in 1960 and later — according to a new narrative review.
Birth cohort CRC is associated with increasing rectal cancer (greater than colon cancer) diagnosis and distant-stage (greater than local-stage) CRC diagnosis, and a rising incidence of early-onset CRC (EOCRC), defined as occurring before age 50.
Recognizing this birth cohort effect could improve the understanding of CRC risk factors, etiology, mechanisms, as well as the public health consequences of rising rates.
“The changing epidemiology means that we need to redouble our efforts at optimizing early detection and prevention of colorectal cancer,” Samir Gupta, MD, the review’s lead author and professor of gastroenterology at the University of California, San Diego, California, told this news organization. Dr. Gupta serves as the co-lead for the cancer control program at Moores Cancer Center at UC San Diego Health.
This requires “being alert for potential red flag signs and symptoms of colorectal cancer, such as iron deficiency anemia and rectal bleeding, that are otherwise unexplained, including for those under age 45,” he said.
We also should make “sure that all people eligible for screening — at age 45 and older — have every opportunity to get screened for colorectal cancer,” Dr. Gupta added.
The review was published online in Clinical Gastroenterology and Hepatology.
Tracking Birth Cohort Trends
CRC rates have increased in the United States among people born since the early 1960s, the authors wrote.
Generation X (individuals born in 1965-1980) experienced an increase in EOCRC, and rates subsequently increased in this generation after age 50. Rates are 1.22-fold higher among people born in 1965-1969 and 1.58-fold higher among those born 1975-1979 than among people born in 1950-1954.
Now rates are also increasing across younger generations, particularly among Millennials (individuals born in 1981-1996) as they enter mid-adulthood. Incidence rates are 1.89-fold higher among people born in 1980-1984 and 2.98-fold higher among those born in 1990-1994 than among individuals born in 1950-1954.
These birth cohort effects are evident globally, despite differences in population age structures, screening programs, and diagnostic strategies around the world. Due to this ongoing trend, physicians anticipate that CRC rates will likely continue to increase as higher-risk birth cohorts become older, the authors wrote.
Notably, four important shifts in CRC incidence are apparent, they noted. First, rates are steadily increasing up to age 50 and plateauing after age 60. Rectal cancers are now predominant through ages 50-59. Rates of distant-stage disease have increased most rapidly among ages 30-49 and more slowly decreased among ages 60-79 compared with those of local-stage disease. In addition, the increasing rates of EOCRC have been observed across all racial and ethnic groups since the early 1990s.
These shifts led to major changes in the types of patients diagnosed with CRC now vs 30 years ago, with a higher proportion being patients younger than 60, as well as Black, Asian or Pacific Islander, American Indian/Alaska Native, and Hispanic patients.
The combination of age-related increases in CRC and birth cohort–related trends will likely lead to substantial increases in the number of people diagnosed with CRC in coming years, especially as Generation X patients move into their 50s and 60s, the authors wrote.
Research and Clinical Implications
Birth cohort CRC, including increasing EOCRC incidence, likely is driven by a range of influences, including demographic, lifestyle, early life, environmental, genetic, and somatic factors, as well as interactions among them, the authors noted. Examples within these broad categories include male sex, food insecurity, income inequality, diabetes, alcohol use, less healthy dietary patterns, in utero exposure to certain medications, and microbiome concerns such as early life antibiotic exposure or dysbiosis.
“From a research perspective, this means that we need to think about risk factors and mechanisms that are associated with birth cohorts, not just age at diagnosis,” Dr. Gupta said. “To date, most studies of changing epidemiology have not taken into account birth cohort, such as whether someone is Generation X or later versus pre-Baby Boomer.”
Although additional research is needed, the epidemiology changes have several immediate clinical implications, Dr. Gupta said. For those younger than 45, it is critical to raise awareness about the signs and symptoms of CRC, such as hematochezia, iron deficiency anemia, and unintentional weight loss, as well as family history.
For ages 45 and older, a major focus should be placed on increasing screening participation and follow-up after abnormal results, addressing disparities in screening participation, and optimizing screening quality.
In addition, as CRC incidence continues to increase, health systems and policymakers should ensure every patient has access to guideline-appropriate care and innovative clinical trials, the authors wrote. This access may be particularly important to address the increasing burden of rectal cancer, as treatment approaches rapidly evolve toward more effective therapies, such as neoadjuvant chemotherapy and radiation prior to surgery, and with less-morbid treatments on the horizon, they added.
‘An Interesting Concept’
“Birth cohort CRC is an interesting concept that allows people to think of their CRC risk according to their birth cohort in addition to age,” Shuji Ogino, MD, PhD, chief of the Molecular Pathological Epidemiology program at Brigham & Women’s Hospital, Boston, Massachusetts, told this news organization.
Dr. Ogino, who wasn’t involved with this study, serves as a member of the cancer immunology and cancer epidemiology programs at the Dana-Farber Harvard Cancer Center. In studies of EOCRC, he and colleagues have found various biogeographical and pathogenic trends across age groups.
“More research is needed to disentangle the complex etiologies of birth cohort CRC and early-onset CRC,” Dr. Ogino said. “Tumor cells and tissues have certain past and ongoing pathological marks, which we can detect to better understand birth cohort CRC and early-onset CRC.”
The study was funded by several National Institutes of Health/National Cancer Institute grants. Dr. Gupta disclosed consulting for Geneoscopy, Guardant Health, Universal Diagnostics, InterVenn Bio, and CellMax. Another author reported consulting for Freenome, Exact Sciences, Medtronic, and Geneoscopy. Dr. Ogino reported no relevant financial disclosures.
A version of this article appeared on Medscape.com .
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Annular Erythematous Plaques on the Back
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
.")
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
.")
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
.")
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
.")
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
An 84-year-old man presented to the clinic for evaluation of a pruritic rash on the back of 6 months’ duration that spread to the neck and chest over the past 2 months and then to the abdomen and thighs more recently. His primary care provider prescribed a 1-week course of oral steroids and steroid cream. The oral medication did not help, but the cream alleviated the pruritus. He had a medical history of coronary artery disease, hypertension, and diabetes mellitus. He also had a rash on the forearms that had waxed and waned for many years but was not associated with pruritus. He had not sought medical care for the rash and had never treated it. Physical examination revealed pink to violaceous annular plaques with central clearing and raised borders that coalesced into larger plaques on the trunk (top). Dusky, scaly, pink plaques were present on the dorsal forearms. Three punch biopsies—2 from the upper back (bottom) and 1 from the left forearm—all demonstrated consistent findings.


New Guidelines: Start PSA Screening Earlier in Black Men
Lowering the recommended age for baseline prostate-specific antigen (PSA) would reduce prostate cancer deaths by about 30% in Black men without significantly increasing the rate of overdiagnosis, according to new screening guidelines from the Prostate Cancer Foundation.
Specifically, baseline PSA testing in Black men should begin at age 40-45, sooner than current guidelines recommend, and should be followed by regular screening intervals, preferably annually, at least until age 70, a multidisciplinary panel of experts and patient advocates determined based on a comprehensive literature review.
The panel’s findings were presented in a poster at the ASCO Genitourinary Symposium.
“Black men in the United States are considered a high-risk population for being diagnosed with and dying from prostate cancer,” lead author Isla Garraway, MD, PhD, of the University of California, Los Angeles, and colleagues wrote. Specifically, Black men are about two times more likely to be diagnosed with and die from prostate cancer than White men. But, the authors continued, “few guidelines have outlined specific recommendations for PSA-based prostate cancer screening among Black men.”
The US Preventive Services Taskforce recommendations, which are currently being updated, set the PSA screening start age at 55. The task force recommendations, which dictate insurance coverage in the United States, acknowledged “a potential mortality benefit for African American men when beginning screening before age 55 years” but did not explicitly recommend screening earlier.
Current guidelines from the American Cancer Society call for discussions about screening in average-risk men to begin at age 50-55. The recommendations do specify lowering the age to 45 for those at a high risk for prostate cancer, which includes Black men as well as those with a first-degree relative diagnosed with prostate cancer before age 65. In some cases, screening can begin at age 40 in the highest risk men — those with more than one first-degree relative who had prostate cancer at a young age.
The Prostate Cancer Foundation “wanted to address the confusion around different guideline statements and the lack of clarity around screening recommendations for Black men,” said William K. Oh, MD, of The Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York City, who chaired the panel for the new guidelines. “We thus convened a distinguished panel of experts from diverse backgrounds and expertise to create six guidelines statements to help Black men, their families, and their healthcare providers to consider options for prostate cancer screening based on the best available evidence.”
After reviewing 287, the expert panel developed six new guideline statements, reaching at least 80% consensus among panel members, addressing screening for Black men:
Because Black men are at a high risk for prostate cancer, the benefits of screening generally outweigh the risks.
PSA testing should be considered first line for prostate cancer screening, although some providers may recommend an optional digital rectal exam in addition to the PSA test.
Black men should engage in shared decision-making with their healthcare providers and other trusted sources of information to learn about the pros and cons of screening.
For Black men who elect screening, a baseline PSA test should be done between ages 40 and 45, and annual PSA screening should be strongly considered based on the PSA value and the individual’s health status.
Black men over age 70 who have been undergoing prostate cancer screening should talk with their healthcare provider about whether to continue PSA testing and make an informed decision based on their age, life expectancy, health status, family history, and prior PSA levels.
Black men who are at even higher risk due to a strong family history and/or known carriers of high-risk genetic variants should consider initiating annual PSA screening as early as age 40.
These statements are based on “the best available evidence, which overwhelmingly supports the conclusion that Black men in the US could benefit from a risk-adapted PSA screening,” the investigators concluded, noting that the latest evidence “warrants revisiting current recommendations for early [prostate cancer] detection in Black men from other national guideline groups.”
“We believe that the outcome of these more directed guidelines will be to give clarity to these men,” Dr. Oh added.
This research was funded by the Prostate Cancer Foundation, National Cancer Institute, Veterans Affairs, Jean Perkins Foundation, and Department of Defense. Dr. Garraway reported having no disclosures.
{kind=link}
A version of this article appeared on Medscape.com.
Lowering the recommended age for baseline prostate-specific antigen (PSA) would reduce prostate cancer deaths by about 30% in Black men without significantly increasing the rate of overdiagnosis, according to new screening guidelines from the Prostate Cancer Foundation.
Specifically, baseline PSA testing in Black men should begin at age 40-45, sooner than current guidelines recommend, and should be followed by regular screening intervals, preferably annually, at least until age 70, a multidisciplinary panel of experts and patient advocates determined based on a comprehensive literature review.
The panel’s findings were presented in a poster at the ASCO Genitourinary Symposium.
“Black men in the United States are considered a high-risk population for being diagnosed with and dying from prostate cancer,” lead author Isla Garraway, MD, PhD, of the University of California, Los Angeles, and colleagues wrote. Specifically, Black men are about two times more likely to be diagnosed with and die from prostate cancer than White men. But, the authors continued, “few guidelines have outlined specific recommendations for PSA-based prostate cancer screening among Black men.”
The US Preventive Services Taskforce recommendations, which are currently being updated, set the PSA screening start age at 55. The task force recommendations, which dictate insurance coverage in the United States, acknowledged “a potential mortality benefit for African American men when beginning screening before age 55 years” but did not explicitly recommend screening earlier.
Current guidelines from the American Cancer Society call for discussions about screening in average-risk men to begin at age 50-55. The recommendations do specify lowering the age to 45 for those at a high risk for prostate cancer, which includes Black men as well as those with a first-degree relative diagnosed with prostate cancer before age 65. In some cases, screening can begin at age 40 in the highest risk men — those with more than one first-degree relative who had prostate cancer at a young age.
The Prostate Cancer Foundation “wanted to address the confusion around different guideline statements and the lack of clarity around screening recommendations for Black men,” said William K. Oh, MD, of The Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York City, who chaired the panel for the new guidelines. “We thus convened a distinguished panel of experts from diverse backgrounds and expertise to create six guidelines statements to help Black men, their families, and their healthcare providers to consider options for prostate cancer screening based on the best available evidence.”
After reviewing 287, the expert panel developed six new guideline statements, reaching at least 80% consensus among panel members, addressing screening for Black men:
Because Black men are at a high risk for prostate cancer, the benefits of screening generally outweigh the risks.
PSA testing should be considered first line for prostate cancer screening, although some providers may recommend an optional digital rectal exam in addition to the PSA test.
Black men should engage in shared decision-making with their healthcare providers and other trusted sources of information to learn about the pros and cons of screening.
For Black men who elect screening, a baseline PSA test should be done between ages 40 and 45, and annual PSA screening should be strongly considered based on the PSA value and the individual’s health status.
Black men over age 70 who have been undergoing prostate cancer screening should talk with their healthcare provider about whether to continue PSA testing and make an informed decision based on their age, life expectancy, health status, family history, and prior PSA levels.
Black men who are at even higher risk due to a strong family history and/or known carriers of high-risk genetic variants should consider initiating annual PSA screening as early as age 40.
These statements are based on “the best available evidence, which overwhelmingly supports the conclusion that Black men in the US could benefit from a risk-adapted PSA screening,” the investigators concluded, noting that the latest evidence “warrants revisiting current recommendations for early [prostate cancer] detection in Black men from other national guideline groups.”
“We believe that the outcome of these more directed guidelines will be to give clarity to these men,” Dr. Oh added.
This research was funded by the Prostate Cancer Foundation, National Cancer Institute, Veterans Affairs, Jean Perkins Foundation, and Department of Defense. Dr. Garraway reported having no disclosures.
A version of this article appeared on Medscape.com.
Lowering the recommended age for baseline prostate-specific antigen (PSA) would reduce prostate cancer deaths by about 30% in Black men without significantly increasing the rate of overdiagnosis, according to new screening guidelines from the Prostate Cancer Foundation.
Specifically, baseline PSA testing in Black men should begin at age 40-45, sooner than current guidelines recommend, and should be followed by regular screening intervals, preferably annually, at least until age 70, a multidisciplinary panel of experts and patient advocates determined based on a comprehensive literature review.
The panel’s findings were presented in a poster at the ASCO Genitourinary Symposium.
“Black men in the United States are considered a high-risk population for being diagnosed with and dying from prostate cancer,” lead author Isla Garraway, MD, PhD, of the University of California, Los Angeles, and colleagues wrote. Specifically, Black men are about two times more likely to be diagnosed with and die from prostate cancer than White men. But, the authors continued, “few guidelines have outlined specific recommendations for PSA-based prostate cancer screening among Black men.”
The US Preventive Services Taskforce recommendations, which are currently being updated, set the PSA screening start age at 55. The task force recommendations, which dictate insurance coverage in the United States, acknowledged “a potential mortality benefit for African American men when beginning screening before age 55 years” but did not explicitly recommend screening earlier.
Current guidelines from the American Cancer Society call for discussions about screening in average-risk men to begin at age 50-55. The recommendations do specify lowering the age to 45 for those at a high risk for prostate cancer, which includes Black men as well as those with a first-degree relative diagnosed with prostate cancer before age 65. In some cases, screening can begin at age 40 in the highest risk men — those with more than one first-degree relative who had prostate cancer at a young age.
The Prostate Cancer Foundation “wanted to address the confusion around different guideline statements and the lack of clarity around screening recommendations for Black men,” said William K. Oh, MD, of The Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York City, who chaired the panel for the new guidelines. “We thus convened a distinguished panel of experts from diverse backgrounds and expertise to create six guidelines statements to help Black men, their families, and their healthcare providers to consider options for prostate cancer screening based on the best available evidence.”
After reviewing 287, the expert panel developed six new guideline statements, reaching at least 80% consensus among panel members, addressing screening for Black men:
Because Black men are at a high risk for prostate cancer, the benefits of screening generally outweigh the risks.
PSA testing should be considered first line for prostate cancer screening, although some providers may recommend an optional digital rectal exam in addition to the PSA test.
Black men should engage in shared decision-making with their healthcare providers and other trusted sources of information to learn about the pros and cons of screening.
For Black men who elect screening, a baseline PSA test should be done between ages 40 and 45, and annual PSA screening should be strongly considered based on the PSA value and the individual’s health status.
Black men over age 70 who have been undergoing prostate cancer screening should talk with their healthcare provider about whether to continue PSA testing and make an informed decision based on their age, life expectancy, health status, family history, and prior PSA levels.
Black men who are at even higher risk due to a strong family history and/or known carriers of high-risk genetic variants should consider initiating annual PSA screening as early as age 40.
These statements are based on “the best available evidence, which overwhelmingly supports the conclusion that Black men in the US could benefit from a risk-adapted PSA screening,” the investigators concluded, noting that the latest evidence “warrants revisiting current recommendations for early [prostate cancer] detection in Black men from other national guideline groups.”
“We believe that the outcome of these more directed guidelines will be to give clarity to these men,” Dr. Oh added.
This research was funded by the Prostate Cancer Foundation, National Cancer Institute, Veterans Affairs, Jean Perkins Foundation, and Department of Defense. Dr. Garraway reported having no disclosures.
A version of this article appeared on Medscape.com.
FROM ASCO GU 2024